

Abstract
AMP-activated protein kinase (AMPK) is a molecular sensor that is critical for the maintenance of cellular energy homeostasis, disruption of which has been indicated in multiple neurodegenerative diseases including Alzheimer’s disease (AD). Mammalian AMPK is a heterotrimeric complex and its enzymatic α subunit exists in two isoforms: AMPKα1 and AMPKα2. Here we took advantage of a recently characterized non-human primate (NHP) model with sporadic AD-like neuropathology to explore potential relationships between AMPK signaling and AD-like neuropathology. Subjects were nine female vervet monkeys aged 19.5 to 23.4 years old. Subjects were classified into three groups, control lacking AD pathology (n=3), moderate AD pathology (n=3), and more severe AD Pathology (n=3). We found increased activity (assessed by phosphorylation) of AMPKα2 in hippocampi of NHP with AD-like neuropathology, compared to the subjects without AD pathology, with no alterations of AMPKα1 activity. Across all subjects, CSF Abeta42 was inversely associated with cerebral amyloid plaque density. Further, Aβ plaque burden is correlated with levels of either soluble or insoluble brain Aβ measurement. Unbiased mass spectrometry based proteomics studies combined with bioinformatics analysis revealed that many of the dysregulated proteins characteristic of AD neuropathology are associated with AMPK signaling. Our findings on the AMPK molecular signaling cascades provide further support for use of the NHP model to investigate new therapeutic strategies and development of novel biomarkers for Alzheimer’s disease.
Keywords: AMPK, non-human primate, Alzheimer’s disease, AMPK isoform, protein synthesis
Introduction
Despite decades of intensive research, the etiology of Alzheimer’s disease (AD) is still unclear and there is no effective cure for this devastating neurodegenerative disease. While many therapeutic strategies are effective in alleviating cognitive syndromes and other pathologies in various AD transgenic mouse models, translation of those findings from pre-clinical research to clinical practice has not been successful to date (Franco and Cedazo-Minguez, 2014; Güell-Bosch et al., 2016). Because of their many similarities to humans (e.g. in neuroanatomy, physiology, development, cognition), nonhuman primates (NHPs) are excellent models for study of mechanisms and therapies for various human diseases associated with aging including AD (Phillips et al., 2014; Van Dam and De Deyn, 2017). In a recent study of old versus middle-aged vervet/African green monkeys (Chlorocebus aethiops sabeus) we observed several features in multiple modalities similar to human AD. These include Aβ plaques in the older vervets, age and plaque density correlations with CSF Aβ42 and physical function observation, and correlations between greater plaque and paired helical filament (PHF)-tau burden and reduced volumes and cerebral metabolic rates of glucose utilization in several brain regions determined by MRI and PET (Latimer et al., 2019). These results strongly support use of the aged vervet monkeys to investigate disease mechanisms, novel therapeutic strategies and biomarkers for AD, particularly sporadic AD, which accounts for 95% of all AD cases (Alzheimer’s Assocition, 2020).
Disruption of energy metabolism is linked to cognitive impairment in AD and several other neurodegenerative diseases (Lin and Beal, 2006; Ma and Klann, 2012; Pedrós et al., 2014; Wang et al., 2019; Yaffe et al., 2004). At the cellular and molecular level, homeostasis of energy metabolism is critically controlled by the AMP-activated protein kinase (AMPK) (Hardie, 2014; Hardie and Carling, 1997). AMPK senses the ratio of AMP/ATP and accordingly regulates catabolic and anabolic processes to adjust the energy metabolism to equilibrium (Hardie, 2011; Hardie et al., 2012). Mammalian AMPK is a heterotrimeric complex composed of a catalytic/enzymatic subunit (α) and two regulatory subunits (β and γ), with multiple isoforms for each subunit (Hardie et al., 2012). The catalytic subunit has two isoforms: α1 and α2, and they are encoded by different genes (PRKAA1 and PRKAA2) that are located in chromosome 5 and 1, respectively (Ross et al., 2016). Both AMPKα1 and α2 isoforms are expressed in most tissues including brain to regulate cellular energy homeostasis. Binding of AMP with the regulatory subunit and phosphorylation of α subunit at the Thr172 site results in AMPK activation (Hardie et al., 2012). AMPK is connected to numerous signaling pathways involved in some fundamental cellular activities (Hardie, 2004; Hardie, 2014), one of which is protein synthesis (mRNA translation). Decades of neuroscience research has established that de novo protein synthesis is indispensable for memory formation and long-term synaptic plasticity (a cellular model for learning and memory) (Costa-Mattioli et al., 2009; Klann and Dever, 2004; Malenka and Bear, 2004; Richter and Klann, 2009). Consistently, impairments of protein synthesis are linked to AD pathophysiology (Beckelman et al., 2019; Ma and Klann, 2012; Ma et al., 2013). Previous studies, mostly in non-neuronal systems, indicate that activation of AMPK leads to inhibition of protein synthesis via two main mechanisms. First, AMPK inhibits the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, which controls cap-dependent mRNA translation initiation. Second, AMPK activation results in phosphorylation of the translational elongation factor eEF2 and consequent disruption of the translation elongation step (Steinberg and Kemp, 2009). Mounting evidence indicates that AMPK signaling dysregulation is involved in AD pathogenesis (Ma et al., 2014; Salminen et al., 2011; Vingtdeux et al., 2011; Wang et al., 2020; Wang et al., 2019; Zimmermann et al., 2020). Nevertheless, whether there exist isoform-specific roles of AMPK in AD pathogenesis is unclear. Here, taking advantage of the vervet NHP model of AD-like neuropathology described above, we investigated brain AMPKα isoform regulation/dysregulation and its potential relationship to AD neuropathology and biomarkers.
Materials and Methods
Animals
Nine aged (mean=22.1 year, range = 19.5 to 23.4 years old) female vervet monkeys (Chlorocebus aethiops sabaeus) from the Vervet Research Colony (VRC) were included in this study. The VRC is a multi-generational, pedigreed and genomically sequenced colony of Caribbean-origin vervets/African green monkeys, originally founded in 1975. The animals are raised in matrilineal social groups which reflect the natural order of vervet social groups in the wild. All the monkeys had free access to standard commercial monkey chow (Purina LabDiet) and water. All procedures were conducted in accordance with state and federal laws, standards of the Department of Health and Human Services, and approved by the Institutional Animal Care and Use Committee (IACUC) at Wake Forest School of Medicine
Behavior assessments
Behavior assessments of physical function were conducted approximately 1 month prior to necropsy. For each monkey, four 15 min focal animal observations were completed. The behaviors assessed included alert, moving, climbing and jumping. For each behavior, the rate (frequency per hour) and time percentage (time spent on the behavior during observation) were recorded.
Tissue collection
Monkeys were euthanized according to the guidelines of IACUC. Animals were sedated on the day of necropsy, and CSF was collected by inserting a 22-gauge needle into the cisterna magnaand stored at −70°C to determine Aβ42 level. Animals were flushed with a lactated ringer’s solution. Brains were rapidly removed and sectioned into 4 mm thick coronal slabs, frozen on metal plates with dry ice, and stored at −80°C. Dissections from either left or right frontal cortex, hippocampus, or cerebellum were included in western blot assay. Tissues from frontal cortex, temporal cortex, parietal cortex, occipital cortex, insular cortex, striatum, thalamus, hippocampus, cerebellum, midbrain, pons and medulla were fixed in 4% paraformaldehyde, embedded in paraffin, 5μm sectioned slices were cut and mounted on positively charged microscope slides for Aβ and tau immunohistochemistry. The slides were kept at −20°C.
Sample preparation for soluble and insoluble Aβ
Frozen prefrontal cortices containing white and gray matter were weighted, mixed with freshly prepared, ice-cold TBS consisting of 25 mM Tris-HCl, 130 mM NaCl, 2.7 mM KCl, pH 7.4 at a ratio of 5:1 (TBS volume / brain wet weight) and homogenized with ultrasonic homogenizer on ice. The homogenate was centrifuged at 22,100g for 90min at 4°C. The resultant supernatant was collected and stored at −80 °C (called TBS extract), and the pellet was re-homogenized in TBS plus 1% Triton X-100 (5:1 volume / brain wet weight) and the homogenate was centrifuged as above. The resultant supernatant was collected and stored at −80 °C (called TBS-TX extract), and the pellet was re-homogenized in 88% formic acid (1:2 volume / brain wet weight) and incubated on a shaker overnight at 4°C. The resultant supernatant was collected (formic acid extract) and neutralized with 1M Tris solution (un-buffered) to pH 7.4. The neutralized solution was stored at −80 °C and ready for future experiments.
Measurement of Aβ42 level in CSF
Aβ42 levels in CSF were measured on first thawed CSF using a Luminex-based INNO-BIA Alzbio3 assay according to manufacturer instructions (Fujirebio, Malvern, PA) as previously described (Latimer et al., 2019).
Western blot
Frontal cortex, hippocampus and cerebellum samples were sonicated in cold lysis buffer (T-PER, Thermo Scientific) with proteinase and phosphatase inhibitors. Protein concentration in each sample was determined by BCA protein assay. Samples containing equal amounts of protein lysates and loading buffer were boiled at 100°C for 5 min. Then samples were loaded to 4-12% or 16.5% Tris-glycine SDS-PAGE gels (Mini-Protean, Bio-Rad) for electrophoresis. After transfer, nitrocellulose membranes were blocked for 10min in blocking buffer (Superblock, Thermo Scientific) and then probed with primary antibodies for Aβ (1:500, 6E10, Covance), P-AMPKα1 (1:1000, Abcam), AMPKα1 (1:1000, Abcam), P-AMPKα2 (1:1000, Abcam), AMPKα2 (1:1000, Abcam), P-AMPKα (1:1000, Cell Signaling), AMPKα (1:1000, Cell Signaling), P-mTORC1 (Ser2448) (1:1000, Cell Signaling), mTORC1 (1:1000, Cell Signaling), P-eEF2 (1:1000, Cell Signaling), eEF2 (1:1000, Cell Signaling), P-TSC2 (Thr1462) (1:1000, Cell Signaling), TSC2 (1:1000, Cell Signaling), P-4EBP1 (1:1000, Cell Signaling), 4EBP1 (1:1000, Cell Signaling) and GAPDH (1:1000, Cell Signaling) in 5% nonfat milk TBS-T buffer overnight at 4°C. The membranes were probed with corresponding secondary antibodies (1:5000, Bio-Rad) in 5% nonfat milk TBS-T buffer at room temperature for 1 hour Proteins were imaged using the ChemiDoc MP imaging system (Bio-Rad). Densitometry analysis was conducted using the ImageJ software (NIH).
Immunohistochemistry
Slides prepared from brain regions mentioned above were baked at 60°C for 30 minutes, then deparaffinized in xylene and rehydrated through a graded alcohol series. For antigen retrieval, slides were boiled in citrate buffer at pH 6.0 for 10 mins. Endogenous peroxidase activity was blocked using 3% hydrogen peroxide for 15 mins. Slides were incubated in a humid chamber in primary antibody for NeuN (1:400, Cell Signaling), Aβ (1:500 for 6E10 antibody, Covance; 1:300 for Aβ42 antibody, EMD Millipore) and p-tau (1:100, AT8, Pierce) overnight at 4°C. Slides were incubated in biotinylated secondary antibody (1:200, Vector Labs) for 30 mins at room temperature followed by incubation in ABC reagent (Vectastain, Vector Labs) for another 30 mins at room temperature. Primary and secondary antibodies and ABC reagent were diluted in 1% normal goat serum. Slides were developed in diaminobenzidine solution (ImmPACT, Vector Labs) for minutes depending on the primary antibodies and counterstained in hematoxylin solution for 30 secs. Negative controls were incubated in 1% goat serum without primary antibodies. Slides then were dehydrated in a grated alcohol series and xylene, cover-slipped and dried overnight. Images were taken by a Keyence BZ-X710 microscope.
To assess the Aβ plaque burden of each animal, area within each brain region with the highest Aβ plaque density was identified, then a single 100X field was taken on that area, Aβ plaque density in each brain region was semi-quantified as follows. Regions without Aβ plaque density were scored as 0, regions with Aβ plaque density between 0 and 6 plaques/mm2 were scored as 1, regions with Aβ plaque density between 6 and 20 plaques/mm2 were scored as 2, regions with Aβ plaque density more than 20 plaques/mm2 were scored as 3. Then scores from all the regions were summarized to represent the Aβ plaque burden of the animal.
To assess neurodegeneration of each animal, five consecutive 40X fields images (0.1 mm2 each) were taken from CA1 and CA3 area respectively (so 10 images in total) in NeuN staining slide of each animal. Total area analyzed for each animal was 1 mm2. Neurons were counted from the field and neuron density was calculated by dividing the number of neurons with the area of the field.
ELISA for Aβ measurement
Aβ42 levels were measured using ELISA kit purchased from Invitrogen (Cat# KHB3544). In brief, standard samples were prepared according to the instructions immediately before experiment. 50 μl of sample and 50 μl of detector antibody were added into each well and incubated for 3 hours at room temperature. All samples were duplicated. After incubation, the plate was washed with washing buffer for 4 times and 100 μl of secondary antibody was added into each well to incubate for 30min at room temperature. After incubation, the plate was washed with washing buffer for 4 times and 100 μl of stabilized chromogen was added into each well. The plate was kept in dark to develop for 30min at room temperature and then 100 μl of stop solution was added into each well. The plate was read in a microplate reader (Bio-Rad) and Aβ42 level of each sample was calculated based on the standard curve.
Proteomic analysis
Hippocampal tissues were dissected in the cold PBS and lysed immediately in 500 μL of PBS with protease/phosphatase inhibitors using a Bead Mill Homogenizer (Bead Ruptor, Omni International). 500 μL of 2X radioimmunoprecipitation (RIPA) buffer was added and the mixture was incubated on ice for 30 minutes. Tubes were centrifuged at 18,000 x g for 10 minutes then supernatant was transferred to a clean tube. Protein concentration was measured by BCA analysis and 100 μg of protein was subjected to tryptic digestion.
Reducing alkylation was performed in the presence of 10 mM dithiothreitol and 30 mM iodoacetamide. Four times the sample volume of cold (−20 °C) acetone was added to the tube which was then incubated at −20 °C overnight. Protein was pelleted by centrifugation at 14,000 x g for 10 minutes. After removal of supernatant, the pellet was dried by evaporation of residual acetone for 10 minutes at room temperature, and then was suspended in 50 mM ammonium bicarbonate. The protein suspension was incubated with trypsin at 1:50 enzyme-to-substrate ratio at 37 °C overnight. The resulting peptides were cleaned up by desalting using a C18 spin column. Purified peptide mixture is prepared in 5% (v/v) ACN containing 1% (v/v) formic acid for LC-MS/MS analysis.
The LC-MS/MS system consisted of a Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, MA) and a Dionex Ultimate-3000 nano-UPLC system (Thermo Scientific, Waltham, MA) employing a Nanospray Flex Ion Source (Thermo Scientific, Waltham, MA). An Acclaim PepMap 100 (C18, 5 μm, 100 Å, 100 μm x 2 cm) trap column and an Acclaim PepMap RSLC (C18, 2 μm, 100 Å, 75 μm x 50 cm) analytical column were used for the stationary phase. Peptides were separated using a linear gradient consisting of mobile phases A (water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid) where the gradient was from 5% B at 0 min to 40% B at 170 min. MS spectra were acquired by data dependent scans consisting of MS/MS scans of the twenty most intense ions from the full MS scan with dynamic exclusion option which was 10 seconds.
To identify proteins, spectra were searched using Sequest HT algorithm within the Proteome Discoverer v2.1 (Thermo Scientific, Waltham, MA) in combination with the combined UniProt protein FASTA database of human, and rhesus monkey (total 20,621 annotated entries, Feb 2018). Search parameters were as follow: FT-trap instrument, parent mass error tolerance of 10 ppm, fragment mass error tolerance of 0.02 Da (monoisotopic), variable modifications of 16 Da (oxidation) on methionine and fixed modification of 57 Da (carbamidomethylation) on cysteine.
The numbers of peptide spectrum matches in each group were averaged and fold change was calculated as mean of AD-like group / mean of normally aged group. P value was calculated using the student’s t-test. Proteins with fold change >1.15 and p<0.08 were considered upregulated and proteins with fold change <0.85 and p<0.08 were considered downregulated. A heat map was generated based on the data by R (V3.3.1). PANTHER (Protein Annotation through Evolutionary Relationship) program V10.0 (http://www.pantherdb.com) was used to classify these upregulated or downregulated proteins according to their molecular function or roles in biological processes. Then STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) program V10.0 (http://string-db.org) was used to perform functional interaction networks between upregulated or downregulated proteins and AMPKα2. Prediction methods were neighborhood, co-expression, gene fusions, experiments, co-occurrence, databases and text mining using medium confidence (0.4).
Statistics
Unpaired independent Student t test was performed for continuous variables between the two age groups. Linear regression was performed to determine the correlation between Aβ42 levels and Aβ plaque burden. All the statistical analyses were conducted using the GraphPad Prism 5.0, and the significance level was set at p<0.05.
Results
The characteristics of the monkeys were summarized in Table 1. They were all female with average age 22.1 ± 1.5 years. Average BMI was 60.5 ± 7.5 kg/m2. Diabetic status and the hemisphere from which brain tissue was taken were indicated in the table.
Table 1.
Characteristics of the monkeys.
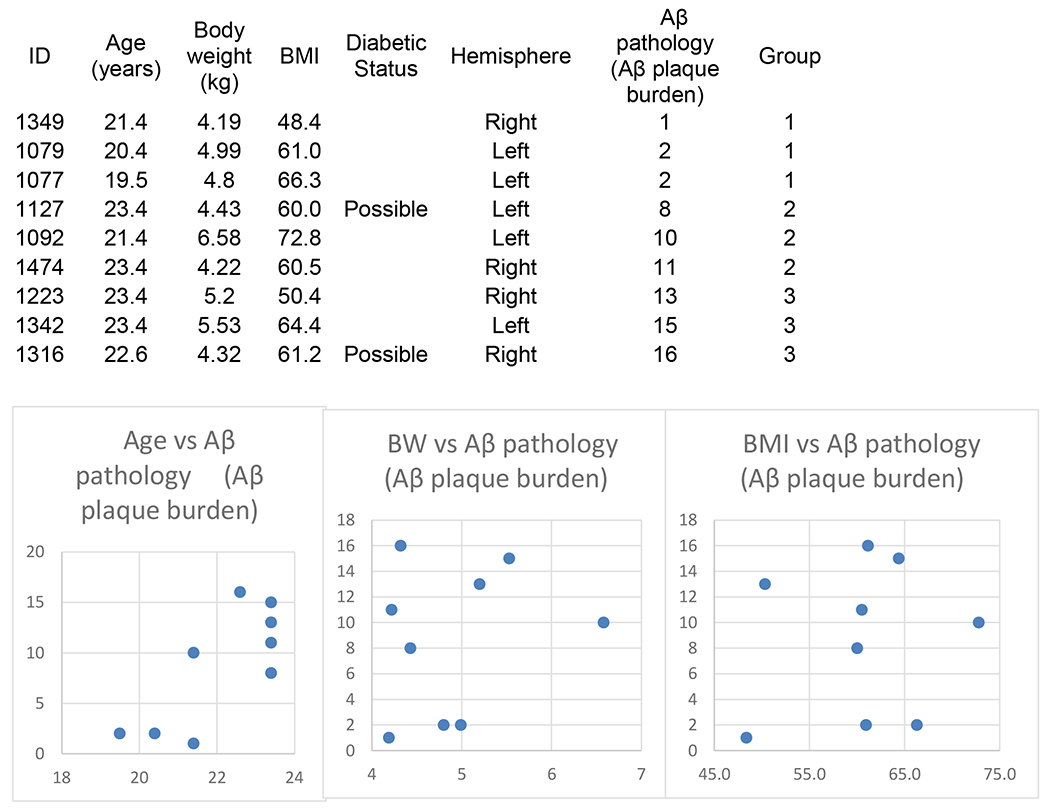
|
Group 1: Control, No AD Pathology.
Group 2: Modest AD Pathology.
Group 3: AD Pathology.
We first performed immunohistochemical experiments to characterize the AD-associated Aβ and phospho-tau (p-tau) neuropathological changes in the brains of these monkeys. In agreement with previous studies (Latimer et al., 2019), typical Aβ plaques were identified in neocortex but rarely in hippocampus (Fig. 1A–C). The brain Aβ plaque burden in each monkey was summarized in Table 1. Neurofibrillary tangles were absent, intra-neuronal p-tau immunostaining was observed in neocortex and hippocampus of all the animals, although neurofibrillary tangles were rarely identified (Fig. 1D–F). Such findings were also consistent with the previous report (Latimer et al., 2019). Using the ELISA method, we determined that CSF Aβ42 levels were inversely related to brain Aβ plaque burden in these animals (Fig. 1G), which is similar to the pattern seen in human AD patients (Tapiola et al., 2009).
Fig. 1. Aβ and p-tau pathologies of the monkeys.
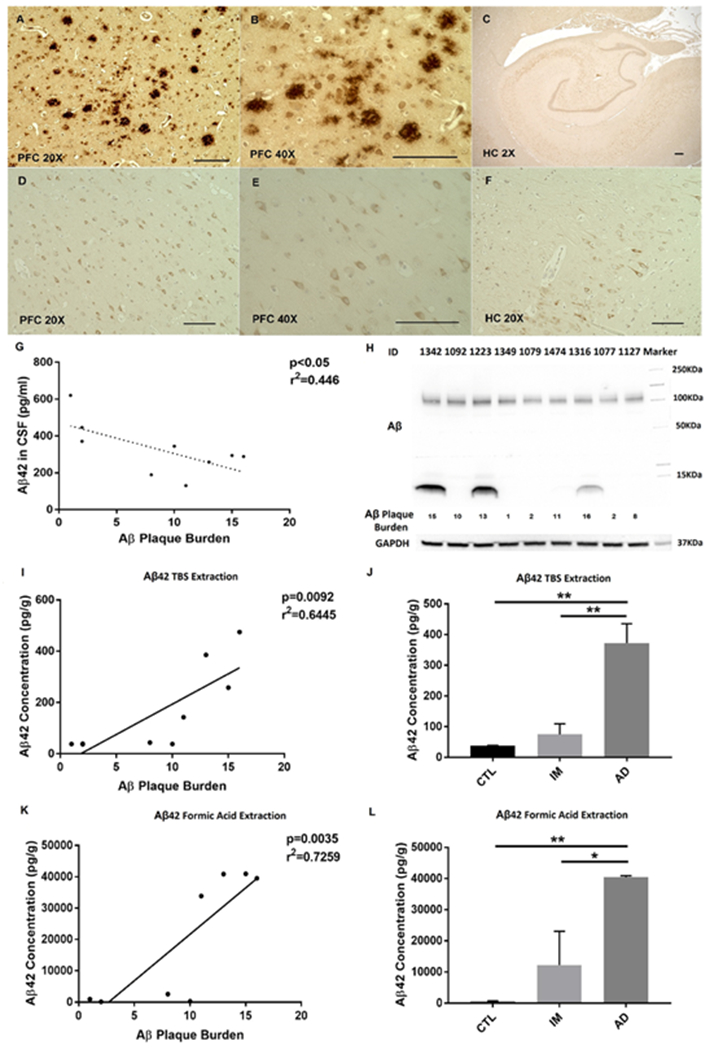
A-B Aβ plaques in 20X and 40X magnification in prefrontal cortex. C absence of plaque in hippocampus. D-E p-tau in 20X and 40X magnification in prefrontal cortex. F p-tau in hippocampus. Scale bars are 100μm in 20X and 40X images, and 300μm in 2X image. (Abbreviation: PFC Prefrontal Cortex, HC Hippocampus). G relationship between Aβ42 levels in CSF and Aβ plaque burden. Aβ level in CSF is significantly correlated with Aβ plaque burden. H Aβ identified in the PFC of three monkeys with the highest Aβ plaque burden. I relationship between soluble Aβ (TBS extract) levels and Aβ plaque burden. J Levels of soluble Aβ are increased in AD-like group, compared to control or intermediate group. K relationship between insoluble Aβ (formic acid extract) levels and Aβ plaque burden. L Levels of insoluble Aβ are increased in AD-like group, compared to control or intermediate group. Pearson’s correlation analysis was applied. *p<0.05, **p<0.01, one-way ANOVA followed by Tukey’s post hoc test. Note: Two data points superimposed in I and K.
We further examined Aβ oligomers in the cortex and hippocampus of these monkeys. Aβ oligomers were found only in the monkeys with the highest Aβ plaque burden, while those with intermediate or lowest Aβ plaque burden had no detectable Aβ oligomers in their brains (Fig. 1H). We didn’t detect Aβ oligomers in the hippocampus (data was not shown).
Synthesizing results from neuropathological examination, and biochemical assays (e.g. CSF Aβ, Aβ oligomers measurement) (Latimer et al., 2019; McKhann et al., 2011), we categorized the monkeys into three groups, AD-like (AD), AD-possible intermediate (IM), and normally aged control (CTL). Moreover, we investigated the soluble (TBS extract) and insoluble (formic acid extract) Aβ in brain using the ELISA method. We found that levels of either soluble or insoluble Aβ were correlated with Aβ plaque burden (Fig. 1I, K). Levels of both soluble and insoluble Aβ were significantly increased in the AD-like group, compared to control of the intermediate group (Fig. 1J, L). Using an Aβ42-specific antibody, we also examined “intra-neuronal” Aβ expression in both PFC and hippocampus. Briefly, we observed increased staining of intra-neuronal Aβ in PFC of AD-like monkeys (Fig. 2A–D). In contrast, Aβ staining was unaltered in hippocampus among all groups (Fig. 2E–H). Such results are consistent with the findings described above (Fig. 1). We focus on AD and CTL groups in this study for the purpose of simplicity.
Fig. 2. Intra-neuronal Aβ42 staining in PFC and hippocampus (D-F) of aged monkeys.
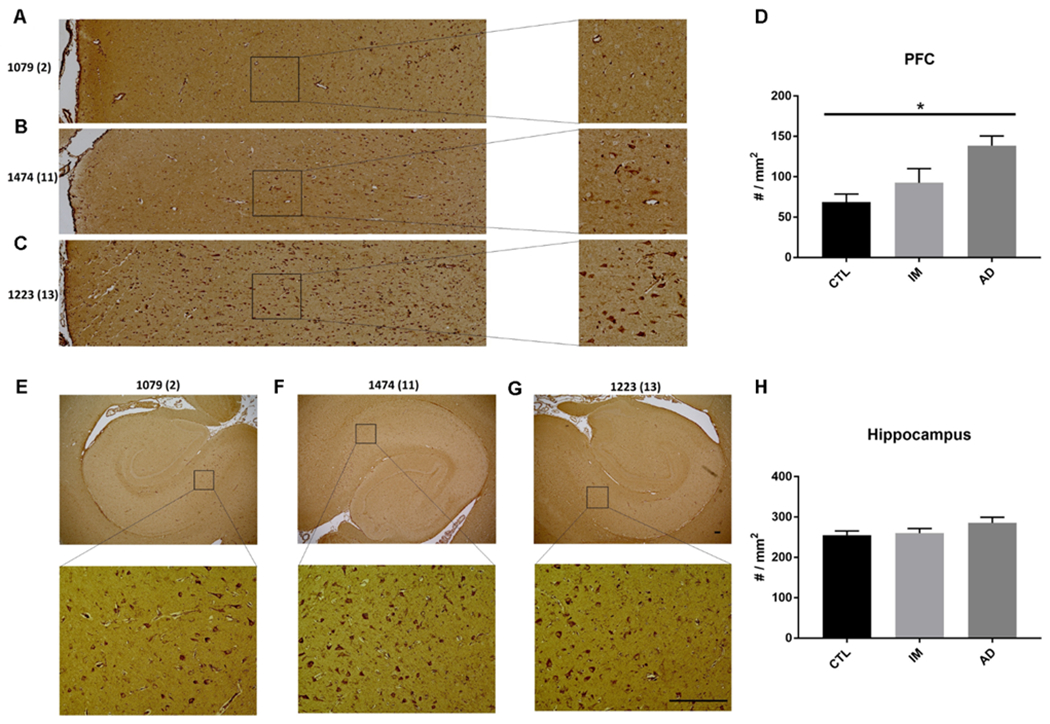
A-C Representative images from PFC staining. D Quantification of PFC staining. E-G Representative images from hippocampal staining. H Quantification of hippocampal staining. Numbers of Aβ plaque were indicated by the monkey IDs. Scale bar=200 αm. n=3 monkeys for each group. *p<0.05, one-way ANOVA followed by Tukey’s post hoc test.
Several behavioral assessments of physical function were performed to evaluate indirectly the cognitive status of these monkeys. Rates of alert, moving, climbing and jumping were decreased in a tiered way in monkeys with intermediate and highest Aβ plaque burden as compared to those with lowest Aβ plaque burden (Fig. 3). The same pattern could be found in percentage of time moving, climbing and jumping, but not alert (Fig. 3). Though the decreases were not statistically significant probably due to small sample size in each group, trends of behavioral differences were consistent with the categorization proposed above.
Fig. 3. Behavioral assessment of the monkeys.
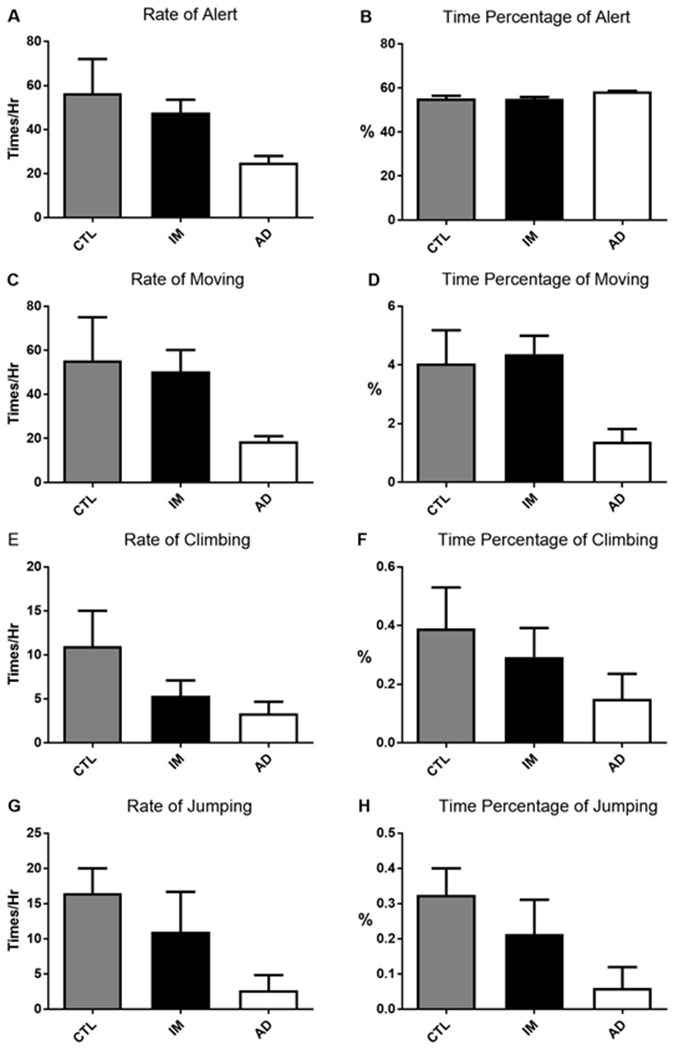
CTL represents the group with lowest Aβ plaque burden, which are considered to be normally aged controls, IM represents the group with intermediate Aβ plaque burden, while AD represents the group with highest Aβ plaque burden, which are deemed as AD-like. Rate=frequency per hour. Percent time= percentage of time performing the behavior. No significant difference for any of behavioral assessment is found among these groups. n=3 monkeys for each group, p>0.05, one-way ANOVA.
One limitation of most if not all AD mouse models is the absence of neurodegeneration. We assessed neurodegeneration in the aged vervet hippocampus using immunohistochemical staining for NeuN, a widely used neuron-specific antibody (Mullen et al., 1992). Notably, neuron density in area CA1 of hippocampus was significantly decreased in the AD-like monkeys as compared to control group (Fig. 4A, B, and E). Neuron density in area CA3 of hippocampus was unaltered between the two groups (Fig. 4C, D, and F).
Fig. 4. Neuron density evaluated by NeuN staining in the hippocampi of AD-like and normally aged monkeys.
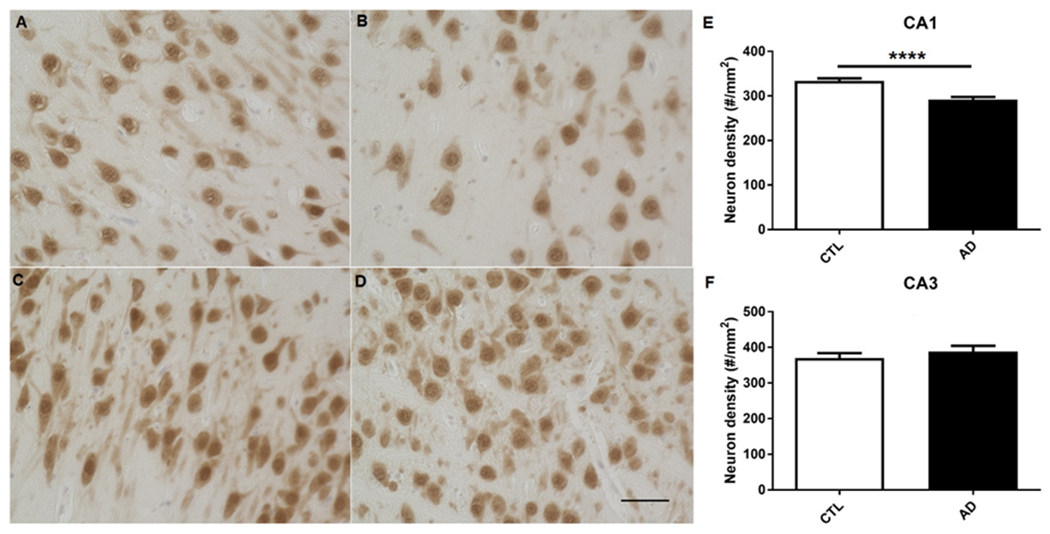
A representative images of neurons in hippocampal CA1 of normally aged monkeys. B representative image of neurons in hippocampal CA1 of AD-like monkeys. C representative images of neurons in hippocampal CA3 of normally aged monkeys. D representative images of neurons in hippocampal CA3 of AD-like monkeys. E quantification of neuronal density in CA1 of the two groups, indicating neuron density is decreased in CA1 of hippocampi of AD-like monkeys as compared to normally aged monkeys. ****p<0.0001, unpaired independent t test. F quantification of neuronal density in CA3 of the two groups. p>0.05, unpaired independent t test. 40x, Scale bar: 50 μm
We next investigated potential AMPKα signaling alterations in AD-like and control groups by Western blot assay. AMPKα1 activity (assessed by phosphorylation) was unaltered in hippocampus of AD-like monkeys (Fig. 5A–B). In comparison, levels of AMPKα2 phosphorylation were significantly increased in hippocampus of AD-like monkeys (Fig. 5C–D). Phosphorylation of the pan-AMPKα was unaltered (Fig. 5E–F). A correlation analysis between the AMPKα2 activity (measured by phosphorylation) and brain Aβ burden was performed (Fig. 5G). We also examined activity of AMPKα isoforms in prefrontal cortex (PFC) and cerebellum, and did not observe significant difference between AD-like and control groups (Fig. S1). AMPK signaling is involved in protein synthesis and impaired mRNA translation is implicated in AD (Beckelman et al., 2019; Ma et al., 2014; Salminen et al., 2011; Zimmermann et al., 2020). We thus further examined downstream signaling of AMPK involved in protein synthesis including the mTORC1 and eEF2K/eEF2 signaling cascade, but did not detect any significant difference between AD-like and control groups (Fig. S2).
Fig. 5. Analysis of the AMPKα signaling in the hippocampus of AD-like and normally aged monkeys.
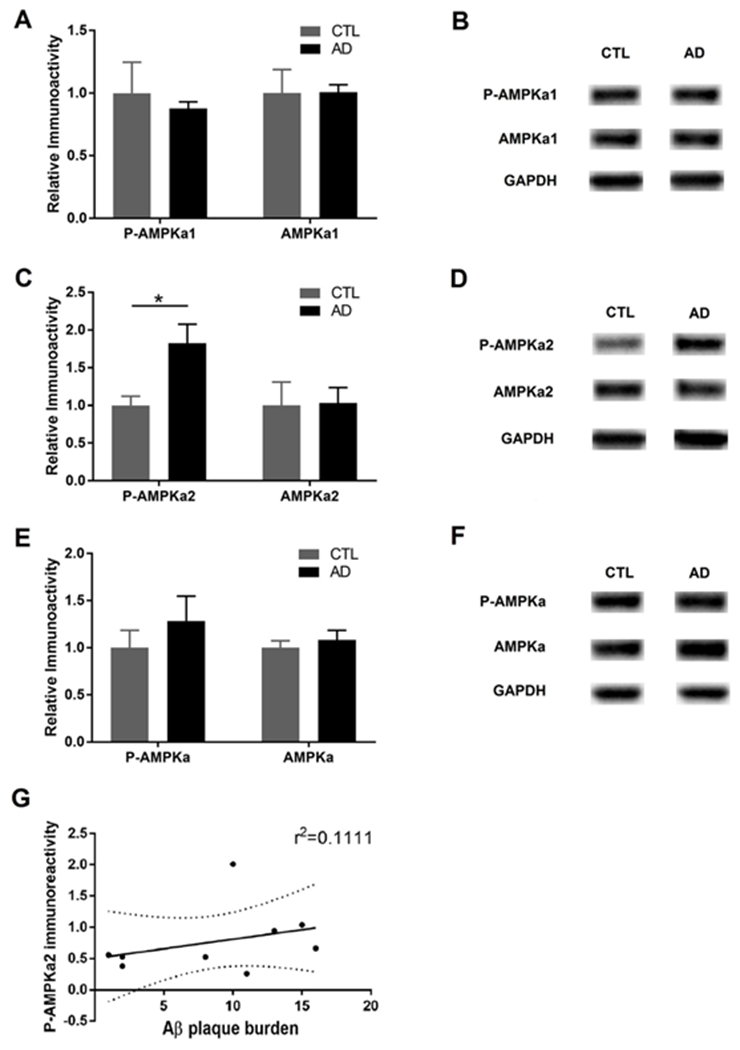
A quantification of phospho-AMPKα1 and total AMPKα1 levels. B representative Western blot of p-AMPKα1 and AMPKα1 assay. C quantification of phospho-AMPKα2 and total AMPKα2 levels. D representative Western blot of p-AMPKα2 and AMPKα2 assay. E quantification of phospho-AMPKα and total AMPKα levels. F representative Western blot of p-AMPKα and AMPKα assay G Correlation analysis between AMPKα2 activity (measured by levels of phosphorylation) and brain Aβ plaque burden. Data demonstrate that levels of AMPKα2 were significantly elevated in the AD group. n=3 monkeys for each group, *p<0.05, unpaired independent t test.
To determine potential alterations of protein profiling in AD-like monkeys, we performed a mass spectrometry (MS)-based proteomic study on hippocampal tissues of AD-like and normally aged monkeys. In total, 2708 proteins were detected in these samples. Of all the proteins identified, 31 were up-regulated (Table 2) and 46 were down-regulated (Table 3) in the hippocampus of AD-like monkeys as compared to normal aged monkeys. Then we conducted the protein-protein interaction analysis using the online program STRING (http://www.string-db.org). The results showed that AMPKα2 could be related to most of the up-regulated or down-regulated proteins through a network, suggesting that AMPKα2 may played a pivotal role in the dysregulation of protein synthesis phenotype associated with the NHP model (Fig. 6A–B). Those up-regulated or down-regulated proteins were further classified according to molecular function or biological process using the online program PANTHER (Protein ANalysis THrough Evolutionary Relationships, http://www.pantherdb.org). Up-regulated proteins were classified into 6 categories with 31 hits according to the molecular function (Fig. 7A). The categories with the most hits were proteins with binding and catalytic activity. By biological process, up-regulated proteins could be classified into 9 categories with 50 hits (Fig. 7B). The categories with the most hits were proteins involved in the cellular and metabolic process. Down-regulated proteins were classified into 4 categories with 29 hits according to molecular function (Fig. 7C). The category with the most hits was proteins with the catalytic activity. By biological process, down-regulated proteins could also be classified into 9 categories with 56 hits (Fig. 7D). The categories with the most hits were also proteins involved in the cellular and metabolic process. A heat map was generated by unsupervised hierarchical clustering (Fig. 7E).
Table 2.
Up-regulated proteins in the hippocampus of AD-like monkeys.
| Protein Name | Gene Name | Accession # | MW (kDa) | mean #PSM CTL | mean #PSM AD | Fold (AD/CTL) |
|---|---|---|---|---|---|---|
| Dynamin-2 | DNM2 | P50570 | 98.0 | 11.67 | 13.67 | 1.17 |
| Ras-related protein Ral-A | RALA | P11233 | 23.6 | 6.00 | 7.33 | 1.22 |
| Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial | IDH3A | P50213 | 39.6 | 13.33 | 16.33 | 1.23 |
| Dynein light chain 2, cytoplasmic | DYNLL2 | Q96FJ2 | 10.3 | 5.67 | 7.00 | 1.24 |
| Ras-related protein Rap-1A | RAP1A | P62834 | 21.0 | 6.67 | 8.67 | 1.30 |
| Guanine nucleotide-binding protein G(q) subunit alpha | GNAQ | P50148 | 42.1 | 9.67 | 13.00 | 1.34 |
| Profilin-1 | PFN1 | P07737 | 15.0 | 8.33 | 11.67 | 1.40 |
| GTP-binding nuclear protein Ran | RAN | P62826 | 24.4 | 4.67 | 7.00 | 1.50 |
| Cell adhesion molecule 4 | CADM4 | Q8NFZ8 | 42.8 | 6.67 | 10.00 | 1.50 |
| Transaldolase | TALDO1 | P37837 | 37.5 | 3.00 | 4.67 | 1.56 |
| S-phase kinase-associated protein 1 | SKP1 | P63208 | 18.6 | 8.33 | 13.33 | 1.60 |
| Translationally-controlled tumor protein | TPT1 | P13693 | 19.6 | 5.00 | 8.33 | 1.67 |
| Sorting nexin-1 | SNX1 | Q13596 | 59.0 | 1.33 | 2.67 | 2.00 |
| Nuclear transport factor 2 | NUTF2 | P61970 | 14.5 | 2.67 | 5.67 | 2.13 |
| Ubiquitin-conjugating enzyme E2 D3 | UBE2D3 | P61077 | 16.7 | 1.67 | 3.67 | 2.20 |
| Receptor expression-enhancing protein 5 | REEP5 | Q00765 | 21.5 | 1.33 | 3.33 | 2.25 |
| AP-1 complex subunit mu-1 | AP1M1 | Q9BXS5 | 48.6 | 1.33 | 3.00 | 2.25 |
| Voltage-gated potassium channel subunit beta-2 | KCNAB2 | Q13303 | 41.0 | 1.67 | 4.00 | 2.40 |
| Keratin, type II cytoskeletal 2 epidermal | KRT2 | P35908 | 65.4 | 3.00 | 8.33 | 2.78 |
| Tumor protein D52 | TPD52 | P55327 | 24.3 | 1.00 | 3.00 | 3.00 |
| Glutathione S-transferase omega-1 | GSTO1 | P78417 | 27.5 | 1.00 | 3.33 | 3.33 |
| Chondroitin sulfate proteoglycan 5 | CSPG5 | O95196 | 60.0 | 0.67 | 2.33 | 3.50 |
| Neural Wiskott-Aldrich syndrome protein | WASL | O00401 | 54.8 | 0.67 | 2.33 | 3.50 |
| Leucine-rich PPR motif-containing protein, mitochondrial | LRPPRC | P42704 | 157.8 | 1.00 | 4.67 | 4.67 |
| IQ and AAA domain-containing protein 1 | IQCA1 | Q86XH1 | 95.3 | 0.33 | 1.67 | 5.00 |
| Heterogeneous nuclear ribonucleoprotein A0 | HNRNPA0 | Q13151 | 30.8 | 0.33 | 1.67 | 5.00 |
| L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase | AASDHPPT | Q9NRN7 | 35.8 | 0.33 | 2.00 | 6.00 |
| Very-long-chain enoyl-CoA reductase | TECR | Q9NZ01 | 36.0 | 0.33 | 2.00 | 6.00 |
| Protein phosphatase 1E | PPM1E | Q8WY54 | 84.9 | 0.33 | 2.00 | 6.00 |
| E3 ubiquitin-protein ligase CHIP | STUB1 | Q9UNE7 | 34.8 | 0 | 1.67 | ∞ |
| Guanine nucleotide-binding protein G(I)/G(S)/G(O) subunit gamma-12 | GNG12 | Q9UBI6 | 8.0 | 0 | 1.33 | ∞ |
Abbreviation: MW molecular weight, #PSM number of peptide spectrum matches, CTL control, AD Alzheimer’s disease.
Table 3.
Down-regulated proteins in the hippocampus of AD-like monkeys.
| Protein Name | Gene Name | Accession # | MW (kDa) | mean #PSM CTL | mean #PSM AD | Fold (AD/CTL) |
|---|---|---|---|---|---|---|
| Glutamate decarboxylase 1 | GAD1 | Q99259 | 66.9 | 2.33 | 0.00 | 0.00 |
| Calcium-binding protein 39 | CAB39 | Q9Y376 | 39.8 | 1.67 | 0.00 | 0.00 |
| Calpain-13 | CAPN13 | Q6MZZ7 | 76.6 | 1.33 | 0.00 | 0.00 |
| Histidine triad nucleotide-binding protein 3 | HINT3 | Q9NQE9 | 20.3 | 1.33 | 0.00 | 0.00 |
| Vesicle-associated membrane protein 1 | VAMP1 | P23763 | 12.9 | 1.33 | 0.00 | 0.00 |
| Disintegrin and metalloproteinase domain-containing protein 23 | ADAM23 | O75077 | 91.9 | 2.33 | 0.00 | 0.00 |
| V-type proton ATPase subunit G 1 | ATP6V1G1 | O75348 | 13.7 | 2.33 | 0.33 | 0.14 |
| NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 11 | NDUFA11 | Q86Y39 | 14.8 | 2.33 | 0.33 | 0.14 |
| COP9 signalosome complex subunit 8 | COPS8 | Q99627 | 23.2 | 2.00 | 0.33 | 0.17 |
| Sodium/potassium-transporting ATPase subunit beta-3 | ATP1B3 | P54709 | 31.5 | 2.00 | 0.33 | 0.17 |
| Vesicular inhibitory amino acid transporter | SLC32A1 | Q9H598 | 57.4 | 3.00 | 0.67 | 0.22 |
| Mitochondrial pyruvate carrier 2 | MPC2 | O95563 | 14.3 | 4.33 | 1.00 | 0.23 |
| Proteasome subunit beta type-5 | PSMB5 | P28074 | 28.5 | 4.33 | 1.00 | 0.23 |
| KH domain-containing, RNA-binding, signal transduction-associated protein 1 | KHDRBS1 | Q07666 | 48.2 | 2.67 | 0.67 | 0.25 |
| Inorganic pyrophosphatase 2, mitochondrial | PPA2 | Q9H2U2 | 37.9 | 2.33 | 0.67 | 0.29 |
| Contactin-associated protein-like 2 | CNTNAP2 | Q9UHC6 | 148.1 | 3.00 | 1.00 | 0.33 |
| Prolyl endopeptidase-like | PREPL | Q4J6C6 | 83.9 | 7.67 | 2.67 | 0.35 |
| Histidine triad nucleotide-binding protein 2, mitochondrial | HINT2 | Q9BX68 | 17.2 | 3.67 | 1.33 | 0.36 |
| ATP synthase subunit delta, mitochondrial | ATP5D | P30049 | 17.5 | 2.67 | 1.00 | 0.38 |
| Heterogeneous nuclear ribonucleoprotein M | HNRNPM | P52272 | 77.5 | 2.67 | 1.00 | 0.38 |
| Mitochondrial import inner membrane translocase subunit Tim8 A | TIMM8A | O60220 | 11.0 | 3.33 | 1.33 | 0.40 |
| Methylglutaconyl-CoA hydratase, mitochondrial | AUH | Q13825 | 35.6 | 2.33 | 1.00 | 0.43 |
| 40S ribosomal protein S16 | RPS16 | P62249 | 16.4 | 2.33 | 1.00 | 0.43 |
| Interferon-inducible double-stranded RNA-dependent protein kinase activator A | PRKRA | O75569 | 34.4 | 6.67 | 3.00 | 0.45 |
| Acylpyruvase FAHD1, mitochondrial | FAHD1 | Q6P587 | 24.8 | 3.33 | 1.67 | 0.50 |
| Vacuolar protein sorting-associated protein 29 | VPS29 | Q9UBQ0 | 20.5 | 2.67 | 1.33 | 0.50 |
| Secretogranin-2 | SCG2 | P13521 | 70.9 | 6.67 | 3.33 | 0.50 |
| ATP-dependent RNA helicase DDX1 | DDX1 | Q92499 | 82.4 | 3.67 | 2.00 | 0.55 |
| Synaptic vesicle membrane protein VAT-1 homolog-like | VAT1L | Q9HCJ6 | 45.9 | 9.67 | 5.33 | 0.55 |
| DnaJ homolog subfamily A member 1 | DNAJA1 | P31689 | 44.8 | 4.67 | 2.67 | 0.57 |
| Phosphatidylinositol transfer protein alpha isoform | PITPNA | Q00169 | 31.8 | 4.00 | 2.33 | 0.58 |
| NADH-cytochrome b5 reductase 3 | CYB5R3 | P00387 | 34.2 | 5.00 | 3.00 | 0.60 |
| Putative heat shock protein HSP 90-beta-3 | HSP90AB3P | Q58FF7 | 68.3 | 20.67 | 13.00 | 0.63 |
| Gamma-synuclein | SNCG | O76070 | 13.3 | 5.67 | 3.67 | 0.65 |
| Destrin | DSTN | P60981 | 18.5 | 5.00 | 3.33 | 0.67 |
| NADH dehydrogenase [ubiquinone] flavoprotein 1, mitochondrial | NDUFV1 | P49821 | 50.8 | 19.33 | 13.33 | 0.69 |
| MICOS complex subunit MIC60 | IMMT | Q16891 | 83.6 | 23.33 | 17.00 | 0.73 |
| ATP-citrate synthase | ACLY | P53396 | 120.8 | 11.67 | 8.67 | 0.74 |
| 2-iminobutanoate/2-iminopropanoate deaminase | HRSP12 | P52758 | 14.5 | 7.33 | 5.67 | 0.77 |
| Protein NipSnap homolog 1 | NIPSNAP1 | Q9BPW8 | 33.3 | 10.33 | 8.00 | 0.77 |
| Malate dehydrogenase, cytoplasmic | MDH1 | P40925 | 36.4 | 37.33 | 29.33 | 0.79 |
| Phosphate carrier protein, mitochondrial | SLC25A3 | Q00325 | 40.1 | 20.33 | 16.00 | 0.79 |
| ATP synthase subunit O, mitochondrial | ATP5O | P48047 | 23.3 | 8.00 | 6.33 | 0.79 |
| Microtubule-associated protein 6 | MAP6 | Q96JE9 | 86.5 | 14.67 | 11.67 | 0.80 |
| Fructose-bisphosphate aldolase C | ALDOC | P09972 | 39.4 | 80.67 | 67.00 | 0.83 |
| Ras-related protein Rab-14 | RAB14 | P61106 | 23.9 | 14.33 | 12.00 | 0.84 |
Abbreviation: MW molecular weight, #PSM number of peptide spectrum matches, CTL control, AD Alzheimer’s disease.
Fig. 6.
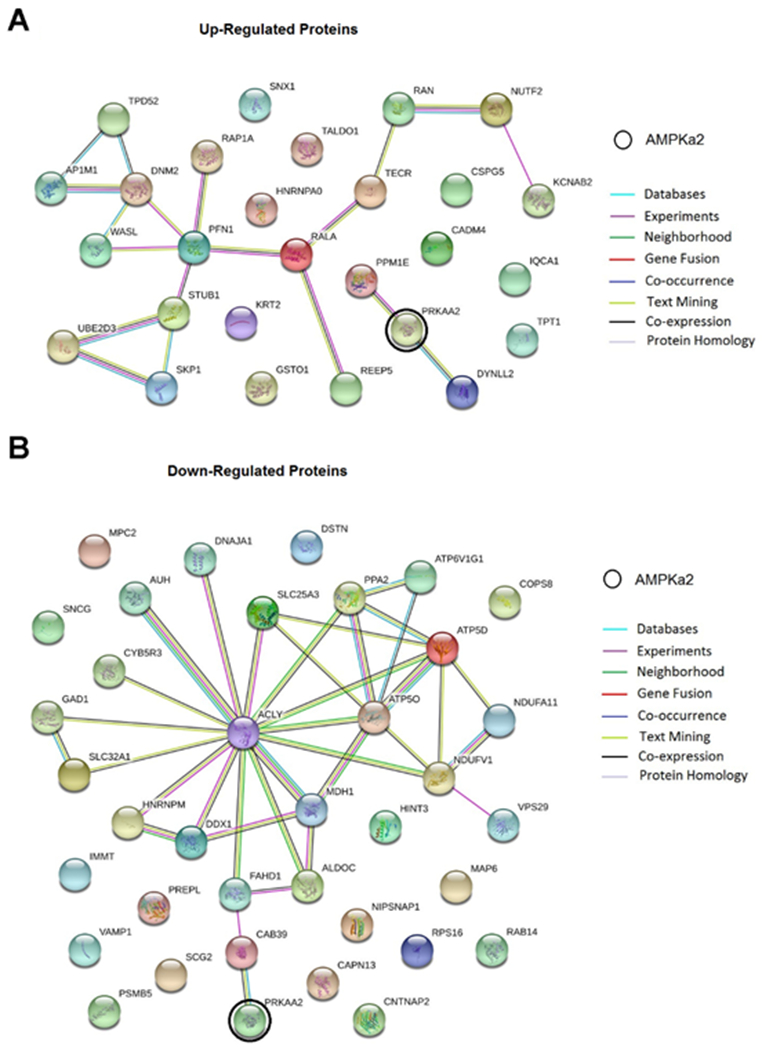
Protein-protein interaction pathways analyses. A interactions between AMPKα2 (PRKAA2) and up-regulated proteins. B interactions between AMPKα2 (PRKAA2) and down-regulated proteins. Each node represents a protein and is marked by the name of gene which encodes the protein. The gene for AMPKα2 is circled in black. Figures next to nodes represent different biological processes that the proteins might be involved in. Lines in color represent different evidence for each identified interaction.
Fig. 7. Classification and heat map of the up-regulated and down-regulated proteins.
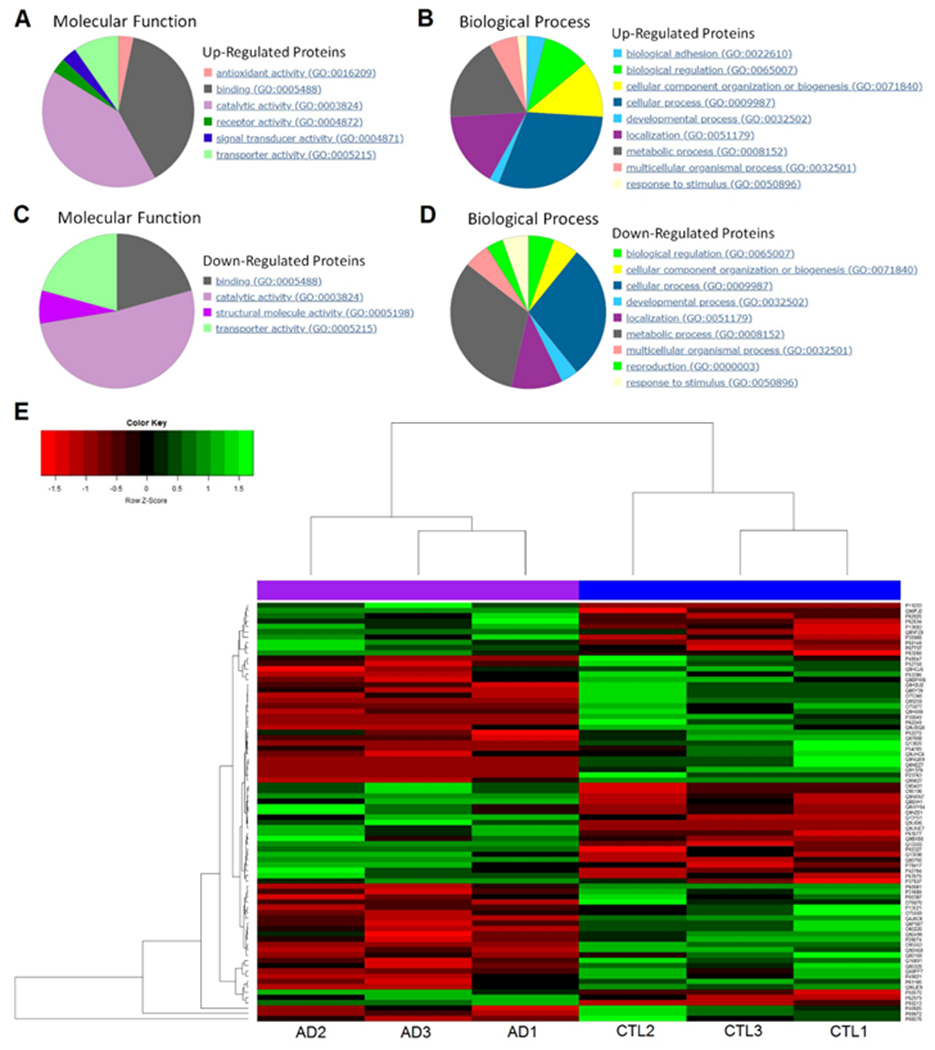
A classification of up-regulated proteins according to molecular function. B classification of up-regulated proteins according to biological process. C classification of down-regulated proteins according to molecular function. D classification of down-regulated proteins according to biological process. E heat map of the significantly altered proteins from 6 individual samples after unsupervised hierarchical clustering. Left 3 columns are the AD-like monkeys, right 3 columns are the normally aged monkeys. Far right column is the accession numbers of these proteins.
Discussion
Non-human primates have long been valuable models for the study of human diseases because of their close phyletic relationships with human beings. Many studies carried out in different species of non-human primates found AD-like pathologies in the brain of these animals when they aged, which resembled the pattern of sporadic Alzheimer’s disease in human (Latimer et al., 2019; Walker and Jucker, 2017). In this study, we confirmed previous findings of AD-like neuropathological change in brains of aged vervet monkeys, an Old World monkey (Latimer et al., 2019). In correlation with the AD pathology, we observed dysregulation of the AMPK signaling, particularly activity of AMPKα2. To identify monkeys that are “AD-like” or “normally aged”, we adopted an approach similar to what is used in the diagnosis of human AD patients, combining pathological, biochemical and behavioral assessments (McKhann et al., 2011). For Aβ pathology, we took multiple measurements into consideration including brain Aβ plaque burdens, Aβ42 levels in CSF, and levels of soluble Aβ oligomers, which are probably the toxic forms of Aβ and are more closely related to the pathogenesis of Alzheimer’s disease (Shankar et al., 2008; Walsh and Selkoe, 2007).
In clinical practice, pathological and biochemical alterations are usually evaluated in the context of cognitive impairment (for disease diagnosis, etc.). Unfortunately, there were no data available on formal cognitive tests in these monkeys, so behavioral assessments were used as surrogates for cognitive measurement. We found that rates and time percentages of alert, moving, climbing, jumping and hanging decreased in AD-like monkeys (also in those with intermediate Aβ plaque burden). Interestingly, rates and time percentages of the behaviors in those with intermediate Aβ plaque burden were also intermediate between AD-like and normally aged groups, indicating that those monkeys were probably in a status between AD and normal aging (e.g. mild cognitive impairment or MCI).
As in early human AD, we found typical Aβ plaques in the neocortex but not in the hippocampus of AD-like monkeys. Moreover, for tau pathology, in the aged monkey brains p-tau is present, but not generally in the form of neurofibrillary tangles. These findings are consistent with previous studies in different species of non-human primates (Heilbroner and Kemper, 1990; Heuer et al., 2012; Sani et al., 2003; Toledano et al., 2012). While Aβ plaques are not identified in hippocampus, we observed significant neurodegeneration in area CA1 of hippocampus in AD-like monkeys. Future studies are warranted to elucidate the possible link between cortical Aβ and hippocampal neurodegeneration. Lack of the neurodegeneration phenotype is a common concern for most, if not all rodent models of AD. In correlation with hippocampal neurodegeneration, AMPKα2 signaling was dysregulated in the hippocampus, but not in PFC of the AD-like monkeys.
As a key molecular sensor to regulate cellular energy metabolism, AMPK activity and its associated signaling pathway are also implicated in cognitive function and regulation of long-term potentiation (LTP), a major form of synaptic plasticity and an established cellular model for learning and memory (Ma et al., 2011; Potter et al., 2010). For instance, over-activation of AMPK results in LTP impairment and conversely inhibition of AMPK activity leads to facilitation of LTP (Potter et al., 2010). In our study, we observed increased activity (assessed by phosphorylation) of AMPKα2 in hippocampi of AD-like monkeys, which is consistent with the aforementioned roles of AMPK in synaptic plasticity and memory formation. Of note, we did not detect a significant increase in levels of the pan-AMPKα phosphorylation in AD-like monkeys, in contrast to previous studies in mouse models of AD and post mortem human brain tissue (Ma et al., 2014; Vingtdeux et al., 2011). The specific targets and roles of AMPKα isoforms in neurons are unclear. A recent study in mouse models indicates a critical role of AMPKα2 in cognition and long-term synaptic plasticity. In particular, brain-specific reduction of the AMPKα2 but not AMPKα1 resulted in decreased hippocampal post-synaptic density formation, abnormal spine morphology, and correlated synaptic plasticity deficits (Yang et al., 2020). Whether there exists isoform-specific role of AMPK and the underlying mechanisms associated with pathophysiology (e.g. synaptic impairments) in NHP models of AD is an interesting topic for future studies.
A plethora of studies have firmly demonstrated that maintenance of long-term synaptic plasticity and memory is dependent on de novo protein synthesis (Costa-Mattioli et al., 2009; Klann and Dever, 2004; Malenka and Bear, 2004; Richter and Klann, 2009). Furthermore, impairments of mRNA translation are linked to AD pathophysiology (Beckelman et al., 2019; Ma and Klann, 2012; Ma et al., 2013). AMPK is known to be a nexus for multiple signaling pathways controlling protein synthesis, and upregulation of the AMPK signaling leads to inhibition of general protein synthesis (Ma et al., 2014; Yoon et al., 2012; Zimmermann et al., 2020). Consistently, here we found increased activity of the AMPKα2 isoform in AD-like monkeys, which is indicative of protein synthesis deficits. We did not observe correlated alterations of the mTORC1 and eEF2 signaling cascades (Fig. S2), both are involved in protein synthesis. The results from the unbiased proteomics study suggest dysregulated protein expression in AD-like monkeys. It is possible that protein synthesis is altered through other signaling pathways (other than mTORC1 or eEF2) that are linked to AMPK. Interestingly, many up-regulated or down-regulated proteins identified in the proteomic study are related to energy metabolism, contributing to existing evidence suggesting a mechanistic connection between disruption of energy metabolism homeostasis and AD pathology. Moreover, through the protein-protein interaction analysis, we revealed that most of those up-regulated or down-regulated proteins are related to the AMPK signaling network. Those findings imply that AMPK might be in a pivotal position in the pathogenesis of Alzheimer’s disease. Elucidation of specific roles of dysregulated proteins (identified in the mass spectrometry experiments) in NHP models of AD is warranted for future studies. We noticed that quite a few AD-related dysregulated proteins were not strongly connected to AMPKα2 (PRKAA2) as shown in the protein-protein interaction maps (Fig.6). Several factors could contribute to such results including limitation on the ability of the MS technique to identify proteins that are not so enriched in the brain tissue. Moreover, AMPK is a master kinase and regulation of mRNA translation is one of the many downstream effects of AMPK. It is possible that increased the AMPKα2 activity in the AD-like monkeys (as shown in Fig. 5) is associated with other effects such as protein phosphorylation alterations. Future in-depth studies using comprehensive approaches such as phosphoproteomics analysis shall provide important insights into the roles of the AMPK signaling dysregulation in pathophysiology of the NHP AD model.
Supplementary Material
Fig. S1 Western blot assays of the AMPKα signaling in PFC and cerebellum of AD-like and normally aged monkeys. A-B levels of phospho-AMPKα1 and total AMPKα1 in PFC. C-D levels of phospho-AMPKα2 and total AMPKα2 in PFC. E-F levels of phospho-AMPKα1 and total AMPKα1 in cerebellum. G-H levels of phospho-AMPKα2 and total AMPKα2 in cerebellum. n=3 monkeys for each group, p > 0.05, unpaired independent t test.
Fig. S2 Western blot assays of proteins involved in the downstream signaling of AMPK in the hippocampus of AD-like and normally aged monkeys. A-B levels of phospho-mTORC1 and total mTORC1. C-D levels of phospho-TSC2 and total TSC2. E-F levels of phospho-eEF2 level and total eEF2. G-H levels of phospho-4EBP1and total 4EBP1. n=3 monkeys for each group, p > 0.05, unpaired independent t test.
Highlights.
Hippocampal AMPKα2 activity is increased a non-human primate (NHP) model of AD.
CSF Aβ42 levels are inversely associated with Aβ plaque in the NHP model of AD.
Aβ plaque burden is correlated with levels of either soluble or insoluble Aβ42.
Proteomics analysis reveals association of protein dysregulation and AMPK signaling.
Acknowledgement
We would like to acknowledge the support provided by the Proteomics and Metabolomics Shared Resource of the Wake Forest Baptist Comprehensive Cancer Center (NIH/NCI P30 CA12197). This work was supported by National Institutes of Health grants R01 AG055581, R01 AG056622 (T.M.), P40-OD010965 (C.A.S.), P30 AG049638 (Wake Forest Alzheimer’s Disease Research Center, S.C.), P50 AG005136 (C.D.K.), the Alzheimer’s Association grant NIRG-15-362799 (T.M.), the BrightFocus Foundation grant A2017457S (T.M.), and the Nancy and Buster Alvord Endowment (C.D.K.). The authors have declared no conflicts of interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit Author Statement
X.W. conceptualized the experiments, collected and analyzed data, and wrote the manuscript. X.Z., B.U., J.L., C.S.L., C.D.K. collected data and provided technical help. C.M.F. helped with the analysis of the proteomics data. T.J.M., T.C.R, S.C., C.A.S. advised on the NHP model. T.M. conceptualized experiments and wrote the manuscript. All authors contributed to the writing of the manuscript.
References
- Alzheimer’s Association. 2020Alzheimer’s disease facts and figures. Alzheimers Dement. [DOI] [PubMed] [Google Scholar]
- Beckelman BC, et al. , 2019. Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J Clin Invest. 129, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, et al. , 2009. Translational control of long-lasting synaptic plasticity and memory. Neuron. 61, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Cedazo-Minguez A, 2014. Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans? Frontiers in pharmacology. 5, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell-Bosch J, et al. , 2016. Aβ immunotherapy for Alzheimer’s disease: where are we? Neurodegenerative Disease Management. 6, 179–81. [DOI] [PubMed] [Google Scholar]
- Hardie DG, 2004. The AMP-activated protein kinase pathway–new players upstream and downstream. Journal of cell science. 117, 5479–5487. [DOI] [PubMed] [Google Scholar]
- Hardie DG, 2011. AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes & development. 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, 2014. AMPK—sensing energy while talking to other signaling pathways. Cell metabolism. 20, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Carling D, 1997. The AMP-activated protein kinase: Fuel gauge of the mammalian cell? European journal of biochemistry. 246, 259–273. [DOI] [PubMed] [Google Scholar]
- Hardie DG, et al. , 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology. 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbroner P, Kemper T, 1990. The cytoarchitectonic distribution of senile plaques in three aged monkeys. Acta neuropathologica. 81, 60–65. [DOI] [PubMed] [Google Scholar]
- Heuer E, et al. , 2012. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Current pharmaceutical design. 18, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, Dever TE, 2004. Biochemical mechanisms for translational regulation in synaptic plasticity. Nature Reviews Neuroscience. 5, 931–942. [DOI] [PubMed] [Google Scholar]
- Latimer CS, et al. , 2019. A nonhuman primate model of early Alzheimer’s disease pathologic change: Implications for disease pathogenesis. Alzheimer’s & Dementia. 15, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Ma T, et al. , 2014. Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. Journal of Neuroscience. 34, 12230–12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Klann E, 2012. Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer’s disease. Journal of neurochemistry. 120, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, et al. , 2013. Suppression of eIF2α kinases alleviates Alzheimer’s disease–related plasticity and memory deficits. Nature neuroscience. 16, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, et al. , 2011. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. Journal of Neuroscience. 31, 17537–17546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF, 2004. LTP and LTD: an embarrassment of riches. Neuron. 44, 5–21. [DOI] [PubMed] [Google Scholar]
- McKhann GM, et al. , 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia. 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, et al. , 1992. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 116, 201–211. [DOI] [PubMed] [Google Scholar]
- Pedrós I, et al. , 2014. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 1842, 1556–1566. [DOI] [PubMed] [Google Scholar]
- Phillips KA, et al. , 2014. Why primate models matter. American journal of primatology. 76, 801–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter WB, et al. , 2010. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PloS one. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Klann E, 2009. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes & development. 23, 1–11. [DOI] [PubMed] [Google Scholar]
- Ross FA, et al. , 2016. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. The FEBS journal. 283, 2987–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, et al. , 2011. AMP-activated protein kinase: a potential player in Alzheimer’s disease. Journal of neurochemistry. 118, 460–474. [DOI] [PubMed] [Google Scholar]
- Sani S, et al. , 2003. Distribution, progression and chemical composition of cortical amyloid-β deposits in aged rhesus monkeys: similarities to the human. Acta neuropathologica. 105, 145–156. [DOI] [PubMed] [Google Scholar]
- Shankar GM, et al. , 2008. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 14, 837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE, 2009. AMPK in Health and Disease. Physiol Rev. 89, 1025–78. [DOI] [PubMed] [Google Scholar]
- Tapiola T, et al. , 2009. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 66, 382–9. [DOI] [PubMed] [Google Scholar]
- Toledano A, et al. , 2012. [Does Alzheimer’s disease exist in all primates? Alzheimer pathology in non-human primates and its pathophysiological implications (I)]. Neurologia. 27, 354–69. [DOI] [PubMed] [Google Scholar]
- Van Dam D, De Deyn PP, 2017. Non human primate models for Alzheimer’s disease-related research and drug discovery. Expert opinion on drug discovery. 12, 187–200. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, et al. , 2011. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 121, 337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Jucker M, 2017. The Exceptional Vulnerability of Humans to Alzheimer’s Disease. Trends Mol Med. 23, 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ, 2007. A beta oligomers - a decade of discovery. J Neurochem. 101, 1172–84. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. , 2020. Decreased levels of blood AMPKα1 but not AMPKα2 isoform in patients with mild cognitive impairment and Alzheimer’s disease: a pilot study. Journal of Alzheimer’s Disease. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. , 2019. Therapeutic Potential of AMP-Activated Protein Kinase in Alzheimer’s Disease. J Alzheimers Dis. 68, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, et al. , 2004. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology. 63, 658–63. [DOI] [PubMed] [Google Scholar]
- Yang W, et al. , 2020. Brain-specific suppression of AMPKα2 isoform impairs cognition and hippocampal LTP by PERK-mediated eIF2α phosphorylation. Molecular Psychiatry. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, et al. , 2012. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron. 75, 824–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann HR, et al. , 2020. Brain-specific repression of AMPKalpha1 alleviates pathophysiology in Alzheimer’s model mice. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Western blot assays of the AMPKα signaling in PFC and cerebellum of AD-like and normally aged monkeys. A-B levels of phospho-AMPKα1 and total AMPKα1 in PFC. C-D levels of phospho-AMPKα2 and total AMPKα2 in PFC. E-F levels of phospho-AMPKα1 and total AMPKα1 in cerebellum. G-H levels of phospho-AMPKα2 and total AMPKα2 in cerebellum. n=3 monkeys for each group, p > 0.05, unpaired independent t test.
Fig. S2 Western blot assays of proteins involved in the downstream signaling of AMPK in the hippocampus of AD-like and normally aged monkeys. A-B levels of phospho-mTORC1 and total mTORC1. C-D levels of phospho-TSC2 and total TSC2. E-F levels of phospho-eEF2 level and total eEF2. G-H levels of phospho-4EBP1and total 4EBP1. n=3 monkeys for each group, p > 0.05, unpaired independent t test.