

Abstract
Purpose:
Improved resolution of molecular diagnostic technologies enabled detection of smaller sized exonic level copy number variants (CNVs). The contribution of CNVs to autosomal recessive (AR) conditions would be better recognized using a large clinical cohort.
Methods:
We retrospectively investigated the CNVs’ contribution to AR conditions in cases subjected to chromosomal microarray analysis (CMA, N=~70,000) and/or clinical exome sequencing (ES, N=~12,000) at Baylor Genetics; most had pediatric onset neurodevelopmental disorders.
Results:
CNVs contributed to biallelic variations in 87 cases, including 81 singletons and three affected sibling pairs. Seventy cases had CNVs affecting both alleles, and seventeen had a CNV and an SNV/indel in trans. 94.3% of AR-CNVs affected one gene; among these 41.4% were single-exon and 35.0% were multi-exon partial-gene events. 69.0% of homozygous AR-CNVs were embedded in homozygous genomic intervals. Five cases had large deletions unmasking an SNV/indel on the intact allele for a recessive condition, resulting in multiple molecular diagnoses.
Conclusions:
AR-CNVs are often smaller in size, transmitted through generations, and underrecognized due to limitations in clinical CNV detection methods. Our findings from a large clinical cohort emphasized integrated CNV and SNV/indel analyses for precise clinical and molecular diagnosis especially in the context of genomic disorders.
Keywords: Autosomal recessive, copy number variants, SNV/Indel, clinical molecular diagnoses, multiple molecular diagnoses
Introduction
Autosomal recessive (AR) conditions constitute a subgroup of human genetic disorders that are caused by defects of both copies of a gene located on the autosomes. Individuals affected with an AR disorder often inherit disease-causing alleles from asymptomatic carrier parents. Such disease-causing alleles mostly occur as a de novo variant ancestrally, forming a “founder mutation” for a specific population by escaping purifying selection that often exerts strong effect against deleterious variant alleles causing dominant diseases 1.
AR conditions are caused by complete or near-complete loss-of-function of the gene product, which are largely attributed to single nucleotide variants (SNVs), small insertions/deletions (indels), or copy number variants (CNVs), and rarely copy neutral events such as balanced chromosome translocations/inversions and uniparental disomy 2,3 (UPD). Advancing molecular technologies enables genome-wide SNV/indels detection. In the past decade, expanded next generation sequencing (NGS)-based carrier screening, with a focus on reproductive medicine, has screened apparently healthy individuals for reproductive risks of recessive disorders 4.
CNVs range from hundreds to millions of base pairs (bp). CNVs can cause genetic defects by exon-level events disrupting the reading frame of one disease gene (genic AR-CNV) or deletions of a genomic interval involving two or more disease genes with the potential to cause more than one disorders (genomic AR-CNV) (Figure 1A). Chromosome microarray analysis (CMA), representing the first-tier diagnostic method for developmental disorders and congenital anomalies by clinical consensus 5, has been widely used for the detection of large CNVs (>400 Kb) causing microdeletion/duplication syndromes. Improved resolution of microarrays with exonic coverage has expanded the recognition of contributions of CNV to single gene disorders 6,7. Although the detection of large CNVs at the megabase (Mb) level has become possible using NGS-based methods, detection of small heterozygous CNVs at the exonic level still largely relies on CMA.
Figure 1 –
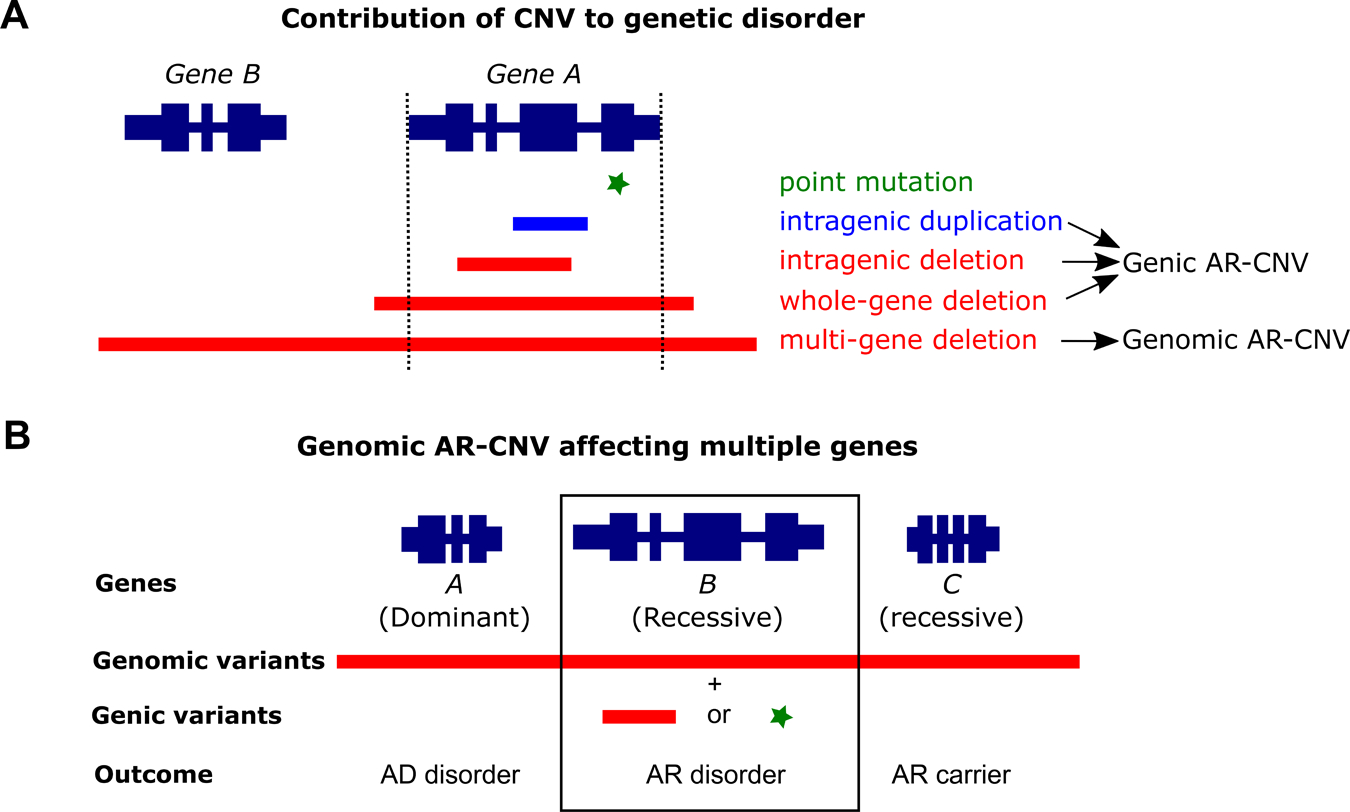
AR-CNVs contribute to diseases by multiple ways. A. A defective gene may have a point mutation, an intragenic duplication or deletion, a whole gene deletion, or a contiguous gene deletion of multiple genes. B. AR-CNVs affecting multiple genes (genomic AR-CNV) may contribute to a more complex outcome. For Panels A and B, genes are presented at the top of the diagrams with the wide dark blue segments representing the exons and the narrow dark blue segments representing the introns. Below the gene diagrams are shown different types of variants: green asterisk, point mutation; blue segment, duplication; red segment, deletion.
CNVs contribute to biallelic variations causing AR conditions by ways that may or may not involve SNV/indels. CNVs affecting a single disease-associated locus may act as the sole variant type to cause an AR disorder by affecting both copies of a gene in homozygous or compound heterozygous configurations. This has been increasingly recognized in individuals with Mendelian diseases 8. Alternatively, single gene events combining an AR-CNV and SNV/indel in trans have also been reported to cause recessive disorders 9. In the context of genomic disorders, large deletions may affect multiple disease-associated loci leading to a complex phenotype: dominant traits may manifest if the deletion causes haploinsufficiency; a recessive genic variant may be uncovered to cause a recessive disorder through compound heterozygosity 10,11 (Figure 1B). Joint CNV and SNV/indel analyses provide extensive utility to identify such events.
We investigated the different ways AR-CNVs contribute to AR conditions by examining a clinical cohort of cases subjected to CMA and/or ES. Our study exemplified the importance of CNV detection in providing accurate molecular diagnoses. The high frequency of homozygous CNVs identified in this study highlighted the transmission of recurrent CNV alleles in healthy populations.
Methods
Samples
We retrospectively analyzed clinical samples submitted for ES (N=~12,000) and CMA (N=~70,000) at Baylor Genetics (BG). The aggregated analyses of anonymized cases were approved by the Baylor College of Medicine Institutional Review Board (protocols H-37568 and H-42680). The variants identified in this study have been submitted to ClinVar (accession numbers SCV001334073—SCV001334129).
CNV detection by CMA
The CMA experimental procedures were performed according to the manufacture’s protocols with minor modifications (Agilent Technologies Inc., Santa Clara, CA, USA) 12. Since 2006, BG has clinically developed and implemented six different versions of customized oligonucleotide arrays (OLIGO V6-V11). OLIGO V8-V11 were designed to interrogate clinically relevant genes with exonic coverage (V8, ~1700 genes; V9-V11, >4,200 genes). SNP probes were also included in the V8.2, V8.3 and V9-V11 OLIGO arrays to enable detection of genomic intervals with absence of heterozygosity (AOH, also referred to as runs of homozygosity). Data analyses were performed using an in-house developed pipeline with published decision-making algorithms 7. Ambiguous CNV calls at the borderline of the cut-off criteria were confirmed by an orthogonal approach, such as PCR or fluorescence in situ hybridization (FISH).
CNV detection by ES and companion SNP array (also called “cSNP array” in this paper)
ES experimental procedures and bioinformatics pipelines were performed according to the previously described methods 13,14. Variant classification followed the American College of Medical Genetics and Genomics (ACMG) guidelines 15. Homozygous/hemizygous deletions were called using an in-house developed pipeline based on exome read-depth analysis as previously described 16.
As part of the quality control measurements, an Illumina SNP array (cSNP array) of two versions (HumanExome-12v1 array, >240K probes, from 2012 to 2016; HumanCoreExome-24v1, >500K probes, since 2016) was run concurrently with ES using aliquots from the same DNA preparation used for ES. The cSNP array data were analyzed for CNV and AOH detection as described 17.
Results
Genomic features of identified AR-CNVs in this study
While genome-wide analyses studies indicated deletion CNV contribute to carrier states at many genic loci, the AR-CNVs in this study only include diagnostic variants contributing to biallelic variation. Among the cases subjected to CMA and/or ES, we identified molecular diagnoses involving clinically relevant AR-CNVs in 87 cases (81 singletons and three pairs of siblings). Among the 174 chromosomes of these 87 cases, 17 contained SNV/indel alleles; the remaining 157 contained AR-CNV alleles.
The majority of AR-CNVs were below 1,000 Kb, except for four CNVs (Figure 2A). The AR-CNVs ranged from a 68 bp deletion of FAM177A1 exon 5 to a 22 Mb deletion of 5p14.3p15.33 encompassing the cri-du-chat region. Homozygous AR-CNVs were identified in 63 cases, containing 126 AR-CNV alleles with size distribution similar to the size distribution of all AR-CNV (Figure 2A). None of the homozygous AR-CNV alleles exceeded 1 Mb (Figure 2A).
Figure 2 –
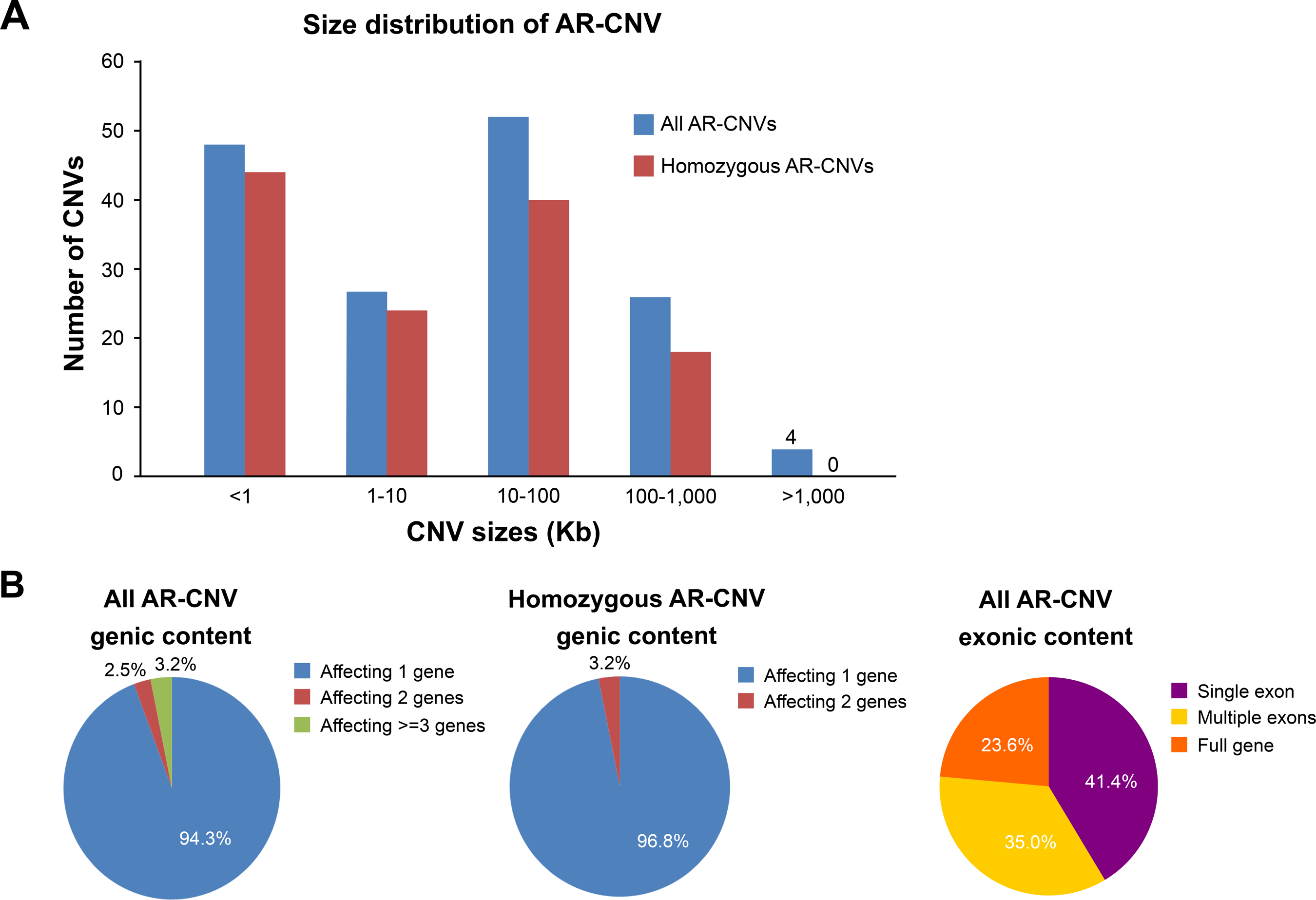
Characteristics of AR-CNVs. A. AR-CNVs had different genomic sizes. The graph compares the genomic sizes of all AR-CNVs and AR-CNVs in a homozygous state. Blue, all AR-CNVs; red, AR-CNVs in a homozygous state. B. AR-CNVs may affect different numbers of disease genes. The left panel shows the distribution of the number of genes affected by AR-CNV in the cohort. The middle panel shows the distribution of gene numbers in cases with homozygous AR-CNV. The right panel shows the distribution of exonic span of the AR-CNV alleles.
Of the 157 AR-CNVs alleles, the majority (94.3%) of the AR-CNVs alleles affected one gene, followed by 2.5% affecting two genes and 3.2% affecting three genes or more (Figure 2B left panel). Homozygous AR-CNV alleles included fewer genes - the majority (96.8%) affected one gene, and the remaining (3.2%) affected two genes (Figure 2B middle panel). This observation was consistent with the increased deleterious impact of larger CNVs. On the exonic level, 41.4% of all AR-CNV alleles affected a single exon, 35.0% spanned multiple exons of a gene, and the remaining 23.6% included the entire gene locus (Figure 2B right panel).
AR-CNVs contribute to diseases in multiple ways
AR-CNVs contribute to recessive disease with or without involvement of SNV/indels. Biallelic AR-CNVs were identified in 70 cases (Figure 3A, 3B). Sixty-three cases had homozygous deletion (N=62) or duplication (N=1). Seven cases had biallelic CNVs in compound heterozygous states involving AR-CNVs that may or may not overlap. Among these seven cases, six had one deletion either partially overlapping with or being nested within the other deletion, resulting in homozygous loss of the overlapping region and biallelic loss of the affected gene; one had two non-overlapping deletions, which were determined to be in trans by parental testing.
Figure 3 – Different mechanisms of AR-CNV contributing to diseases in the current cohort.
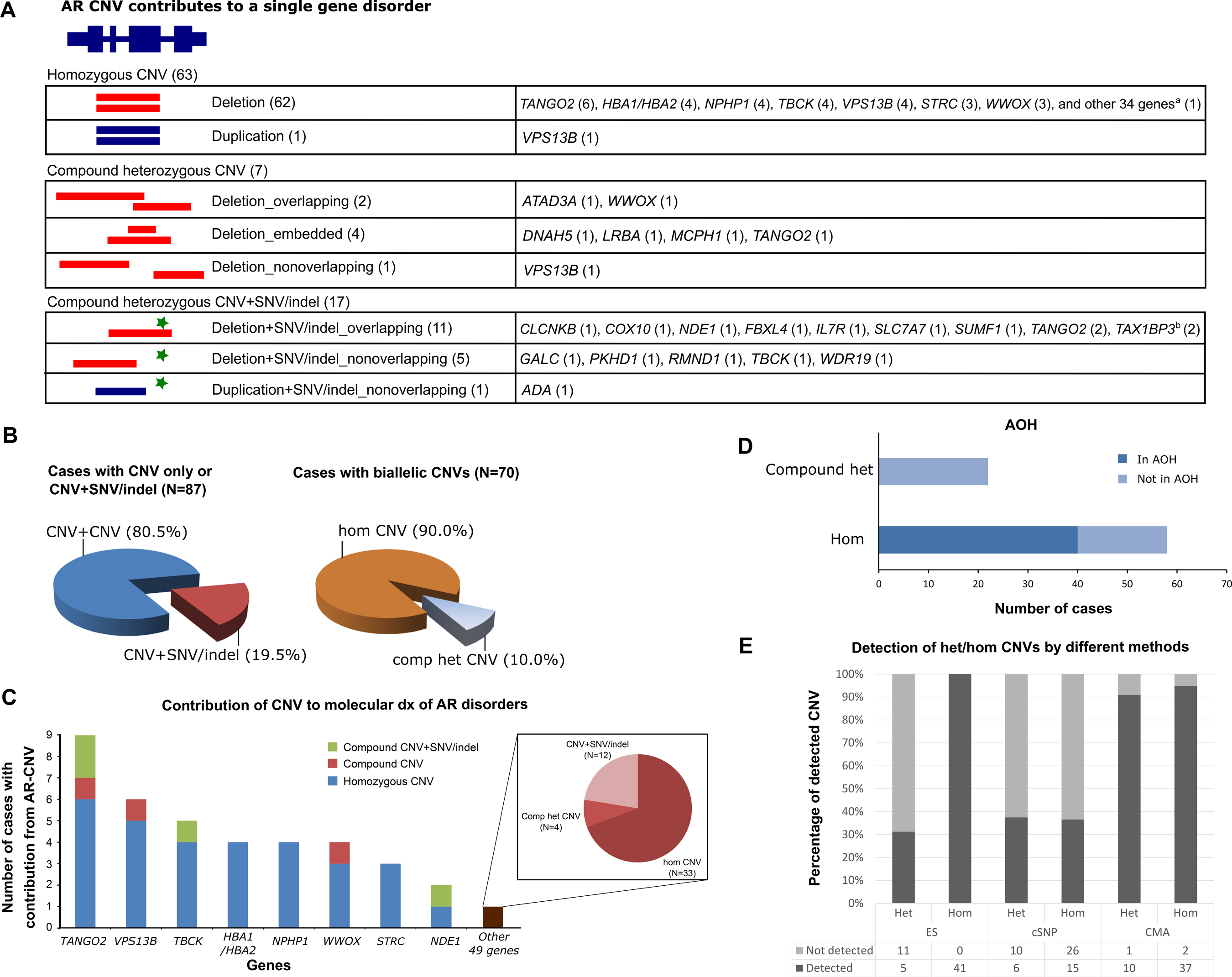
A. AR-CNVs may contribute to recessive disorders in forms of homozygous CNV (62 cases with deletion and one case with duplication), compound heterozygous CNVs with overlapping boundaries (two cases with deletion), one embedded in the other (four cases with deletion), or nonoverlapping boundaries (one case with deletion). AR-CNV may also contribute to recessive disorders in combination with an SNV/indel either inside (11 cases with overlapping deletion and SNV/indel) or outside of the CNV (five cases with nonoverlapping deletion and SNV/indel, one case with nonoverlapping duplication and SNV/indel). The genes affected by the AR-CNV events are noted behind each category. a The 34 genes include ABCB11, ACBD5, ADCK3, ARSB, C12orf65, CFAP52, CLDN1 (2), CLN3, CNTNAP2, CRX, DDR2, DIAPH1, ECE1 (2), ETHE1, FAM177A1, FBP1, GJB6, IFT140, ITGB4, LARGE, NDE1, PLA2G6, PRDM12, SERAC1, SLC3A1, PREPL, SLCO1B3/SLCO1B1, SMN1, SPTA1, SRD5A2, TNNT1, TNNI3, TRAPPC9, and TRIM37. CLDN1 and ECE1 each had AR-CNVs in two patients, who were siblings. Two AR-CNVs involved two independent disease-associated genes (SLC3A1 and PREPL, TNNT1 and TNNI3) in one event, respectively. b TAX1BP3 had AR-CNV in two patients, who were siblings. B. AR-CNVs contributed to the molecular diagnoses in variable ways. The left pie chart compares the percentages of cases with pure CNV contribution (CNV+CNV) versus those with combined CNV and SNV/indel contributions (CNV+SNV/indel). The right pie chart compares the percentages of homozygous CNV (hom CNV) versus compound heterozygous CNV (comp het CNV) within the cases with pure CNV contribution. Blue, percentage of cases with pure contribution from AR-CNVs; maroon, percentage of cases with contributions from both AR-CNV and SNV/indel; brown, percentage of cases with homozygous AR-CNVs; grey, percentage of cases with compound heterozygous AR-CNVs. C. Genes are variably affected by AR-CNVs. The genes recurrently affected by AR-CNVs are presented along the x-axis. The last bar (brown) represents the genes (N=49) that are affected by AR-CNV once in our cohort. The height of each bar represents the total number of cases with contribution from AR-CNVs of the corresponding gene on the x-axis. The colored portions of each bar represent different forms of AR-CNV contribution. The right panel details the different forms of AR-CNV contribution for the uniquely affected genes. D. Most homozygous AR-CNVs were embedded in a region exhibiting absence of heterozygosity (AOH). The graph compares the AR-CNVs in a homozygous state versus those in a compound heterozygous state. E. Comparison of heterozygous or homozygous CNV detection by ES, cSNP array and CMA. The bar graph shows percentage (Y-axis) of detected versus non-detected CNVs by each method (X-axis). Heterozygous and homozygous CNVs are show in separate bars for each method. Light grey, percentage of CNV not detected; dark grey, percentage of CNV detected. The number of CNVs not detected versus detected by each method is listed under the bar graph.
Seventeen cases had AR-CNVs in trans with SNV/indels (Figure 3A, 3B). These include 11 cases with overlapping deletion and SNV/indels, five cases with nonoverlapping deletion and SNV/indels, and one case with nonoverlapping tandem duplication and SNV/indel alleles. Parental studies were performed to determine the phase of variants except for the WDR19 variants.
Genes were variably affected by AR-CNVs
A total number of 57 AR loci were affected by AR-CNVs in forms of homozygous CNV, compound heterozygous CNVs, and compound heterozygous “CNV+SNV/indel” biallelic genotypes in our cohort. TANGO2 was the most frequently affected gene by AR-CNV (N=9). Homozygous deletions affecting TANGO2 were identified in six cases, along with compound heterozygous deletions in one case and compound heterozygous “deletion+SNV/indel” in two cases (Figure 3A, 3C). Other recurrently affected genes include VPS13B (N=6), TBCK (N=5), HBA1/HBA2 locus (N=4), NPHP1 (N=4), WWOX (N=4), STRC (N=3), and NDE1 (N=2). Similar to TANGO2, more biallelic CNVs were detected than “CNV+SNV/indel” for recurrently affected genes, except for NDE1 (Figure 3C). Forty-nine other genes were non-recurrently affected by AR-CNVs: 33 genes had homozygous CNVs, four genes had compound heterozygous CNVs, and 12 genes had “CNV+SNV/indel” genotypes (Figure 3C). The details of genes affected by AR-CNVs can be found in Figure 3C.
Homozygous AR-CNVs were associated with AOH
Homozygous pathogenic variants may be embedded in AOH regions formed by haplotype blocks transmitted in a specific population, a result of identity-by-descent (IBD) 18. AOH has also been used to guide identification of variant or discovery of new disease gene 8,19–22. Recurrent AR-CNV alleles tend to be less frequently detected than SNV/indel due to the limited resolution of genomic assays. We identified homozygous AR-CNVs in 63 patients, 61 of which had homozygous AR-CNVs affecting a single gene. The remaining two cases had homozygous AR-CNVs concurrently affecting two disease-associated genes, resulting in dual molecular diagnoses. Fifty-eight cases with homozygous deletions had homozygosity mapping data available. Among those, forty cases (69.0%, 39 deletions and one duplication) had homozygous AR-CNVs embedded within an AOH interval with variable sizes (minimum <1 Mb, maximum 46 Mb) (Figure 3D). This is significantly different from the 22 cases harboring compound heterozygous biallelic CNVs or CNV+SNV/indel, none of which were identified in an AOH region (Chi-square p < 0.0001) (Figure 3D).
The sum of AOH regions identified in the 40 cases ranged from a single genomic region of 7 Mb to multiple genomic regions summing 367 Mb in each personal genome (Supplemental Table 1). Three cases had a single stretch of AOH with sizes <10 Mb encompassing homozygous AR-CNVs of TBCK, ACBD5 and SPTA1, respectively. Although consanguinity was not indicated for these cases, the observation of the genetic defects being embedded in an AOH region was suggestive of a result of IBD, or a de novo event of UPD. Multiple AOH regions totaling 50 Mb and above were identified in 29 cases, indicating consanguinity between second cousins or closer relatives.
Homozygous recurrent CNVs were identified in unrelated cases. These recurrent homozygous CNVs included TANGO2 exons 3–9 deletion in five cases, VPS13B exons 18–23 deletion in two cases, TBCK exon 23 deletion in four cases, and WWOX exon 7 deletion in two cases. These recurrent AR-CNVs, lacking extensive homologous sequences surrounding the breakpoint region, were unlikely to be events resulting from nonallelic homozygous recombination (NAHR) and may instead be ancestral variants transmitted through generations.
Homozygous AR-CNVs may simultaneously provide more than one diagnosis
Five cases had genomic AR-CNVs affecting both haploinsufficient genes and AR disease genes (Cases 1–8, 8–1, 21, 25, and 50), resulting in genomic disorders as well as recessive conditions (Supplemental Table 1 and 2). Homozygous AR-CNVs affecting three or more genes were not identified among the 63 cases with homozygous AR-CNVs, supporting the expectation that multi-gene CNVs are more likely to reduce reproductive fitness. We identified two homozygous deletions concurrently affecting two genes, potentially providing dual molecular diagnoses for these two cases (Table 1). The first case (Case 44) had a 77 Kb homozygous deletion encompassing the entire SLC3A1 gene (OMIM* 104614), causing cystinuria (OMIM# 220100) in either an AD or AR manner, and exons 5–15 of PREPL (OMIM* 609557), causing congenital myasthenic syndrome 22 (OMIM# 616224) in an AR manner. The parents were heterozygous for this deletion. SLC3A1 and PREPL map on the human genome in close physical proximity with the last exons overlapping, increasing the chance for a deletion to affect both genes. In fact, deletions with variable genomic span encompassing both genes were reported in individuals with hypotonia-cystinuria syndrome 23. Close gene proximity is associated with a higher frequency of contiguous gene deletion. In the Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home?ref=hg19), 11/48531 individuals were found to carry nonrecurrent deletions involving both genes. Consistently, in the BG databases where ~70,000 cases were tested by CMA, six unrelated cases were reported as carriers for such deletions.
Table 1.
Deletions affecting consecutive disease-associated genes.
| Deletion coordinates (hg19) | Genomic content | Gene 1: Disorder 1 | Gene 2: Disorder 2 | Cases in BG CMA database | Carrier frequency in DGV |
|---|---|---|---|---|---|
| Chr2:44494834–44571747 | SLC3A1 entire gene and PREPL exons 5–15 | SLC3A1 (OMIM* 104614, AD/AR): Cystinuria (OMIM# 220100) | PREPL (OMIM* 609557, AR): Congenital myasthenic syndrome 22 (OMIM# 616224) | Case 44 (homozygous), six other cases (heterozygous) | 11/48531 |
| Chr19:55652251–55663286 | TNNT1 exon 1–9 and TNNI3 exon 8 | TNNT1 (OMIM* 191041, AR): Amish type Nemaline myopathy 5 (OMIM# 605355) | TNNI3 (OMIM* 191044, AD/AR): cardiomyopathy, dilated or hypertrophic (OMIM# 115210, 611880, 613286, 613690) | Case 52 (homozygous), two other cases (heterozygous) | 0 |
The second case 24 (Case 52) had an 11 Kb homozygous deletion encompassing exons 1–9 of TNNT1 (OMIM* 191041), causing Amish type nemaline myopathy 5 (OMIM# 605355) in an AR manner, and exon 8 of TNNI3 (OMIM* 191041), causing cardiomyopathies (OMIM# 115210, 611880, 613286, 613690) in either AD or AR manner (Table 2). Although TNNT1 and TNNI3 are separated by ~2.5 Kb, deletions involving both genes were rare in the general population. Such deletions were not observed in either DGV or BG internal database. Interestingly, this case had multiple AOH regions totaling 18 Mb, and this deletion was embedded in a stretch of a 3 Mb AOH region, indicating a result of IBD. The parental genotypes were unknown.
Table 2.
Contiguous gene deletions unmasked a recessive disease locus.
| Chromosome bands | Genomic coordinates (hg19) | Microdeletion syndromes | Unmasked recessive variants |
|---|---|---|---|
| 3p26.3p26.1 | Chr3:107776–5257572 | 3p- syndrome (OMIM# 613792) | c.836C>T (p.A279V) VUS* in SUMF1 |
| 5p15.33p14.3 | Chr5:71904–22078969 | cri-du-chat syndrome (OMIM# 123450) | DNAH5 exon 32 deletion |
| 16p13.11 | Chr16:15521713–16292235 | 16p13.11 deletion syndrome (PMID 20398883) | c.872C>T (p.S291F) VUSa in NDE1 |
| 17p12 | Chr17:14128550–15422557 | Hereditary neuropathy with liability to pressure palsies (OMIM# 162500) | c.1277_1282dup (p.M426_L427dup) VUS in COX10 |
| 22q11.21 | Chr22:18912403–21431174 | DiGeorge/Velocardiofacial syndrome (OMIM# 188400) | TANGO2 exons 3–9 deletion |
VUS: variant of unknown significance
Other multiple molecular diagnoses involving AR-CNV
Among the 87 cases with molecular diagnoses attributed to AR-CNV, 17 (19.5%) cases had more than one molecular diagnosis (Supplemental Table 2). These included cases with molecular diagnoses involving additional loci in the mitochondrial and/or nuclear genomes. More importantly, seven cases had AR-CNV related multiple molecular diagnoses. In addition to the two cases described above with homozygous deletion affecting two adjacent disease-associated genes, five large deletions caused haploinsufficiency in the meanwhile unmasked a recessive pathogenic variant allele, resulting in possible dual molecular diagnoses (Table 2). These deletions included a 5.2 Mb deletion of 3p26.3p26.1 causing 3p minus syndrome (OMIM# 613792), a 22.0 Mb deletion of 5p15.33p14.3 causing cri-du-chat syndrome (OMIM# 123450), a 0.77 Mb deletion of 16p13.11 causing 16p13.11 deletion syndrome 25, a 1.3 Mb deletion of 17p12 causing hereditary neuropathy with liability to pressure palsies (OMIM# 162500), and a 2.5 Mb deletion of 22q11.21 causing DiGeorge/Velocardiofacial syndrome (OMIM# 188400). These deletions unmasked variants including three SNV/indels (SUMF1, NDE1, and COX10) and two AR-CNVs (DNAH5 and TANGO2) at the corresponding loci (Table 2).
Discussion
We focused on the CNV events including deletions and intragenic tandem duplications that are predicted to cause reduced dosage or functional defects of genes associated with AR disorders in a clinical cohort assembled from cases with CMA and/or ES analyses. We identified 87 cases with molecular diagnoses of AR conditions involving CNVs, emphasizing the important contribution of CNVs to disease etiologies of AR diseases. Our data demonstrate the clinical utility of integrated CNV and SNV/indel analyses for a more comprehensive molecular diagnostic evaluation.
This study suggested that AR-CNVs may be under-recognized for AR conditions. Nine loci (TANGO2, VPS13B, TBCK, HBA1/HBA2, NPHP1, WWOX, STRC, and NDE1) were affected by AR-CNVs in more than one case in our cohort (Figure 3C). CNV alleles of these loci out-numbered SNV/indels, indicating remarkable contribution of CNV to disorders associated with these loci. However, this observation may be biased by our cohort being more likely to contain cases with heterogeneous and clinically unrecognizable phenotypes and by the diagnostic techniques. In fact, the deletion alleles of NPHP1 and HBA1/HBA2 are well-known major pathogenic alleles for the NPHP1-associated ciliopathies and α-thalassemia, respectively, and the high carrier frequencies of these deletions in the general population have demanded extensive carrier screening 6,9,26–28. The recurrent NPHP1 deletion is mediated by NAHR and no ethnic specificity is reported 27, while deletions involving HBA1/HBA2 are known to include multiple types highly specific to ethnicities 29. Defects of VPS13B cause Cohen syndrome (OMIM# 216550) in an AR manner. Numerous SNV/indels or gene-disrupting CNVs resulting in VPS13B defect have been reported in individuals with Cohen syndrome, among which deletions or duplications of VPS13B were observed at high frequency 30. TANGO2, TBCK, WWOX, and NDE1 are recently identified AR disease genes that were associated with disorders with extensive phenotypic heterogeneity. Although a limited number of disease-causing alleles were identified for these genes, CNV alleles appeared to constitute a relatively large fraction of the mutant alleles. The TANGO2 exons 3–9 deletion was recurrently observed in the Hispanic/European descent, while the exons 4–6 deletion allele was confined to the Arabic population to-date 16. Another recurrently identified deletion in our cohort is the TBCK exon 23 deletion. Among the five cases with ethnicity information, three were European Caucasian, one was Middle Eastern, and one was South Asian (Supplemental Table 1). Such diversity may represent a genetic drift event after the origin of the mutation allele in the ancestral population, or de novo events in diverse populations due to locus-specific genomic instability. Our data is limited due to the heterogeneous nature of the clinical cohort, thus a comprehensive analysis of the correlation between the CNV alleles and ethnicity was not performed. The gnomAD and BG internal CMA databases contained heterozygous AR-CNVs in the above loci (Supplemental Table 3 and 4). Further analysis is warranted especially for genes with recurrent AR-CNVs.
The contribution of AR-CNV may be underrepresented in our cohort due to the technical limitation of CNV detection methods. SNP array, CMA, and ES were the three major assays used for CNV detection in this study. The number of probes on a SNP array can range from hundreds of thousands to millions, which potentially provides high resolution CNV detection with larger number of SNP probes. However, the design of probe coverage depends on the SNP distribution, hence non-customizable coverage design. CMA at BG used the array comparative genomic hybridization (aCGH) platform, allowing customized probe design at regions of interest. This provides ultra-high resolution at targeted regions and enables CNV detection at the exonic level, which is less achievable by SNP arrays 22. Numerous computational algorithms have been developed to improve the CNV detection using capture-based ES data 31, which remains challenging for clinical usage due to sub-optimal sensitivity and specificity and requires extensive confirmation by secondary methods such as aCGH or multiplex ligation-dependent probe amplification (MLPA) 3. The sensitivity of ES in clinically relevant CNV detection has been demonstrated to be higher with larger (e.g. Mb sized) events 32,33. Homozygous or hemizygous deletions can be readily detected by ES 8. Inter-assay comparison among ES, CMA and cSNP array used in this study shows that ES has a higher sensitivity for detecting homozygous CNVs than CMA (Figure 3E). The missed CNV detection by CMA may be caused by lack of probe coverage due to novel disease loci or unavailability of appropriate probes in that region. cSNP array has the lowest sensitivity for homozygous CNVs, because many such CNVs in our cohort are small, exonic CNVs, which are beyond the resolution of the cSNP array. For heterozygous CNVs, CMA apparently has the highest sensitivity because of exon-by-exon coverage of the CMA design, which allows detection of ultra-small CNVs. Intra-assay comparison of three assays suggests that ES has a higher sensitivity for homozygous events than heterozygous ones, while cSNP array and CMA have comparable CNV detection capability for either heterozygous or homozygous events (Figure 3E). cSNP array has overall low sensitivity for both heterozygous and homozygous events, which may be caused by the lack of SNP probe coverage for certain regions, especially small ones, of the genome.
Small heterozygous duplications/deletions involving few or partial exons remain challenging for ES. For scenarios where a deletion overlaps another deletion or SNV/indel in trans, the resulting call of a homozygous deletion or SNV/indel may trigger alert for an overlapping deletion event. However, for nonoverlapping CNVs or SNV/indels, the CNV events may potentially be missed if they are below the resolution of detection, and therefore underrecognized due to assay limitation. The vast majority of AR-CNVs detected in our study are deletions. Only two duplications were detected in our cohort, consistent with the technical challenges of detecting small duplication events.
Concurrent analyses of both SNV/indels and CNV are needed for a more comprehensive evaluation of the genetic changes underlying the personal genome of a clinically affected individual. For example, in our cohort, one case had a COX10 SNV/indel in trans with the 17p12 deletion. The COX10 gene spans one end of the breakpoint of 17p12 deletion, and therefore one copy of the gene is disrupted in individuals with such deletion. The identified hemizygous p.M426_L427dup variant in exon 7 on the intact allele in combination of the deletion resulted in COX10 deficiency. Notably, more than 20 years ago, COX10 deficiency had been predicted for individuals with a more complicated clinical phenotype involving mitochondrial myopathy in addition to a neuropathy 34.
CMA serves as the first-tier genome-wide assay for individuals with neurodevelopmental disorders, with a 10–20% diagnostic yield 5. ES can effectively provide potential molecular diagnoses for 25% or more of individuals affected with rare genetic disorders 13,14,35. Recent publications have reported an ~2% increase when implementing CNV detection in ES, demonstrating the advantage of integrating both CNV and SNV/indel analyses 17,32,36. In our cohort, homozygous deletions were mostly detectable by exon-focused CMA or ES read-depth analysis. For recessive disorders involving both CNVs and SNV/indel, ES is needed to provide the SNV/indel findings to corroborate with the CNV findings. However, ES, CMA or SNP arrays are not routinely used to detect balanced structural variants, such as inversions and balanced translocations. Genome sequencing (WGS) provides CNV detection capacity comparable to CMA 2, as well as potential detection of balanced structural variants that are not readily detectable by CMA or ES 37, offering a unique opportunity to interrogate both SNV/indel and CNV/SV in one assay.
Molecular diagnoses involving two or more disease loci were reported in 4.9% of cases positive by ES 38. In this study, we identified two or more molecular diagnoses in 19.5% (17/87) of cases (Supplementary Table 2). This percentage is significantly higher than the rate previously observed in 2076 cases with positive molecular findings 38, a population without a pre-selection for CNV contributions (Fisher’s exact test p < 0.0001). This high rate is not unexpected when comparing to the estimated multi-locus diagnoses under Poisson model (14.0%) or independence model (26.4%) 38. The high multi-locus diagnoses rate in our cohort may be largely attributed to the CNV contributions. In the 17 cases identified with multiple molecular diagnoses, 11.8% (2/17) were attributed to homozygous AR-CNVs affecting two disease associated loci, and 29.4% (5/17) were attributed to large genomic deletions that unmasked a recessive disease allele. Therefore, multiple molecular diagnoses may be related to a genomic disorder. The recessive conditions unmasked by a genomic deletion may contribute to a more complicated clinical presentation. Note that some genomic deletions, such as the deletions of 16p13.11 and 17p12, have reported incomplete penetrance or an age-dependent disease manifestation. These deletions may be present in asymptomatic individuals and run through generations, further complicating the genetic counseling and reproductive risk assessment. If we exclude the cases with multi-locus molecular diagnoses due to genomic deletions, the multi-locus diagnoses rate (11.5%, 10/87) is still significantly higher than the previous observation of ~5% (Fisher’s exact test p = 0.0119). Although the majority of multiple molecular diagnoses are attributed to SNV/indels, attention needs to be paid to CNV contribution especially for recessive disorders. Nonetheless, it is always recommended to examine for potential CNV alleles for an AR gene found with one pathogenic variant in an individual with related phenotypes.
Currently, CMA and ES are the two most frequently used approaches for genome-wide detection of genetic variants. Combined ES and CMA would provide more informative molecular diagnosis although the combined cost is high. Alternatively, sequential testing can be used. For cases with prior CMA results, ES should be considered for those with negative results, and for those with positive CMA results yet not fully explaining clinical phenotypes based on clinical correlation. For cases with one heterozygous finding in a gene highly specific to clinical phenotypes by ES, additional CMA or targeted CNV analysis should be considered.
In summary, we described different ways by which CNVs may contribute to AR disorders. Similar to SNV/indels, AR-CNVs may occur ancestrally and transmit through generations. Homozygous AR-CNVs may result from IBD. AR-CNVs may be of higher carrier frequencies in the general population for specific AR diseases, suggesting expanded carrier screening for both SNV/indels and CNVs. Since AR-CNVs may contribute to multiple molecular diagnoses via concurrent impact on contiguous disease genes, or by unmasking of recessive disease alleles, a comprehensive genomic evaluation, such as combined CMA and ES analyses or perhaps WGS, should be considered for individuals with complex or atypical phenotypes.
Supplementary Material
Supplementary Table S1. Cases with contributions of CNVs to AR disorders
Supplementary Table S2. Cases with multiple molecular diagnoses involving contributions from AR-CNVs.
Supplemental Table 3. Frequency of deletion CNVs in the gnomAD databases for the genes recurrently found with AR-CNVs in our cohort.
Supplemental Table 4. Heterozygous carrier alleles reported in BG CMA database.
Acknowledgement
This study is partially supported by the NHGRI/NHLBI grant UM1HG006542 to the BHCMG; NINDS grant R35 NS105078-01 to JRL.
References
- 1.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trost B, Walker S, Wang Z, et al. A Comprehensive Workflow for Read Depth-Based Identification of Copy-Number Variation from Whole-Genome Sequence Data. Am J Hum Genet. 2018;102(1):142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao R, Zhang C, Yu T, et al. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol Cytogenet. 2017;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled Fetal Risk of Genetic Diseases Identified by Expanded Carrier Screening. JAMA. 2016;316(7):734–742. [DOI] [PubMed] [Google Scholar]
- 5.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boone PM, Bacino CA, Shaw CA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31(12):1326–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gambin T, Yuan B, Bi W, et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Med. 2017;9(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gambin T, Akdemir ZC, Yuan B, et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45(4):1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindstrand A, Davis EE, Carvalho CM, et al. Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum Genet. 2014;94(5):745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1(6):e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lupski JR. 2018 Victor A. McKusick Leadership Award: Molecular Mechanisms for Genomic and Chromosomal Rearrangements. Am J Hum Genet. 2019;104(3):391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung SW, Shaw CA, Yu W, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7(6):422–432. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalani SR, Liu P, Rosenfeld JA, et al. Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations. Am J Hum Genet. 2016;98(2):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dharmadhikari AV, Ghosh R, Yuan B, et al. Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med. 2019;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet. 2013;132(11):1197–1211. [DOI] [PubMed] [Google Scholar]
- 19.Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10(2):148–161. [DOI] [PubMed] [Google Scholar]
- 20.Karaca E, Harel T, Pehlivan D, et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron. 2015;88(3):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan B, Pehlivan D, Karaca E, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015;125(2):636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiszniewska J, Bi W, Shaw C, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;22(1):79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaeken J, Martens K, Francois I, et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am J Hum Genet. 2006;78(1):38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Streff H, Bi W, Colon AG, Adesina AM, Miyake CY, Lalani SR. Amish nemaline myopathy and dilated cardiomyopathy caused by a homozygous contiguous gene deletion of TNNT1 and TNNI3 in a Mennonite child. Eur J Med Genet. 2019;62(11):103567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinzen EL, Radtke RA, Urban TJ, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86(5):707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shang X, Peng Z, Ye Y, et al. Rapid Targeted Next-Generation Sequencing Platform for Molecular Screening and Clinical Genotyping in Subjects with Hemoglobinopathies. EBioMedicine. 2017;23:150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan B, Liu P, Gupta A, et al. Comparative Genomic Analyses of the Human NPHP1 Locus Reveal Complex Genomic Architecture and Its Regional Evolution in Primates. PLoS Genet. 2015;11(12):e1005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parisi MA, Bennett CL, Eckert ML, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75(1):82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98(1):250–251. [DOI] [PubMed] [Google Scholar]
- 30.Parri V, Katzaki E, Uliana V, et al. High frequency of COH1 intragenic deletions and duplications detected by MLPA in patients with Cohen syndrome. Eur J Hum Genet. 2010;18(10):1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadalayil L, Rafiq S, Rose-Zerilli MJ, et al. Exome sequence read depth methods for identifying copy number changes. Brief Bioinform. 2015;16(3):380–392. [DOI] [PubMed] [Google Scholar]
- 32.Pfundt R, Del Rosario M, Vissers L, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19(6):667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Retterer K, Scuffins J, Schmidt D, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med. 2015;17(8):623–629. [DOI] [PubMed] [Google Scholar]
- 34.Reiter LT, Murakami T, Koeuth T, Gibbs RA, Lupski JR. The human COX10 gene is disrupted during homologous recombination between the 24 kb proximal and distal CMT1A-REPs. Hum Mol Genet. 1997;6(9):1595–1603. [DOI] [PubMed] [Google Scholar]
- 35.Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med. 2018. [DOI] [PubMed] [Google Scholar]
- 36.Bergant G, Maver A, Lovrecic L, Cuturilo G, Hodzic A, Peterlin B. Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: a retrospective survey in 1,059 cases. Genet Med. 2018;20(3):303–312. [DOI] [PubMed] [Google Scholar]
- 37.Dong Z, Wang H, Chen H, et al. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genet Med. 2018;20(7):697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017;376(1):21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Cases with contributions of CNVs to AR disorders
Supplementary Table S2. Cases with multiple molecular diagnoses involving contributions from AR-CNVs.
Supplemental Table 3. Frequency of deletion CNVs in the gnomAD databases for the genes recurrently found with AR-CNVs in our cohort.
Supplemental Table 4. Heterozygous carrier alleles reported in BG CMA database.