
Figure 1.
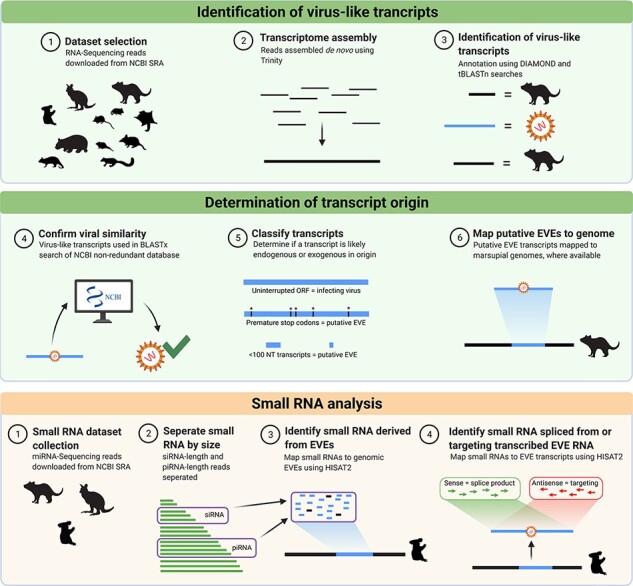
The bioinformatics workflow used in this study to identify viral transcripts from thirty-five marsupial RNA-Seq data sets. Publicly available data sets were downloaded from NCBI SRA and quality checked using FastQC before assembly into transcriptomes, using Trinity. DIAMOND was used to annotate each assembled transcript as host, viral, or other. Viral transcripts were filtered to merge overlapping hits and remove duplicate hits. To confirm the viral origin of each transcript, a reciprocal BLASTx search of the NCBI nr protein database was performed. Transcripts were classified as EVEs if they mapped to the representative marsupial genome (where available), or putative EVEs if they had interrupted reading frames, identity to confirmed EVEs, or were from frequently endogenised viral families. Small RNA analysis was undertaken to identify if any EVEs gave rise to small RNA molecules. Figure created using BioRender.com.