
Fig. 2 |. Phylogenetic inference for retrospective lineage tracing.
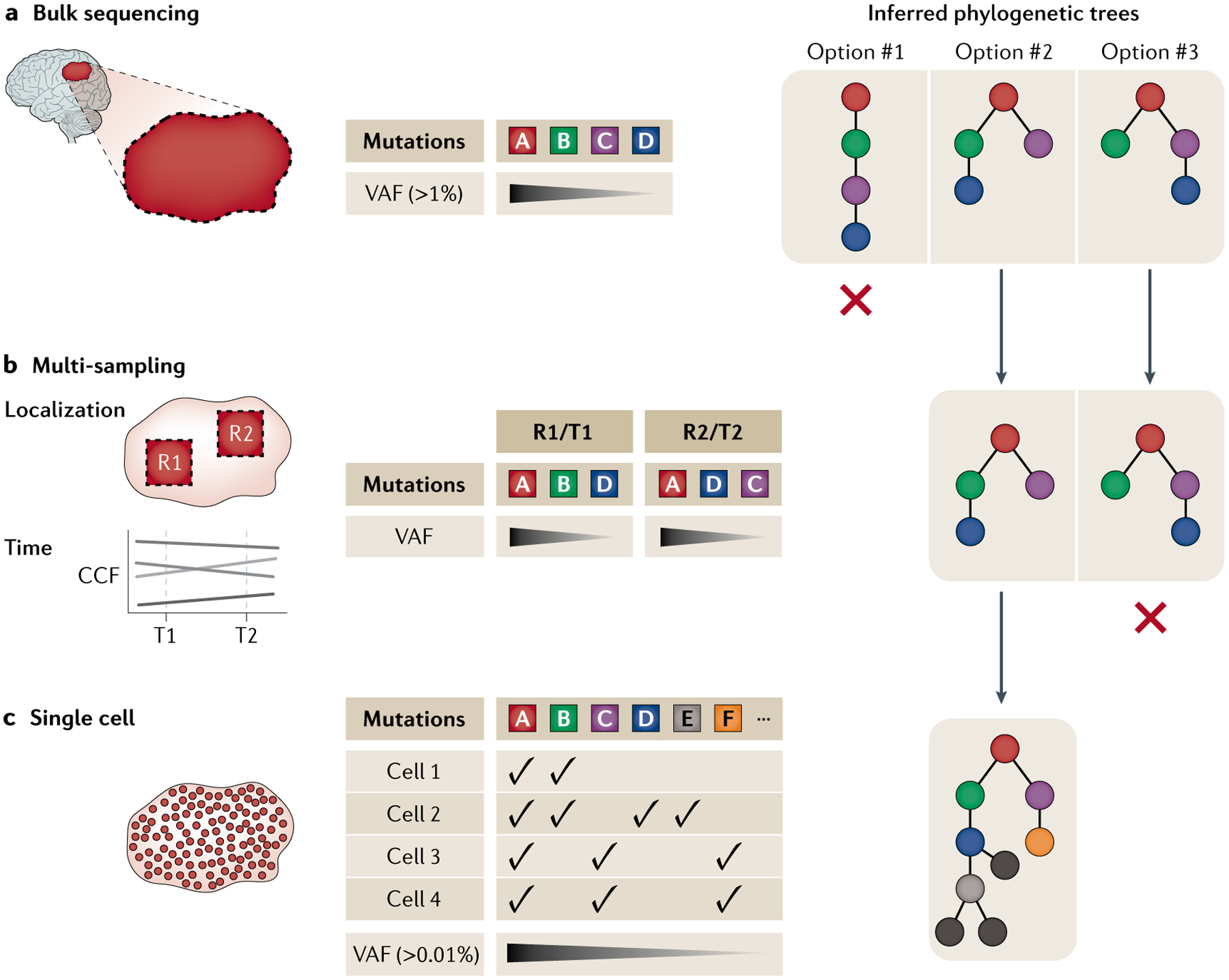
a | Bulk next-generation sequencing allows inference of clonal architecture phylogenetic trees of genetically heterogeneous populations. However, these data can resolve clonal and subclonal relationships to a limited extent by enabling the assessment of the order of acquisition of mutations (A–D) and are limited in their abilities to resolve the phylogenetic relationships of clones, especially at low cancer cell fractions (CCFs). b | Multi-sampling at different time points (T1, T2) during clonal evolution or at different regions (R1, R2) within a tumour (to assess for intratumoural clonal spatial composition) can provide higher-resolution phylogenetic relationships, even for subclones with low CCFs, owing to coordinated patterns of CCF fluctuations over time. c | Even though additional resolution is gained through multi-sampling, resolving phylogeny at single-cell resolution (by single-cell whole-genome sequencing or targeted sequencing) is required to derive the precise clonal dynamics and evolutionary history of a tumour. High-resolution trees pave the way for critical inferences derived directly from primary patient tumours for defining key parameters of somatic evolution. VAF, variant allele frequency.