

Abstract
While CD19-directed CAR T cells can induce remissions in patients with B cell acute lymphoblastic leukemia (ALL), a large subset relapse with CD19-negative disease. Like CD19, CD22 is broadly expressed by B-lineage cells and thus serves as an alternative immunotherapy target in ALL. Here we present the composite outcomes of two pilot clinical trials (NCT02588456 and NCT02650414) of T cells bearing a 41BB-based CD22-targeting CAR in patients with relapsed or refractory ALL. The primary endpoint of these studies was to assess safety, and the secondary endpoint was anti-leukemic efficacy. We observed unexpectedly low response rates, prompting us to perform detailed interrogation of the responsible CAR biology. We found that shortening of the amino acid linker connecting the variable heavy and light chains of the CAR antigen-binding domain drove receptor homo-dimerization and antigen-independent signaling. In contrast to CD28-based CARs, autonomously signaling 41BB-based CARs demonstrated enhanced immune synapse formation, activation of pro-inflammatory genes and superior effector function. We validated this association between autonomous signaling and enhanced function in several CAR constructs, and based on these observations designed a new short-linker CD22 scFv for clinical evaluation. Our findings suggest that tonic 41BB-based signaling is beneficial to CAR function and demonstrate the utility of bedside-to-bench-to-bedside translation in the design and implementation of CAR T cell therapies.
Introduction
Refractory or relapsed B-cell acute lymphoblastic leukemia (ALL) is often resistant to chemotherapy and has a very poor prognosis1,2. Immunotherapy using chimeric antigen receptor (CAR)-engineered T lymphocytes targeting CD19 (CART19) has demonstrated impressive results against ALL, with many patients achieving complete disease remission (CR) after therapy3–6.
Long-term follow up, however, has revealed that a significant fraction of patients who achieve remission ultimately relapse. In 40–60% of relapses, the recurrent leukemia lacks surface expression of CD193,7,8. Target antigen loss can occur by multiple mechanisms9–12, highlighting the need for alternative immunotherapeutic strategies in these patients.
CD22 is a cell-surface molecule that, similarly to CD19, is broadly and uniquely expressed by B-lineage cells. A recent study conducted at the National Cancer Institute (NCI) reported the first clinical experience using CD22-directed CAR T cells (CART22) in patients with ALL, many of whom had relapsed with CD19-negative disease after CART1913,14. In this report, the investigators demonstrated that >70% of patients in the highest-dose cohort achieved CR. The CAR used in this study was composed of variable heavy (VH) and light (VL) chains derived from the m971 monoclonal antibody15,16 connected by a five amino acid linker (GGGGS) to form a single-chain variable fragment (scFv). This scFv was linked to hinge and transmembrane regions originating from the surface molecule CD8α linked to CD3ζ and 4–1BB intracellular signaling domains. Using an anti-CD22 CAR with the same VH and VL but connected by a longer 20 amino acid ((GGGGS)4) scFv linker instead of a short five amino acid sequence, we also performed clinical trials in pediatric and adult patients with ALL at the Children’s Hospital of Philadelphia (CHOP) and the University of Pennsylvania (Penn). Despite in vivo expansion of CART22 and some transient responses, overall clinical activity was surprisingly poor.
Several studies have shown that CAR hinge and transmembrane motifs influence CAR T cell activity17–20, however the scFv linker has not previously been implicated in regulating CART efficacy21. While acknowledging the challenges inherent to inter-trial comparisons, we sought to understand the relationship between CAR22 scFv structure and T cell function responsible for the apparently discrepant outcomes. Here, we report the outcomes of our CD22 CAR pilot clinical trials and elucidate the mechanisms driving the divergent outcomes in patients treated with short and long scFv linker CD22 CAR T cells. We identify that modifications to scFv structure can lead to autonomous CAR signaling which, in the context of 41BB-based co-stimulation, leads to enhanced CAR-driven T cell effector function.
Results
Clinical activity of CART22 cells in pediatric and adult ALL
We conducted two pilot clinical trials in children or adults with relapsed or refractory CD22+ B-cell ALL (ClinicalTrials.org Identifiers NCT02650414 and NCT02588456, respectively). Autologous T cells were collected and engineered as previously described22,23 to express a CAR composed of variable heavy and light chains derived from the m971 antibody connected by the University of Pennsylvania’s standard 20 amino acid linker to create a CD22-targeted scFv3,24. Nine children and five adults were screened for trial enrollment, and ultimately six children and three adults were treated (Figure 1a, Extended Data 1, Methods). Among the nine patients treated, pre-infusion bone marrow assessment demonstrated that one pediatric patient underwent a phenotypic lineage switch from lymphoid to myeloid leukemia and was taken off the study protocol for alternative treatment. All patients had previously undergone CD19-directed therapy and most had CD19-negative leukemia (2/3 adults, 4/5 children; Table 1). Anti-leukemic bridging therapy was allowed during CART22 production at the discretion of treating physician, and lymphodepletion with fludarabine (30mg/m2 daily for four days) and cyclophosphamide (500mg/m2 daily for two days) was performed prior to CART22 infusion (Supplementary Table 1). The planned treatment dose of CART22 cells was 1–10×106 CAR+ cells/kg for pediatric patients <50kg or 5×108 CAR+ total cells for pediatric patients >50kg and adults, and was delivered in a standard split-dosing scheme (10% on day 1, 30% on day 2, 60% on day 3). At time of treatment, three of five pediatric patients had morphologic disease and two were in remission with disease detectable by minimal-residual disease (MRD) evaluation; of the three adult patients, two had morphologic disease and one was in an MRD-negative remission.
Figure 1 |. Clinical trial of CAR22 T cells in adults and children with relapsed ALL.
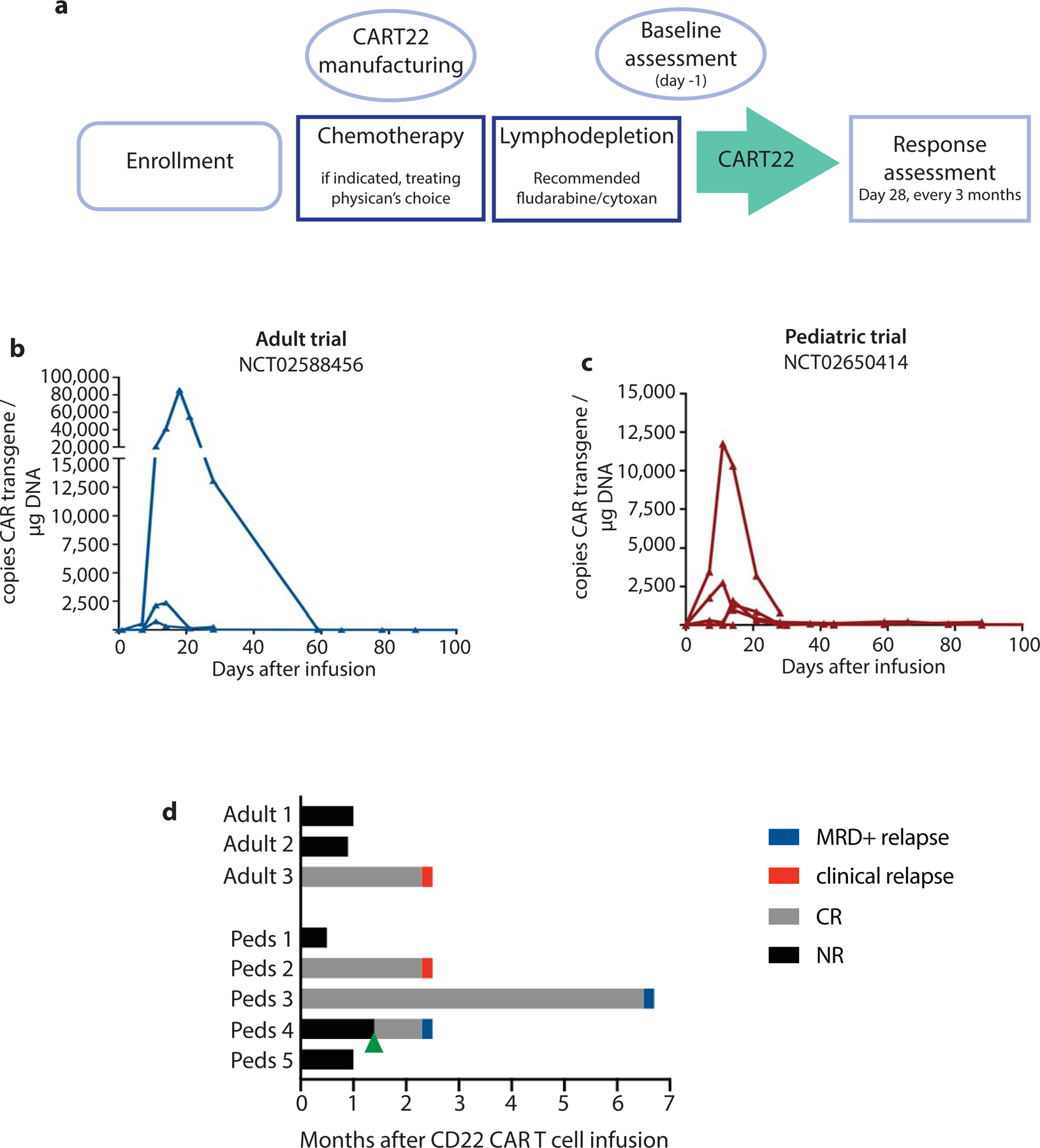
a, Schema of trial design. b-c, Expansion and persistence of CART22 in peripheral blood from treated b, children and c, adults. d, Outcomes after CART22 treatment. Green arrow represents a second CART22 infusion in pediatric patient 4.
Table 1 |.
Characteristics and clinical outcomes of patients treated with CART22.
| Subject | % marrow blasts at time of treatment | CD19 expression at time of treatment | CART22 cells infused (doses of 3) | Peak CART22 expansion (copies/ug DNA) | CRS (grade) | Best disease response |
|---|---|---|---|---|---|---|
| Adult 1 | 97% | Positive | 2.00×108 (2) | 2359.30 | Y (3) | NR |
| Adult 2 | 95% | Negative | 5.00×107 (1) | 85697.83 | Y (2) | NR |
| Adult 3 | 0% (MRD-negative) | Negative | 5.00×108 (3) | 756.15 | Y (1) | CR (MRD-neg) |
| Pediatric 1 | 96% | Positive | 2.77×108 (3) | 0 | N | NR |
| Pediatric 2 | 1% (MRD-positive) | Negative | 5.00×108 (3) | 1323.64 | N | CR (MRD-pos) |
| Pediatric 3 | 2% (MRD-positive) | Negative | 5.00×108 (3) | 2753.89 | Y (2) | CR (MRD-neg) |
| Pediatric 4 | 83.9% | Negative | 3.96×107 (1) | 1594.24 | Y (2) | CRi (MRD-pos) |
| Pediatric 5 | 80% | Negative | 4.32×108 (3) | 11765.17 | Y (2) | NR |
Data from pilot clinical trials of CART22 in adults (NCT02588456) and children (NCT026550414).
Following treatment, most patients developed cytokine release syndrome (CRS) (highest grade 3 per University of Pennsylvania grading system25, Table 1). The patient with severe (grade 3) CRS had significant elevations in serum cytokines compared to other patients, particularly notable for GCSF, IL-6 and MCP1 (Extended Data 2; see Supplementary Table 2 for full toxicity reporting). Significant CART22 expansion was observed in pediatric patient #5 and adult patient #2 (Table 1, Figures 1b–c) but was modest in all other patients.
One of the three pediatric patients with active disease had a transient disease response at day 28 (reduction in marrow blasts from 75% to 10%), at which time the patient received a second dose of CART22 cells and entered a morphologic remission with 2.4% marrow blasts two months after second infusion. Of the two pediatric patients in MRD+ remission at time of infusion, both had a reduction in disease burden (Figure 1d, Table 1). Neither of the two adult patients with active disease demonstrated a response, and the adult patient treated in MRD-negative remission remained MRD-negative. Overall, the complete remission rate was 50% (4/8); of the five patients with morphologic disease, only one achieved an objective disease response (pediatric patient #4), and of the three patients treated in remission only one remained in remission >6 months after treatment (pediatric patient #3, Figure 1d). Of the four patients who achieved or remained in CR, all four progressed with CD22+ disease (2 by MRD assessment and 2 with clinical relapse). While the small sample size of this study limits statistical evaluation, patients with higher disease burdens tended to have a lower likelihood of response: only one of the five patients with morphologic disease experienced a reduction in ALL burden.
In two patients (pediatric patient #2 and adult patient #2) who had previously received CART19, the CART22 products contained detectable CD19 CAR (Extended Data 3a–b). Both of these patients had CD19-negative leukemia at time of CART22 treatment yet demonstrated re-expansion of CAR19 signal during the expansion phase of CART22, as detected by the presence of CAR19 transgene (Extended Data 3c–d). This observation could be explained by inadvertent co-transduction of existing CART19 cells with the CAR22 transgene, or by re-expansion of a CAR19 single-positive population as a bystander driven by CART22-mediated cytokine production. Flow cytometry on adult patient #2’s peripheral blood collected 21 days after infusion of CART22 (using donor T cells that were either unmanipulated or engineered to express both CARs as reference controls, Extended Data 3e–f) revealed only CAR19 and CAR22 single-positive populations (Extended Data 3g). All CAR19+ T cells were CD8 (Extended Data 3h–i), suggesting that these cells may indeed be long-lived memory cells that re-expanded in the pro-inflammatory environment of rapid CAR22+ T cell expansion.
Short scFv linker promotes CAR22 multimerization
While direct comparison of distinct clinical trials at different institutions is inherently limited, the differing outcomes of these two trials of CART22 (NCI and Penn/CHOP) were striking. Patient characteristics and manufacturing processes in these trials were largely similar13, and both CAR constructs were composed of the m971 VH and VL, as well as the same hinge, transmembrane and intracellular signaling domains (Figure 2a). These constructs differed only in the length of the scFv linker; the CD22 CAR evaluated by the NCI connected the VH and VL using a GGGGS amino acid linker (CAR22-short), while the trials at Penn/CHOP connected the variable chains using a 4x repeat of this amino acid sequence ((GGGGS)4, CAR22-long). We therefore hypothesized that scFv linker length influences CAR activity. We generated CARs with either a short or long scFv linker for in vitro interrogation, beginning with biochemical evaluation. Measurement of scFv affinities revealed a negligible difference between short and long linkers (dissociation constants (KD) 6.1nM and 1.7nM, respectively; Figure 2b). Predictive protein structural modeling of the two scFvs suggested that the long linker allowed appropriate pairing of the heavy and light chains from the same molecule, while the short linker precluded intramolecular heavy and light chain pairing (Figure 2c). This modeling is consistent with previous scFv studies demonstrating that linkers exceeding 9 amino acids in length allow appropriate scFv folding26. Linkers shorter than 9 amino acids in length, however, are known to induce a rigid scFv structure that prohibits intramolecular variable chain pairing and instead promotes intermolecular heavy and light chain pairing, which often results in small increases in measured affinity27. Evaluation of scFv oligomerization using size-exclusion chromatography confirmed that CAR22-long existed as a monomer, while CAR22-short homo-dimerized (Figure 2b). To better understand how scFv dimerization impacted activity of CAR molecules, we engineered T cells to express GFP in frame with the CAR polypeptide, allowing us to visualize receptor distribution on the cell surface using confocal microscopy. CAR22-long was found to distribute uniformly across the cell surface (Figure 2d), while CAR22-short appeared to self-aggregate and cluster (Figure 2e).
Figure 2 |. scFv linker influences CAR surface membrane activity.
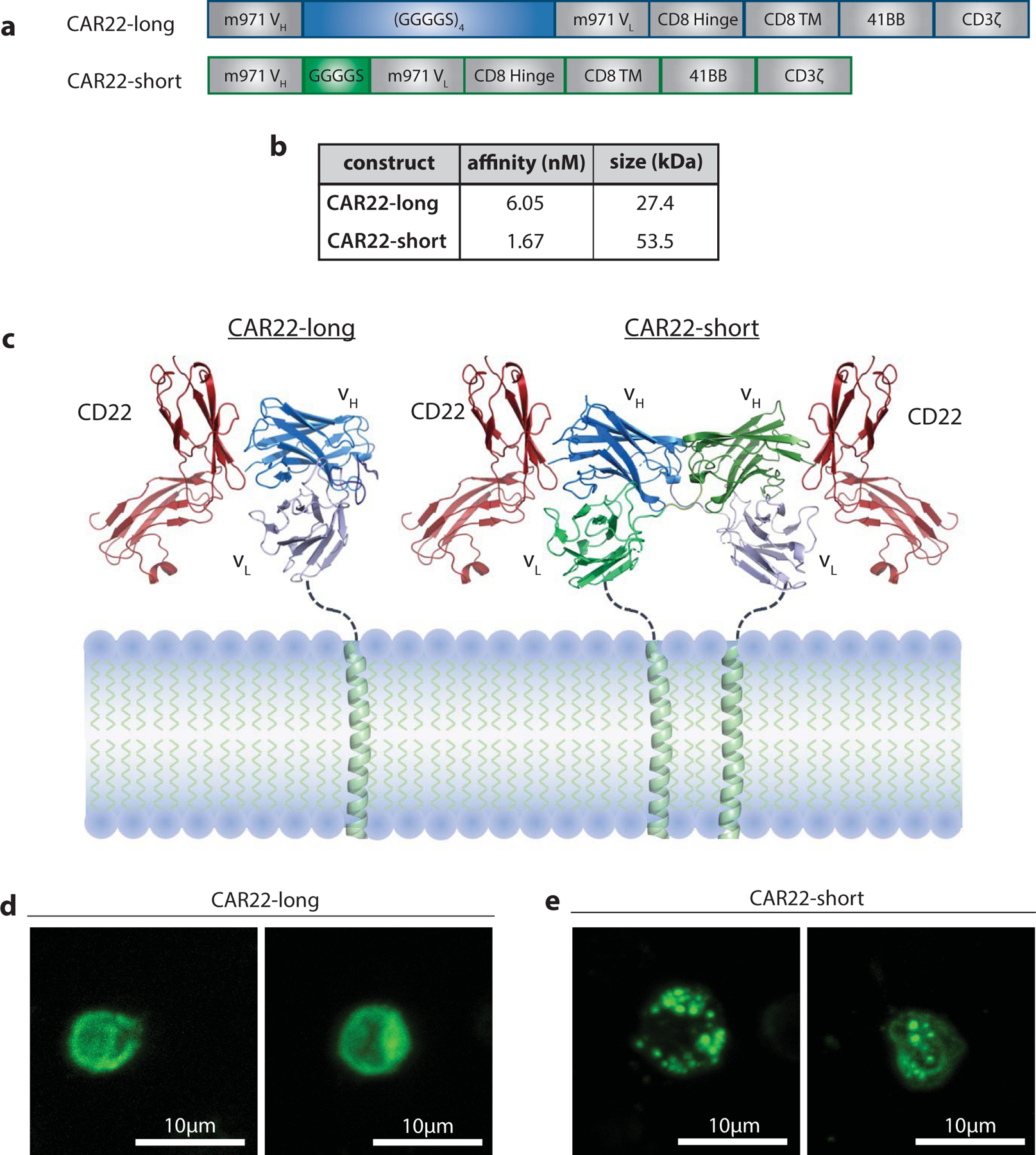
a, Schematic of CAR22-short and long constructs. b, Affinity and size measurements of purified CAR22-short and long scFvs. c, Predictive modeling of scFv structures. d-e, Composite confocal microscopy images of GFP-tagged d, CAR22-long and e, CAR22-short constructs expressed on human T cells.
CAR clustering results in antigen-independent T cell activation
We next interrogated the impact of CAR clustering on antigen-independent receptor activity. Using a phospho-peptide array28, we evaluated differences in signaling protein activation driven by long and short receptors in the absence of target antigen. CAR clustering resulted in greater phosphorylation of critical T cell signaling molecules in the phosphotidyl-inositol-3 kinase (PI3K) and mitogen activated protein kinase (MAPK) signaling pathways (Figure 3a). We also observed increased activation of p53, a key cell-cycle regulatory protein. Notably, no T cell activation-associated proteins demonstrated greater phosphorylation in CAR22-long T cells as compared to CAR22-short cells. We next investigated the transcriptional impact of these observed differences in tonic receptor signaling. Gene set enrichment analysis of sequenced transcripts revealed a significant increase in expression of cell cycle gene programs (FDR = 2.94 ×10−91, Figure 3b). This transcriptional activity was associated with enhanced proliferation of CAR22-short engineered cells during CAR T cell manufacturing (Extended Data 4a). Importantly, autonomous signaling of CAR22-short did not result in sustained expression of exhaustion-associated surface proteins (Extended Data 4b–d).
Figure 3 |. CAR clustering leads to antigen-independent signaling.
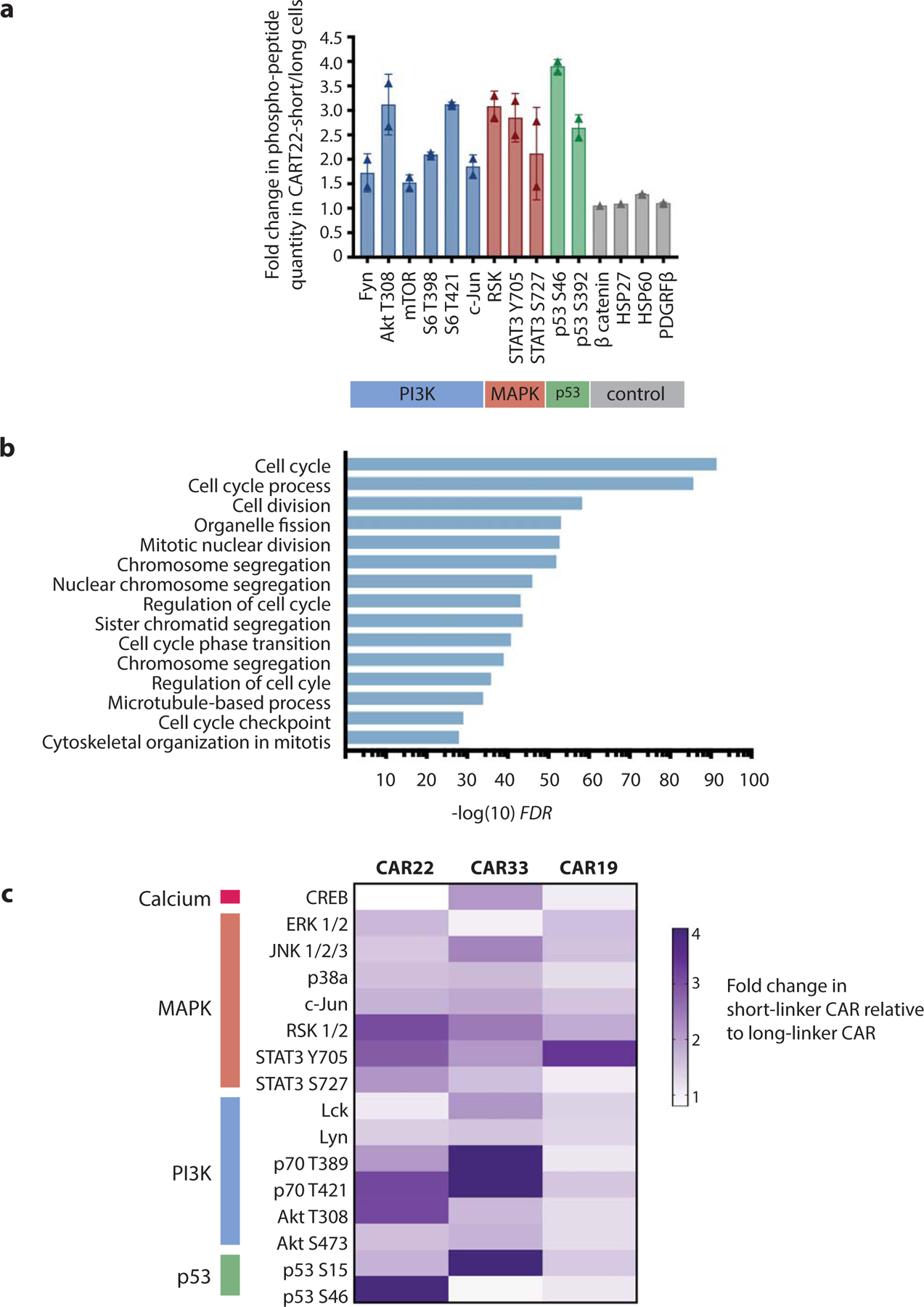
a, Difference in phospho-peptide quantity in resting CAR22-short compared to CAR22-long T cells. Proteins with >1.5-fold difference are shown. b, Upregulated transcriptional programs in resting CAR22-short compared to CAR22-long T cells. c, Heatmap of relative phospho-peptide quantities in short versus long scFv linker versions of CAR19, CAR22 and CAR33 T cells.
To validate this observation in other CAR models, we synthesized long and short version of CD33 and CD19-targeted CARs (CAR33 and CAR19, respectively) and performed similar phospho-array analysis. Similar to CAR22-short, CAR33-short T cells exhibited robust phosphorylation of a broad range of signaling proteins as compared to CAR33-long (Figure 3c). Intriguingly, CAR19-short only exhibited minor increases in phosphorylation of a few signaling proteins compared to CAR19-long. To explore this contrasting observation further, we generated GFP-linked CAR19-short and long and performed fluorescent microscopy of CART19 cells. We found that, unlike CAR22-short, CAR19-short did not induce observable cell-surface receptor clustering (Extended Data 5a–b), consistent with the lack of tonic signaling.
Autonomous CAR signaling results in enhanced T cell function
To explore the impact of antigen-independent signaling and transcriptional activity on T cell function, we performed several studies combining short and long CART22 cells with CD22+ Nalm6 ALL cells. T cell:Nalm6 immune conjugate formation was not impacted by scFv linker length (Extended Data 5c), suggesting that both CARs were able to successfully bind CD22 on target cells. We further investigated the ability of CAR22-short and long T cells to form canonical immune synapses. CAR T cell synapses were imaged using three-dimensional confocal microscopy29,30 (Methods). Measurement of F-actin accumulation at the synapse, a key determinant of synapse stability31, revealed greater polarization in T cells bearing CAR22-short (Figure 4a). Similarly, we observed greater accumulation of perforin at the immune synapse in CART22-short cells (Figure 4b), reflecting enhanced activation of effector function. This approach was also used to evaluate signaling protein phosphorylation upon CAR engagement with CD22, and revealed greater phosphorylation of both CD3ζ and the key signaling regulator Zap-70 in CAR22-short (Extended Data 5d–e), suggesting that CAR22-short mediates both enhanced immune synapse formation and down-stream receptor activation. To explore the implications on in vivo activity of these two CAR products, immunodeficient NOD/SCID/γ−/− (NSG) mice engrafted with Nalm6 were given distinctly labeled CART22-long and short cells, and T cell:Nalm6 interactions were tracked over time using intravital two-photon microscopy. We observed that CART22-short cells were less mobile and remained in contact with target cells longer than CART22-long cells, consistent with more durable synapse formation (Extended Data 5f, Supplementary Movie 1).
Figure 4 |. Functional characterization of CAR22-engineered T cells.
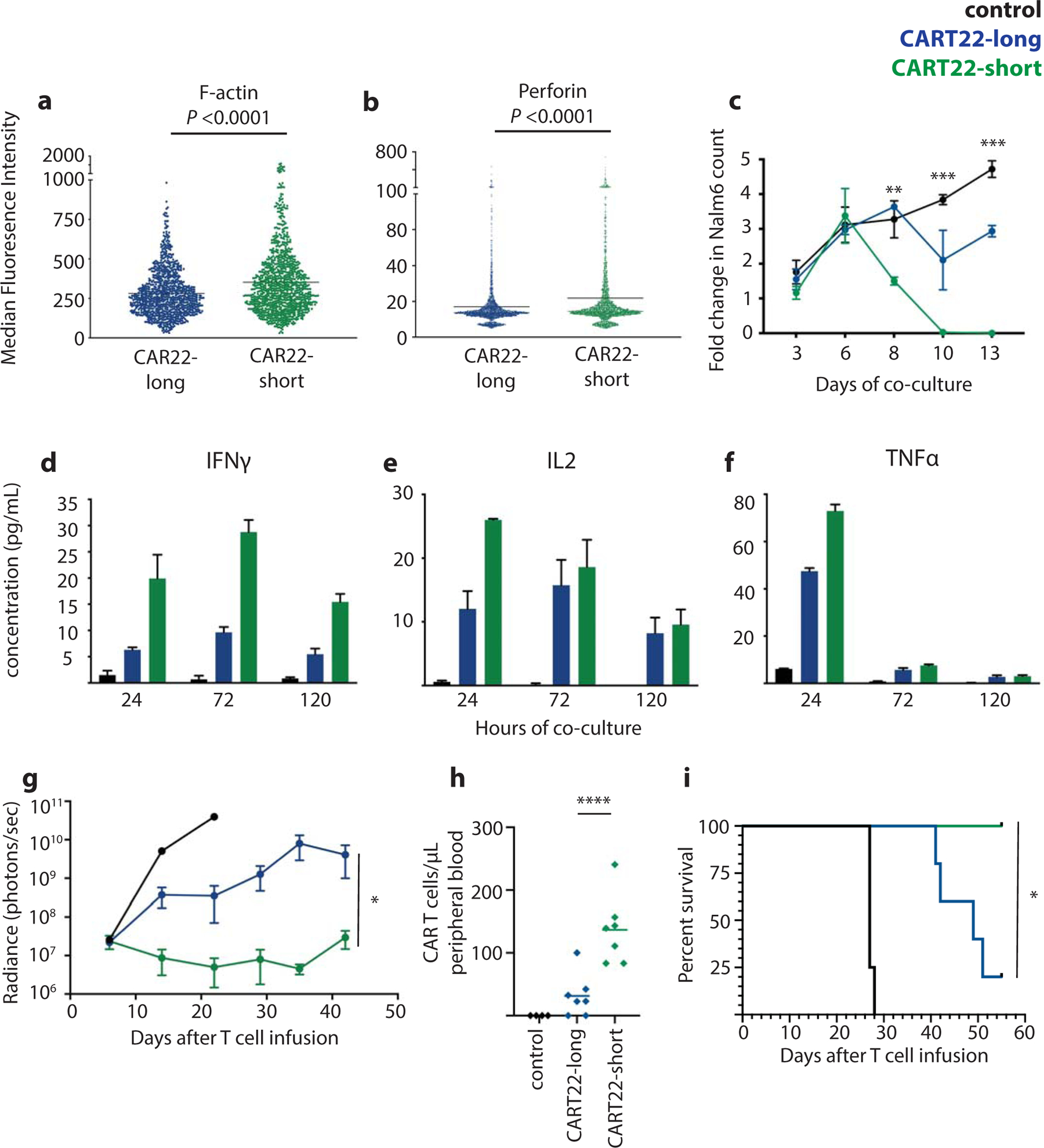
Quantification of a, F-actin (three independent experiments, n=1000 measurements per group) and b, perforin (four independent experiments, n=2400 measurements per group) polarization in CAR T cells engaged with Nalm6. c, Nalm6 survival over time during in vitro co-culture with control (un-engineered), CAR22-long or short T cells. Secretion of d, IFNγ, e, IL-2 and f, TNFα over time during co-culture. g, Leukemia progression over time in xenograft mice bearing Nalm6 treated with control, CAR22-long or short T cells (representative of 4 replicate experiments, n=4–7 mice per condition; see Supplementary Figure 1 for individual animal responses and Supplementary Figure 2 for experimental replicates). h, Quantification of CAR T cells in animal peripheral blood on day 15 after T cell transfer, and i, animal survival over time. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR22-short and long T cells.
We went on to quantify differences in intracellular signaling induced upon receptor engagement (Methods). Consistent with our three-dimensional confocal microscopy findings, phospho-array analysis demonstrated that activated CART22-short had significantly greater phosphorylation of MAPK and PI3K mediators, as well as CREB, a pro-inflammatory transcription factor that is rapidly activated by T-cell receptor-induced calcium influx (Extended Data 6a). To assess how signaling through these two CARs influenced transcriptional activity, CAR T cells were cultured with CD22+ Nalm6 cells overnight and then sorted using flow cytometry for transcriptional profiling. We observed significantly greater expression of immune activation programs in CART22-short (Extended Data 6b), including significantly higher expression of genes directly involved in regulation of T cell effector function, such as TNFSF4, CCR7, TCF7, KLF2 and Eomes, among others (Extended Data 6c). Together, these studies suggest that autonomous CAR signaling leads to a state of heightened responsiveness wherein CAR22-short T cells are “primed” to rapidly initiate intracellular signaling and activate immune response programs upon target engagement.
We next sought to explore the impact of this apparent T cell priming on anti-leukemic efficacy. CART22 cells were combined with Nalm6 cells in long-term co-cultures and leukemic cell death was measured over time by flow cytometry. While anti-tumor activity was not different over the first several days, prolonged in vitro co-culture revealed superior cytotoxicity of CART22-short (Figure 4c). Notably, these functional differences did not result from enhanced activation-induced cell death of CART22-long cells. Indeed, CART22-short was found be slightly more prone to apoptosis in response to target cells, consistent with our findings of enhanced CART22-short antigen-dependent activation broadly (Extended Data 7a). Collection of co-culture supernatants demonstrated higher secretion of IFNγ, IL2 and TNFα by CART22-short (Figures 4d–f, Supplementary Figure 1). Interestingly, these differences were most pronounced early after antigen exposure, further supporting the development of a primed phenotype. We next evaluated the in vivo activity of CAR22-short and long cells in immunodeficient NSG mice bearing ALL (Methods). CART22-short demonstrated significantly enhanced disease control, while CART22-long only partially reduced disease burden as compared to control T cells (Figure 4g, Supplementary Figure 2). These differences were also reflected by in vivo T cell expansion (Figure 4h), and directly translated to enhanced animal survival (Figure 4i).
To directly interrogate the role of 41BB co-stimulation in generating these enhanced effector functions, we replaced the 41BB co-stimulatory domain with the CD28 domain in both long (non-tonically signaling) and short (tonically signaling) CAR22s and performed similar in vitro cytotoxicity studies. In both treatment (high effector:target ratio) and stress (low effector:target ratio) models, CAR22/CD28-short were less efficacious than CAR22/CD28-long (Extended Data 7b–c). These data demonstrating inferiority of CAR22/CD28-short are consistent with several previous reports demonstrating that tonic signaling of CD28-based CARs leads to T cell dysfunction32–34, and highlight the divergent activity driven by tonic signaling of CD28 and 41BB-based CAR T cells.
To validate the relationship between autonomous signaling and efficacy, we evaluated the effector function of short and long versions of 41BB-based CAR33 and CAR19. We found that the tonically-signaling CAR33-short drove enhanced cytotoxicity against the CD33+ acute myeloid leukemia (AML) cell line Molm14, as well as enhanced CART cell expansion upon antigen exposure (Extended Data 8a–b). Further, CART33-short secreted more IFNγ, IL2 and TNFα than CART33-long (Extended Data 8c–e). In contrast, CAR19-short, which does not promote more tonic signaling than its long counterpart, demonstrated equivalent cytotoxicity against CD19+ Nalm6 in both in vitro treatment and stress models, as well as in an in vivo xenograft model (Extended Data 9a–c, Supplementary Figure 3). While CAR19-short did demonstrate a trend towards more secretion of IFNγ, this was the only metric by which it outperformed CAR19-long. These data suggest that shortening of linker length can induce CAR intermolecular pairing, but that this is not a universal phenomenon. However, we observe that when shortening of the linker does lead to tonic autonomous signaling of 41BB-based CARs, this has a beneficial impact on T cell effector function.
Development and evaluation of a novel dimerizing CD22 CAR
Based on these observations demonstrating an association between linker length, tonic signaling and effector function, we sought to develop a new CD22-directed CAR with unencumbered intellectual property as a more effective therapeutic product for patients at our institutions who had previously received CD19 CAR T cells but relapsed with CD19-negative disease. We evaluated the impact of scFv linker length on the function of a de novo designed, high-affinity CAR that could be given to patients who had failed CD19-directed CAR therapy at the University of Pennsylvania and Children’s Hospital of Philadelphia (Figure 5a). Using scFvs derived from a phage-display library, we screened >60 CD22-directed scFvs and chose the molecule with the strongest CD22-directed activity in vitro (CD22-f). CD22-f then underwent affinity enhancement to develop CD22-f2, which was then designed with both long and short linkers (CD22-f2-long and CD22-f2-short, respectively). These molecules had similar affinities and biochemical analysis demonstrated that, like the m971-based CAR22-short, CD22-f2-short homo-dimerized (Figure 5b). CD22-f2-long and short full-length CARs expressed well on the surface of primary T cells (Figure 5c), and in vitro functional characterization demonstrated that CD22-f2-short secreted slightly higher levels of IFNγ as compared to CD22-f2-long (Figure 5d). Direct comparison of CD22-f2-short and long in our in vivo Nalm6 model demonstrated that both CARs had potent anti-leukemic function, but CD22-f2-short was superior (Figure 5e, Supplementary Figures 4). In vivo evaluation of all four CD22-targeted CARs demonstrated similar potent activity of both m971 and CD22-f2-short, with long-term survival of all animals treated. While both long CARs were inferior to their short counterparts (m971 P = 0.0006, CD22-f2 P = 0.0014), CD22-f2-long demonstrated a modest survival benefit over m971-long (Figure 5f, P = 0.004). This was confirmed in a second in vivo model of disseminated ALL, in which both short CARTs demonstrated superior activity to their long counterparts, had similar efficacy to each other, and CD22-f2-long was superior to m971-long (Extended Data 10, Supplementary Figure 5).
Figure 5 |. Development of a novel CD22 CAR with potent pre-clinical activity.
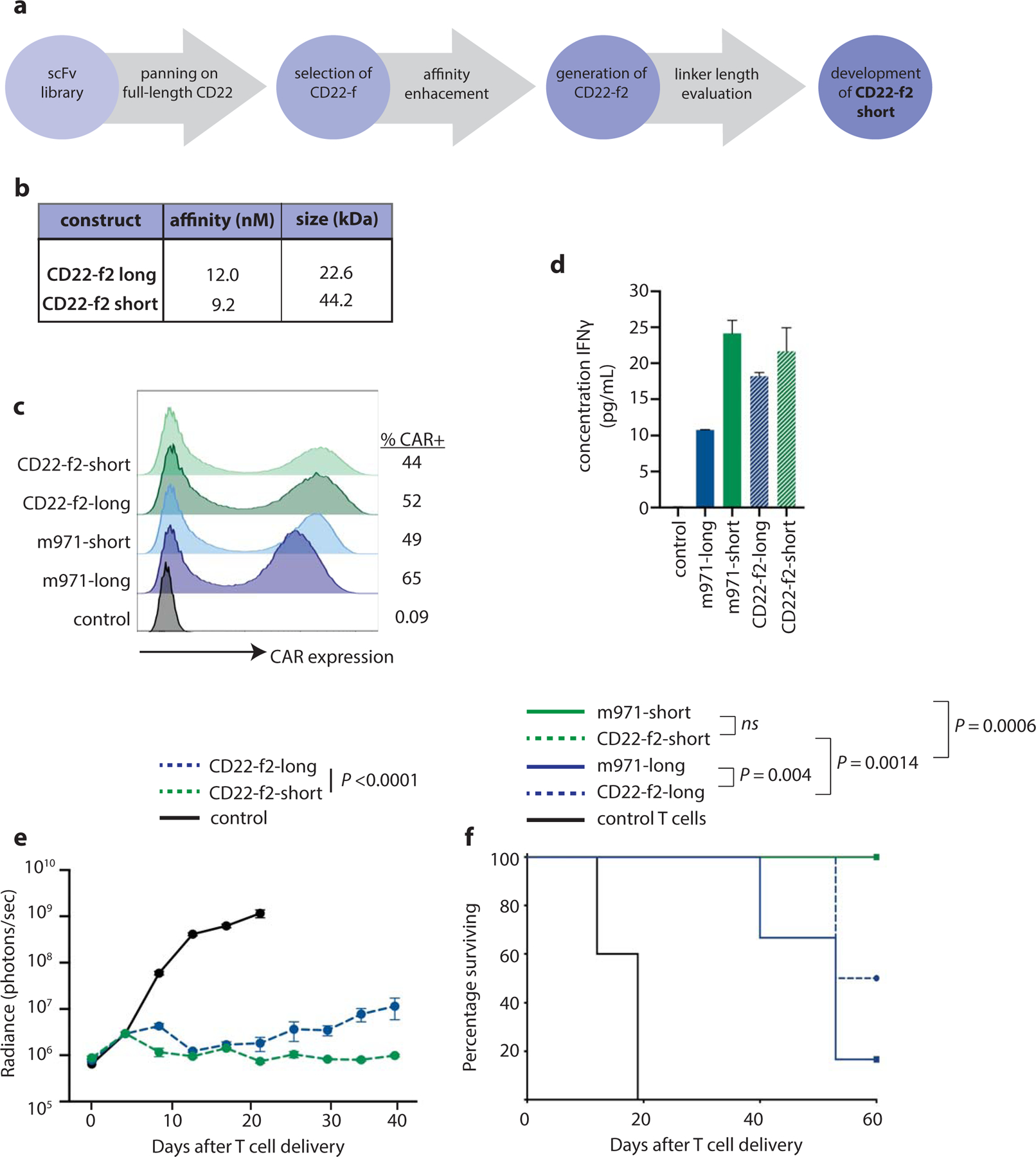
a, Pipeline for development and evaluation of new CD22-f2-short CAR. b, Affinity and size of purified CD22-f2-long and short scFvs. c, Expression of CD22 CARs on primary T cells. d, Measurement of secreted IFNγ by CD22-engineered T cells after 24h exposure to CD22+ target cells. e, Progression of Nalm6 disease burden in xenograft mice treated with CD22-f2-short and long T cells (Representative of 4 replicate experiments, n=4–7 mice per condition; see Supplementary Figure 5 for individual animal responses and Supplementary Figure 6 for experimental replicates). f, Survival of Nalm6-bearing xenograft mice after treatment with m971 or CD22-f2 CAR T cells. Data are presented as mean values +/− standard error of the mean (S.E.M.) Statistics reflect differences between CAR22-short and long T cells.
Based on this work, we have initiated a clinical trial of CD22-f2-short CAR T cells in adults (NCT03620058), as well as amended the original pediatric CART22 trial (NCT02650414) to permit evaluation of CD22-f2-short.
Discussion
Motivated by disparate findings from two independent clinical trials evaluating CD22 CAR T cells, we have identified that tonic signaling enhances the function of 41BB-based CAR T cells. While patient demographics, previous therapies, disease burden and CAR T cell manufacturing practices were largely similar, these studies were both small and thus many other variables may contribute to differences in outcome. However, we were struck by the broadly differing clinical outcomes associated with a singular difference in CAR structure, which led us to hypothesize that the linker length may have contributed to the observed differences in patient responses. We observed that the short scFv linker promoted intermolecular CAR pairing, likely as a result of impaired intramolecular heavy and light chain pairing. We found the short scFv linker promoted antigen-independent receptor signaling and downstream T cell activation, leading to enhanced anti-leukemic function. This enhanced function extended to other CARs models, leading to the conclusion that autonomous signaling and the resultant T cell priming is beneficial for CAR-driven T cell function. Intriguingly, CAR19 did not demonstrate evidence of significantly increased tonic signaling by shortening of the scFv linker. The lack of cell-surface clustering validates the association between clustering and tonic signaling, however the biochemistry responsible for this difference in intermolecular pairing remains unclear and is the subject of ongoing studies. While there are no significant differences in scFv structural motifs between these four constructs, we hypothesize that the divergence in clustering is indeed related to scFv biochemistry itself, given that all observations were made without presence of antigen. The features of the FMC63 VH and VL that may make them less pre-disposed to intermolecular pairing are not known, but these observations do indicate that shortening of linker length does not universally result in clustering. As such, each new CAR should likely be evaluated individually for clustering and tonic signaling.
Previous studies have demonstrated that tonic signaling of CD28-based CARs occurring as a result of distinct receptor biochemistry (not linker-driven clustering) leads to T cell exhaustion32–34. Replacement of the CD28 co-stimulatory domain with the 4–1BB co-stimulatory domain has been shown to prevent the development of exhaustion, suggesting a protective effect of tonic 4–1BB signaling. In this report, we identify that tonic 41BB signaling can both protect against exhaustion and enhance CAR T cell function, pointing to a central role of the co-stimulatory domain in determining T cell fitness in the setting of tonic signaling.
Optimization of CAR structure to improve synthetic receptor-driven T cell function has been a central focus of the field for more than two decades. A great majority of this work has focused on intracellular signaling domain optimization, resulting in two FDA-approved CD19-directed CAR products with different co-stimulatory signaling domains. More recent work has identified that several components of extracellular CAR structure, including regions that do not directly interact with antigen, have an impact on CAR-driven T cell function17–20. We demonstrate that changes in linker length can control CAR structure, which has a direct impact on effector function. Based on these findings, we developed a new CAR22 with a short linker that demonstrated enhanced anti-leukemic efficacy as compared to its long linker counterpart. Notably, the differences in effector function induced by shortening of the scFv linker were not as pronounced for this novel CAR (CD22-f2) as were observed for the original CAR (m971). We speculate that this is a result of the affinity enhancement that occurred for CD22-f prior to its evaluation, resulting in an overall better CAR as compared to m971.
Notably, both the clinical trials done at Penn/CHOP and the NCI revealed limited CAR T cell persistence after infusion, as compared to historical studies of CD19-targeted 41BB-bearing CARs. In our studies, we only observed robust CAR T cell expansion in one patient, a divergence from our experience in targeting CD19. While these studies were small, and thus we hesitate to draw any conclusions, we speculate that these distinctions in pharmacokinetics may be a result of differences in surface antigen density between CD19 and CD22. Most B-ALL cells express significantly lower levels of CD22 as compared to CD1913, which may drive distinct cellular programming involved in regulating T cell persistence. Further work is needed to understand the role of antigen density of CAR T cell efficacy, as well as the etiology of the limited persistence seen with both of these 4–1BB-bearing CARs.
Given that clinical studies directly comparing two CAR products have not yet been performed, it has remained difficult to identify which pre-clinical assessments are most valuable in predicting clinical activity. Historically, early in vitro anti-tumor activity, as assessed by luciferase-based or chromium-based killing assays, has served as a surrogate of function. Our studies, as well as others33, identify the limitations in early CAR T cell function as predictor of overall efficacy. The disparate long-term in vitro efficacy is mirrored by other pre-clinical comparisons, and identifies that early signaling protein activation, early cytokine secretion and durable in vivo activity are most closely associated with clinical outcomes and may be effective predictors of clinical anti-tumor efficacy.
In summary, beginning with observations from CART22 clinical trials, we demonstrate that short scFv linkers can drive intermolecular CAR interactions that promote autonomous 41BB signaling, priming T cells for enhanced anti-leukemic function.
Methods
Clinical trial design and evaluation:
Pilot clinical trials designed to assess the safety and feasibility of CD22 CAR T cell therapy in relapsed/refractory CD22+ malignancies were conducted at the Children’s Hospital of Philadelphia (ClinicalTrials.gov Identifier NCT02650414) and the Hospital of the University of Pennsylvania (ClinicalTrials.gov Identifier NCT02588456). Both study protocols are available in the Supplementary Information. Written informed consent for participation was obtained from patients or their guardians according to the Declaration of Helsinki, and protocols were approved by the institutional review boards of each institution. Patients underwent leukapheresis at the time of relapse or after one or more reinduction attempts. Patients with prior allogeneic hematopoietic cell transplantation were eligible, and T cells were collected from the patient, not the donor. Leukapheresis products were stimulated with anti-CD3/CD28 paramagnetic beads and transduced with the m971-long anti-CD22 CAR transgene. After leukapheresis, patients received interim therapy at the discretion of their treating physician with no restrictions on this bridging therapy (Supplementary Table 1). Lymphodepleting chemotherapy (cyclophosphamide 500mg/m2/day for two days and fludarabine 30mg/m2/day for four days) was given 1 week before CD22 CAR T cell infusion. Patients <50kg were treated on a weight-based dosing scheme of 1–10×106 CAR+ T cells/kg, while patients >50kg received a flat-dose of 5–50×107 T cells in split fractions (10%, 30%, 60%) on days 1–3. This dose range accounts for patients who only received the initial 10% dose. Subsequent doses on days 2–3 were held if fever developed after the first dose. Response was assessed by bone marrow (all patients) and CSF evaluation (pediatric trial only) at 1, 3, 6, 9, and 12 months, after which disease surveillance was limited to analysis of peripheral blood unless there were clinical concerns. Complete remission (CR) was defined by morphologic assessment of the bone marrow as M1 (<5% leukemic blasts) with no evidence of extramedullary disease. Minimal residual disease (MRD) was assessed by multiparametric flow cytometry at the University of Washington with a sensitivity of 0.01% mononuclear cells. Presence of CART22 and CART19 was performed by measurement of CAR transgene in peripheral blood by quantitative PCR.
For NCT02588456, the first patient was enrolled on 11/11/2015 and last patient on 7/6/2016. Inclusion criteria for enrollment were signed informed consent; relapsed B-cell ALL in first or greater relapse, any relapse after allogeneic hematopoietic cell transplantation and >100 days from transplant, or refractory disease in patients <60 years old that have not achieved a complete remission after >2 or more chemotherapy regimens or >60 years old that have not achieved complete remission after 1 prior chemotherapy regimen; patients with Philadelphia chromosome (Ph)-positive disease were eligible if not intolerant to or previously failed tyrosine kinase inhibitor therapy; patients with CNS3 disease were eligible if CNS disease is responsive to therapy; documented CD22 expression on malignant cells at relapse.; adequate organ function defined as (1) creatinine <1.6mg/dL, (2) ALT/AST <3x upper limit of normal range, (3) direct bilirubin <2.0mg/dL, (4) <grade 1 dyspnea, pulse oxygen >92% on room air and DLCO >40% when corrected for anemia, (5) left ventricular ejection fraction >40%; evidence of disease by morphologic or minimal residual disease criteria; male or female age >18 years; ECOG performance status of 0–1; no contraindications for leukapheresis; subjects of reproductive potential must agree to use acceptable birth control methods. Exclusion criteria for enrollment were active hepatitis B or C; HIV infection; NYHA class III/IV cardiovascular disability; subjects with clinically apparent arrhythmia or arrhythmia not stable on management within two weeks of enrollment; active acute or chronic graft-versus-host disease requiring systemic therapy; concurrent use of systemic steroids of immunosuppressant medications; CNS3 disease that is progressive on therapy or with CNS parenchymal lesions; pregnant or nursing women; receipt of a prior investigational study agent within 4 weeks prior to enrollment, except for receipt of CART19/CTL019 therapy; known history or prior diagnosis of optic neuritis or other immunologic or inflammatory disease affecting the central nervous system.
For NCT02650414, the first patient was enrolled on 3/18/2016 and last patient on 3/14/2017. At this time enrollment was paused pending the development of the newly designed CD22-f2 construct, after which enrollment was re-initiated. Inclusion criteria included second or greater bone marrow relapse, any marrow relapse after allogeneic hematopoietic cell transplantation and >6 months from transplant, any marrow relapse after CAR-modified T cell therapy, or refractory disease defined as having not achieved complete remission after >2 chemotherapy regimens; patients with Ph+ ALL were eligible if they were intolerant to or had failed previous tyrosine kinase inhibitor therapy or were ineligible for allogeneic transplant because of comorbid disease, lack of donor, prior transplant, other contraindication or declined to pursue transplant; patients with CNS3 disease will be eligible if CNS disease is responsive to therapy; documented CD22 expression on malignant cells at time of relapse; if the patient has previously received CD22-targeted therapy then a marrow should be obtained to confirm CD22 expression; adequate organ function defined by (1) normal creatinine for gender and age, (2) ALT <500U/L, (3) bilirubin <3x upper limit of normal, (4) ALT and or bilirubin, (5) <grade 1 dyspnea, pulse oxygen >92% on room air and DLCO >40% when corrected for anemia, (5) left ventricular ejection fraction >40% or left ventricular shortening function >28%; evidence of morphologic or minimal residual disease; age 1–29 years; Karnofsky or Lansky performance status score >50; subjects of reproductive potential must agree to use acceptable birth control methods. Exclusion criteria for enrollment were active hepatitis B or C; HIV infection; active acute or chronic graft-versus-host disease requiring systemic therapy; concurrent use of systemic steroids of immunosuppressant medications; CNS3 disease that is progressive on therapy or with CNS parenchymal lesions; pregnant or nursing women; receipt of a prior investigational study agent within 4 weeks prior to enrollment, except for receipt of CART19/CTL019 therapy.
For both studies, the primary study endpoint was safety as measured by grade 3 and higher toxicity. The secondary endpoints included overall survival, duration of remission, relapse free survival, and event free survival. For both NCT02588456 and NCT02650414 the Data and Safety Monitoring Board responsibilities were to evaluate the progress of each trial, including periodic assessments of data quality, timeliness, participant recruitment, accrual and retention, participant risk versus benefit, aspects of trial conduct, study futility, and other factors that can affect study outcome; consider factors external to the study when relevant information becomes available, such as scientific or therapeutic developments that may have an impact on the safety of the participants or the scientific value or ethics of the trial; ensure that the safety of the study participants is not being compromised; make recommendations to the Sponsor concerning continuation, termination, or other modifications of the trial based on the observed beneficial or adverse effects of the treatment under study, or other concerns; consider whether the data exceed any early stopping boundaries which have been clearly defined in advance of data analysis; maintain the confidentiality of the trial data and the results of monitoring; and, assist the sponsor by making recommendations regarding any problems with study conduct, enrollment, and sample size and/or data collection.
scFv modeling:
A single chain model of the Fv region lacking the heavy-light chain linker was first constructed using Repertoire Builder35. Next, the CAR22-long construct was modeled by re-writing the Fv model as a single sequence and aligning it to the Fv sequence that included the 20-residue heavy-light chain linker. A structural model of the complete Fv region was then constructed using Spanner36. In order to model the CAR22-short Fv region as a bivalent diabody, the heavy and light chain structural models were separated and superimposed on those of a bivalent diabody crystal structure (PDB entry 1LMK, chains A C E and G)37. The 5-residue heavy-light chain linker was then added using Spanner, as above. Fv structures were positioned above a TCR transmembrane helix (PDB entry 6JXR) manually. Analysis of the CD8 hinges for the CAR22-long construct and the CAR22-short construct revealed that the hinge was intrinsically disordered, implying that the orientation of both the long- and short-linker Fv models relative to the plasma membrane could not be constrained by modelling.
scFv production:
To assess biophysical and binding characteristics, m971 and CD22–65 variants were produced as recombinant scFv with a short and long linker. The plasmids encoding the scFv constructs with a short linker and a corresponding scFv with a longer linker were synthesized externally using publicly-available patents and previously published data16. The scFvs were produced transiently in HEK293F cells and purified using the HisAvi tag on the C-terminus of the constructs with standard methodology. Briefly, 100 ml of HEK293F cells at 3×106 cells/ml were transfected with 100 μg plasmid and 300 μg polyethylenimine. The cells were incubated at 37°C with 8% CO2 and rotated at 80 rpm. After six days, the cells were harvested by centrifugation at 3500g for 20 minutes. The supernatant was purified by binding the scFv to Ni-NTA agarose beads (Qiagen) overnight at 4°C. The protein was eluted with 300 mM imidazole, and dialyzed against phosphate buffered saline. Identity of scFvs was confirmed via mass spectrometry.
Affinity measurements:
Binding affinity of purified m971 and CD22–65 scFv short and long variants was measured on a Biacore T200 system. Briefly, recombinant human CD22 ECD-Fc was obtained from R&D systems (Cat. # 1968-SL-050). CD22-Fc was immobilized on a CM5 chip surface using standard amine coupling at a target density of 150 RU. Purified scFvs were injected over the chip under constant flow rate at concentrations ranging from 1 to 900 nM. Association and dissociation rates of the protein complex were monitored and double referencing was performed against a blank immobilized flow cell and a buffer blank; the data was fit using a 1:1 Langmuir model.
Size exclusion chromatography:
Size exclusion chromatography was performed to determine the oligomerization state of the scFvs. 15μg were injected onto a TSKGel Super SW3000 4.6 mm x 300 mm column (Tosoh Bioscience) heated to 35°C. The scFv was eluted at 0.3 mL/min in 750 mM arginine, 1 mM EDTA, 20 mM sodium phosphate, 250 mM sodium chloride, pH 7.2; the UV absorbance was monitored at 280 nm.
Lentiviral vector production and transduction of CAR-engineered human T cells:
Replication-defective, third-generation lentiviral vectors were produced using HEK293T cells (ATCC ACS-4500) or Lenti-X 293T cells (Takara). Approximately 8×106 cells were plated in T150 culture vessels in standard culture media and incubated overnight at 37°C. 18–24h later, cells were transfected using a combination of Lipofectamine 2000 (96μL, Invitrogen), pMDG.1 (7μg), pRSV.rev (18μg), pMDLg/p.RRE (18μg) packaging plasmids and 15μg of expression plasmid (CAR). Lipofectamine and plasmid DNA was diluted in 4mL Opti-MEM media prior to transfer into lentiviral production flasks. At both 24 and 48h following transfection, culture media was isolated and concentrated using high-speed ultracentrifugation (25,000 x g for 2.5 hours) or using Lenti-X Concentrator (Takara). For experiments analyzing m971 CARs, human T cells were procured through the University of Pennsylvania Human Immunology Core. CD4 and CD8 cells were combined at a 1:1 ratio, and activated using CD3/CD28 stimulatory beads (Thermo-Fisher) at a ratio of 3 beads/cell and incubated at 37°C overnight. The following day, CAR lentiviral vectors (composed of the respective variable heavy and light chains, CD8α hinge region, CD8α transmembrane domain CD3ζ signaling domain and either 4–1BB or CD28 co-stimulatory domains) were added to stimulatory cultures at an MOI between 4–6. Beads were removed on day 6 of stimulation, and cells were counted daily until growth kinetics and cell size demonstrated they had rested from stimulation. For experiments analyzing CD22–65 CAR variants, T cells were obtained by negative selection (Pan T Cell Isolation Kit, Miltenyi) of a Leukopak purchased from HemaCare. Pan T cells were stimulated and transduced as described above, starting with the donor’s respective CD4/CD8 T cells ratio.
Glass-supported planar lipid bilayer confocal imaging:
Planar lipid bilayers were prepared by fusing small liposome droplets with clean glass coverslips as previously described38. Briefly, the liposome was trapped in a µ-Slide VI 0.4 chamber (Ibidi, Germany). Lipid bilayers were first blocked with 5% Casein for 15 min and then incubated with 6.3 nM Streptavidin (Life Technologies) for 15 min. After being washed extensively with imaging buffer (HEPES-buffered saline), bilayers were incubated with biotinylated antibodies conjugated with Alexa Fluor dyes at room temperature for 30 min. After a second wash with imaging buffer, bilayers were blocked with 2.5 μM D-biotin to saturate the streptavidin-binding sites. Cells were activated on the lipids for 60 min. CAR T cells were stimulated on lipid bilayers containing fluorescently labeled CD22 proteins. Cells were stained by fluorescently conjugated antibodies against perforin (deltaG9, Thermo), pZeta (phosphor-Y83, Abcam), and pZAP-70 (Tyr 319, Cell Signaling), as described previously29. F-actin was stained by Alexa Fluor 405-conjugated phalloidin (Life Technology). A Nikon microscope was used to obtain 3D-confocal image data.
Immune conjugate formation and confocal microscopy:
Immune conjugates were generated as previously described39. Briefly, CAR T cells were stained with CellTrace Violet (CTV) and were combined with GFP+ Nalm6 cells at an effector:target ratio of 2:1. Co-cultures were incubated for either 5 or 15 minutes at 37°C, fixed and conjugate formation (CTV+GFP+ doublets) was quantified by flow cytometry.
For analysis of CAR distribution on the cell membrane, GFP tagged lentiviral constructs were generated by introducing in-frame fusions of GFP at the 3’ end of the CAR construct using standard molecular cloning. T cells were spun at 500rpm for 3 minutes, followed by a 10 minute incubation at 37°C. Cells were then applied to poly-L-lysine coated glass coverslips, washed and fixed with 4% paraformaldehyde. Coverslips were mounted and imaged using a 63x PlanApo 1.4 NA objective on an Axiovert 200M (Zeiss) with a spinning disk confocal system (Ultraview ERS6; PerkinElmer). Z-planes were collected every 0.25 μm, spanning a total of 10 μm with an Orca Flas 4.0 CMOS camera (Hamamatsu). Analysis was done using Volocity v6.3 software and ImageJ open source software was used to prepare representative images.
Intravital multiphoton microscopy:
GFP+ Nalm6 was established in xenograft mice as described below. T cells engineered with CAR22-short were labeled using CellTrace Violet, CAR22-long with CellTrace Orange and 1×106 of each were delivered to mice via tail vein. 48 hours following T cell injection, mice were anesthetized and maintained at core temperatures of 37°C. Bone marrow was imaged after removing the scalp and immobilizing the skull. Imaging was performed using a Leica SP5 2-photon microscope system (Leica Microsystems) equipped with a picosecond laser (Coherent). Each imaging acquisition lasted 20 minutes followed by an assessment of mouse sedation. CellTrace Violet, GFP, and CellTrace Orange were excited using a laser light of 850 nm. Images were obtained using a 20× water-dipping lens. The resulting images were analyzed with Volocity software (PerkinElmer) to quantify number of immobile T cells per field during 20 minute acquisition.
Phospho-protein quantification:
Peptide phosphorylation was quantified using the Human Phospho-Receptor Tyrosine Kinase Array Kit (R&D Systems)28. Briefly, we generated CD22-coated magnetic beads using recombinant CD22 protein (Sino Biologicals) and tosyl-activated M450 DynaBeads (Invitrogen). 1–1.5 × 107 T cells were combined with CD22 beads at a ratio of 4 beads per T cell, spun at 300 x g for 5 minutes and incubated at 37°C for the time period indicated. Cells were then lysed according to the array kit protocol, and phospho-peptides were detected and analyzed using quantitative chemiluminescence. Images were captured using ImageQuant LAS 4010 (GE Healthcare Bio-Sciences) and densitometry analysis was performed with Image Studio Lite Version 5.2 (LI-CORBiotechnology). Relative phospho-peptide quantities were determined by comparison of signal intensity in CAR22-short to CAR22-long, and vice-versa.
RNA sequencing:
2×106 T cells (control, CAR22-long or CAR22-short) were combined with either 2×106 GFP+ Nalm6 cells or media (in triplicate) and incubated for 18 hours at 37°C. T cells were isolated by flow-assisted cell sorting (GFP-negative gate) and RNA was isolated using RNeasy Mini Kit (Qiagen). Total RNA quantity and quality were assayed with an Agilent 2100 Bioanalyzer instrument using the RNA 6000 Pico Kit (Agilent Technologies). Libraries were prepared using NEBNext Ultra II Directional RNA Library Prep Kit (polyA mRNA workflow) for Illumina (New England BioLabs) as per standard protocol. Libraries were assayed for size using DNA 1000 kit of Agilent 2100 Bioanalyzer (Agilent Technologies) and quantified using KAPA Library Quantification Kit for Illumina platforms (KAPABiosystems). 100bp single-read sequencing of multiplexed samples was performed on an Illumina HiSeq 4000 sequencer. Illumina’s bcl2fastq version 2.20.0.422 software was used to convert bcl to fastq files. After sequencing, read quality was evaluated using FastaQC (v0.11.2), and low-quality bases were removed using Trimmomatic (v0.36). The remaining reads were then mapped to human genome (hg38) using STAR (v2.6.0c) with default parameters. Gene count was calculated using featureCounts (v1.6.1), and non-expressed genes with read count < 1 in all samples were removed prior to differential expression analysis. DESeq2 (v1.22) and cutoff values of log2 fold change > or <0.5 and adjusted P-value of <0.1 was used for identifying differentially expressed genes. Gene set enrichment analysis was performed using gene ontology (GO) terms.
General cell culture:
Unless otherwise specified, cells were grown and cultured at a concentration of 1×106 cells/mL of standard culture media (RPMI 1640 + 10% FCS, 1% penicillin/streptomycin, 1% HEPES, 1% non-essential amino acids) at 37°C in 5% ambient CO2. All co-culture studies were performed an effector cell to target cell ratio of 1:4, unless otherwise stated. Cell size and expansion were determined using a BD Multisizer 4 Coulter Counter or Cellometer (Nexcelom) and Scepter Cell Counter (MilliporeSigma).
Flow cytometry:
Cells were resuspended in FACS staining buffer (PBS + 3% fetal bovine serum) using the following antibodies: CD3 (clone OKT3, eBiosciences), PD-1 (clone J105, eBiosciences), TIM3 (clone F38–2E2, BioLegend), Lag3 (clone 3DS223H, Invitrogen). CARs were detected using a PE or APC-conjugated CD22, CD19 or CD33 protein. All changes in overall tumor or T cell counts reflect changes in absolute cell counts, which were determined using CountBright absolute counting beads (ThermoFisher). Cell viability was established using Live/Dead Aqua fixable staining kit (ThermoFisher), and data were acquired on an LSRII Fortessa Cytometer (BD). All data analysis was performed using FlowJo 9.0 or 10 software (FlowJo, LLC). See Supplementary Figure 6 for gating strategy.
Cytokine and cytolytic molecule quantification:
Human cytokine quantification was performed using a custom 30-plex Luminex panel (Life Technologies, Carlsbad, CA) containing the following analytes: IL-1RA, FGF-Basic, MCP-1, G-CSF, IFN-γ, IL-12, IL-13, IL-7, GM-CSF, TNF-α, IL-1β, IL-2, IL-4, IL-5, IL-6, IFN-α, IL-15, IL-10, MIP-1α, IL-17, IL-8, EGF, HGF, VEGF, MIG, RANTES, Eotaxin, MIP-1β, IP-10, IL-2Ra. Cell culture supernatants were flash frozen on dry ice, and thawed at time of cytokine analysis. Assays were established per manufacturer recommendations. Data were acquired on a FlexMAP 3D quantification instrument, and analysis was done using xPONENT software. Data quality was determined by ensuring the standard curve for each analyte had a 5P R2 value > 0.95 with or without minor fitting using xPONENT software. To pass assay technical quality control, the results for two controls in the kit were required to be within the 95% confidence interval provided by the vendor for >25 of the tested analytes. Data were analyzed using Omega Data Analysis software. For analysis of CD22–65 CAR-T cells, culture supernatant was assayed for cytokines using the MSD V-plex (Meso Scale Diagnostics).
Xenograft mouse models:
6–10 week old NOD-SCID-γc−/− (NSG) mice were obtained from the Jackson Laboratory and maintained in pathogen-free conditions. Animals were injected via tail vein with 1×106 Nalm6 or 3×106 SEM cells in 0.2mL sterile PBS. On day 7 after tumor delivery, 1×106 T cells (control or CAR engineered) were injected via tail vein in 0.2mL sterile PBS. Animals were monitored for signs of disease progression and overt toxicity, such as xenogeneic graft-versus-host disease, as evidenced by >10% loss in body weight, loss of fur, diarrhea, conjunctivitis and disease-related hind limb paralysis. Disease burdens were monitored over time using a the Xenogen IVIS bioluminescent imaging system, as previously described.40 All animal studies were approved and supervised by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
scFv design and optimization:
To identify novel binders to human CD22 extracellular domain, three rounds of panning were done against recombinant human CD22 (R&D systems, Cat. # 1968-SL-050) using a fully human derived scFv phage library derived internally. Antigen concentration for the first round was 400 nM, and 200 nM for the second and third rounds. For the second and third rounds, depletion against human IgG1-Fc was performed to deselect Fc binding phages. Potential binders after the third round of panning were screened against phage ELISA and reconfirmed using FACS against CD22-positive (Raji) and negative (K562) cell lines. To assess functionality as chimeric antigen receptors, hits were sub-cloned into chimeric antigen receptor vectors and nucleofected into Jurkat NFAT reporter cells. A molecule (CD22–12) was selected for affinity maturation based upon functional NFAT reporter activity against full-length recombinant CD22 and recombinant truncated forms of CD22 containing different domains and additional screening in primary T-cells. To find binders with improved functional activity, a phage library using CD22–12 was generated via error prone PCR. Panning was done for three rounds. In each round, there was a control step to avoid selection of anti-Fc binders (Fc depletion step). The supernatant (which did not bind the beads) was then used to incubate with the desired antigen, biotinylated CD22-Fc coated on the streptavidin beads.
In round 1, 40 μg of the biotinylated CD22-Fc was added to the beads, and this was incubated with the library for 30 min at RT. In round 2, 4 μg of biotinylated CD22-Fc was added to the beads, and this was incubated with the library for 1 min at RT. This was followed by competition for 5 minutes using 5-fold excess of scFv CD22–12 to the antigen concentration. Identical conditions were used for round 3 and the resulting supernatant was sequenced and top 24 clones assessed for functional activity as described in the original panning.
Statistical analysis:
All comparisons between two groups were performed using either a two-tailed unpaired Student’s t-test or Mann-Whitney test, depending on normality of distribution. Comparisons between more than two groups were performed by two-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons. All results are represented as mean ± standard error of the mean (s.e.m.). Survival data were analyzed using the Log-Rank (Mantel-Cox) test. Data analysis was performed used GraphPad Prism v8.0.
Supplementary Material
Extended Data 1 | CONSORT structure of clinical trials of CD22 CAR T cells in adults and children.
Extended Data 2 | Concentration of serum cytokines in patients treated with CAR22-long T cells. Adult patient 1 had grade 3 CRS with mean GCSF, IL-6 and MCP-1 greater than all other patients (denotated with an *), and significantly higher levels of IL-10, IFNγ, IL-1Ra, IL-2R, IL-8, MIP-1α and VEGF than at least 3 other patients.
Extended Data 4 | CART22 phenotypes over time. a, Cell growth during T cell engineering. Expression of b, PD-1, c, LAG3, and d, Tim3. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR22-short and long T cells. Statistics reflect differences between CAR22-short and long T cells.
Extended Data 3 | Re-expansion of CART19 cells in patients treated with CART22. a-d, CART19 and CART22 composition in apheresis and CART22 products in a, pediatric patient #2 and b, adult patient #2, and expansion of CART19 and CART22 in c, pediatric patient #2 and d, adult patients #2 over time after infusion of CART22. e-i, Evaluation of peripheral blood from adult patient 2 at peak CART19 expansion (day 21). CD3+ peripheral blood mononuclear cells (PBMCs) from a healthy donor were used as either e, negative, untransduced control or f, CAR19 and CAR22 dual-engineered positive control. g, CD3+ PBMCs, h, CD3+CD4+ PBMCs, and i, CD3+CD8+ PBMCs from adult patient 2.
Extended Data 6 | CART22-short cells are primed for activation. a, Difference in phospho-peptide quantity in stimulated CAR22-short compared to CAR22-long T cells. Proteins with >1.5-fold difference are shown. b, Upregulated transcriptional programs in CAR22-short compared to CAR22-long T cells than have been stimulated by CD22-coated beads for 18 hours. c, Volcano plot of differentially expressed transcripts in CAR22-short compared to CAR22-long T cells.
Extended Data 5 | a-b, Fluorescent microscopy of T cells engineered with GFP-linked a, CAR19-short or b, CAR19-long constructs. c-f, CAR22-short T cells form more effective immune synapses. c, % of cells forming CART:tumor cell immune conjugates after 5 and 15 minutes of co-culture. d, phosphorylated CD3ζ (three independent experiments, n=820 measurements per group) and e, phosphorylated Zap70 (n=180 measurements per group) in CAR T cells engaged with Nalm6. f, Quantification of CAR T cell mobility in mice engrafted with Nalm6, as measured by % of cells moving within an intravital imaging field (n=5 per group).
Extended Data 8 | Functional characterization of CAR33-enginereed T cells. a, Growth of CD33+ Molm14 acute myeloid leukemia cells and b, CAR33-bearing T cells during in vitro co-culture. Quantification of c, IFNγ, d, IL-2 and e, TNFα by CART33 cells during an in vitro co-culture with Molm14 cells. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR33-short and long T cells.
Extended Data 7 | a, Measurement of activation-induced cell death by Annexin-V expression. Anti-tumor activity of an in vitro b, treatment or c, stress model of T cells bearing either a short or long CAR composed of the m971 scFv with the CD28 co-stimulatory domain. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR22-short and long T cells.
Extended Data 10 | Anti-tumor activity of all four CD22 CAR T cells in a xenograft model of SEM ALL. n=5 mice per condition; see Supplementary Figure 7 for individual animal responses.
Supplementary Figure 1 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART22-long or CART22-short cells. Summary data represented in Figure 4g.
Supplementary Figure 2 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART22-long or CART22-short cells in three additional replicate experiments.
Supplementary Figure 3 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART19-long or CART19-short cells. Summary data represented in Extended Data 9c.
Supplementary Figure 4 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART19-long or CART19-short cells in an additional replicate experiment.
Supplementary Figure 5 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CD22-f2-long or CD22-f2-short cells. Summary data represented in Extended Data 5e.
Supplementary Figure 6 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CD22-f2-long or CD22-f2-short cells in three additional replicate experiments.
Supplementary Figure 7 | Measurement of radiance over time in individual mice bearing SEM ALL receiving a, control, b, m971-long, c, m971-short, d, CD22-f2-long or e, CD22-f2-short cells. Summary data represented in Extended Data 10.
Supplementary Figure 8 | Flow cytometry gating strategy.
Extended Data 9 | Functional characteristics of CAR19-engineered T cells. Anti-tumor activity in an in vitro a, treatment or b, stress model of T cells bearing either a short or long CD19-targeted CAR. c, Anti-tumor effect of long or short CART19 cells in a xenograft model of Nalm6 ALL (representative of two individual experiments, n=5 mice per condition; see Supplementary Figure 3 for individual animal responses and Supplementary Figure 4 for replicate experiment). Quantification of d, IFNγ, e, IL-2 and f, TNFα by CART33 cells during an in vitro co-culture with Molm14 cells. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR19-short and long T cells.
Supplementary Movie 1
NSG mice were engrafted with GFP+ Nalm6 (green), and injected with 1×106 CAR22-short (red) and 1×106 CAR22-long (blue) T cells 48h later.
Supplementary Table 1: Additional clinical characteristics of subjects enrolled on NCT0288456 and NCT02650414.
Supplementary Table 2: Full toxicity reporting for subjects enrolled on NCT0288456 and NCT02650414. Given the small size of each study, each independent event represents occurrence in one patient (no event is shown more than once per patient), and thus each number reflects both number of patients as well as overall incidence.
Acknowledgements
The authors thank F. Chen and N. Koterba for technical assistance with cytokine quantification assays, and J. Schug for assistance with RNA sequencing. The pediatric trial was supported by the CHOP Immunotherapy Frontier Program, in addition to support from the Emily Whitehead Foundation, V Foundation, and Curing Kids Cancer (S.A.G.). The pre-clinical research was supported by the Society of Immunotherapy for Cancer Holbrook Kohrt Immunotherapy Translational Fellowship (N.S.), Breakthrough Bike Challenge Buz Cooper Scholarship (N.S.), Stand Up To Cancer Innovative Research Grant SU2C-AACR-IRG 12–17 (D.M.B.; Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C), NIH R01GM104867 (J.K.B.), Cancer Research Institute Irvington Fellowship (N.H.R.), NHLBI HL125018 and NIAID AI124769, AI129594 and AI130197 (D.L.), Japan Agency for Medical Research and Development (AMED) grant P20am0101108 (D.M.S.), NCI K08CA194256 (S.G.), American Society of Hematology Scholar Award, NCI 1K99CA212302 and R00CA212302 (M.R.), University of Pennsylvania-Novartis Alliance (S.G. and C.H.J.), NCI 1P01CA214278 and R01CA226983 (C.H.J.).
Footnotes
Competing interests
M.R., S.G. hold patents related to CART22. C.H.J. has received grant support from Novartis, and has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. C.H.J. is also a scientific founder and holds equity in Tmunity Therapeutics. S.A.G. has received support from Novartis, Servier and Kite, and serves as a consultant, member of the scientific advisory board or study steering committee for Novartis, Cellectis, Adaptimmune, Eureka, TCR2, Juno, GlaxoSmithKline, Vertex, Cure Genetics, Humanigen and Roche. B.E., L.Z., L.P., A.P., B.G., M. Ramones and J. B. are employees of Novartis. S.F.L. has received grant support from Novartis, Tmunity and Cabaletta, and has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania, and has acted as a consultant for Kite/Gilead. S.L.M has served as a member of advisory boards or steering committee for Novartis and Kite. All other authors declare no competing interests.
Data availability
All requests for raw and analyzed pre-clinical data and materials will be promptly reviewed by the University of Pennsylvania to determine if they are subject to intellectual property or confidentiality obligations. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement. Sequences for the m971 CAR are publicly available under patent PCT/US2013/060332. Sequences for the CD22-f2 CAR are private property of Novartis. Raw data for Figures 3–5, Extended Data 4–10 and Supplementary Figures 2a–c (located with data for Figure 4g), 3a–c (located with data for ED9c), 4a–c (located with data for Figure 5d) and 5 (located with data for ED10) are available in the submitted Source Data files; Raw data for Supplementary Figures 1, 2d–l, 3d–f and 4d–l are located in the Supplementary Dataset.
References
- 1.Geyer MB et al. Overall survival among older US adults with ALL remains low despite modest improvement since 1980: SEER analysis. Blood 129, 1878–1881, 10.1182/blood-2016-11-749507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma H, Sun H & Sun X Survival improvement by decade of patients aged 0–14 years with acute lymphoblastic leukemia: a SEER analysis. Sci Rep 4, 4227, 10.1038/srep04227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448, 10.1056/NEJMoa1709866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila ML et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6, 224ra225, 10.1126/scitranslmed.3008226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner RA et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129, 3322–3331, 10.1182/blood-2017-02-769208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528, 10.1016/S0140-6736(14)61403-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grupp SA et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368, 1509–1518, 10.1056/NEJMoa1215134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hay KA et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 133, 1652–1663, 10.1182/blood-2018-11-883710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orlando EJ et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 24, 1504–1506, 10.1038/s41591-018-0146-z (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ruella M et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med 24, 1499–1503, 10.1038/s41591-018-0201-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sotillo E et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5, 1282–1295, 10.1158/2159-8290.CD-15-1020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamieh M et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116, 10.1038/s41586-019-1054-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24, 20–28, 10.1038/nm.4441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah NN et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol 38, 1938–1950, 10.1200/JCO.19.03279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao X, Ho M, Zhu Z, Pastan I & Dimitrov DS Identification and characterization of fully human anti-CD22 monoclonal antibodies. MAbs 1, 297–303, 10.4161/mabs.1.3.8113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haso W et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1174, 10.1182/blood-2012-06-438002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudecek M et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3, 125–135, 10.1158/2326-6066.CIR-14-0127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonnalagadda M et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther 23, 757–768, 10.1038/mt.2014.208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alabanza L et al. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol Ther 25, 2452–2465, 10.1016/j.ymthe.2017.07.013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ying Z et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med, 10.1038/s41591-019-0421-7 (2019). [DOI] [PMC free article] [PubMed]
- 21.Richman SA et al. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol Res 6, 36–46, 10.1158/2326-6066.CIR-17-0211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371, 1507–1517, 10.1056/NEJMoa1407222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3, 95ra73, 10.1126/scitranslmed.3002842 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milone MC et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17, 1453–1464, 10.1038/mt.2009.83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter D, Frey N, Wood PA, Weng Y & Grupp SA Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol 11, 35, 10.1186/s13045-018-0571-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudson PJ & Kortt AA High avidity scFv multimers; diabodies and triabodies. J Immunol Methods 231, 177–189 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Schirrmann T et al. Oligomeric forms of single chain immunoglobulin (scIgG). MAbs 2, 73–76, 10.4161/mabs.2.1.10784 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colomb F et al. Galectin-9 Mediates HIV Transcription by Inducing TCR-Dependent ERK Signaling. Front Immunol 10, 267, 10.3389/fimmu.2019.00267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu D, Peterson ME & Long EO The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity 36, 600–611, 10.1016/j.immuni.2012.03.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong W et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol Ther 26, 963–975, 10.1016/j.ymthe.2018.01.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dustin ML & Depoil D New insights into the T cell synapse from single molecule techniques. Nat Rev Immunol 11, 672–684, 10.1038/nri3066 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frigault MJ et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res 3, 356–367, 10.1158/2326-6066.CIR-14-0186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long AH et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590, 10.1038/nm.3838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynn RC et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300, 10.1038/s41586-019-1805-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schritt D et al. Repertoire Builder: High-throughput structural modeling of B and T cell receptors. Mol. Syst. Des. Eng 4, 761–768 (2019). [Google Scholar]
- 36.Lis M et al. Bridging the gap between single-template and fragment based protein structure modeling using Spanner. Immunome Research 7 (2011). [Google Scholar]
- 37.Perisic O, Webb PA, Holliger P, Winter G & Williams RL Crystal structure of a diabody, a bivalent antibody fragment. Structure 2, 1217–1226, 10.1016/s0969-2126(94)00123-5 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Liu D et al. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity 31, 99–109, 10.1016/j.immuni.2009.05.009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed F, Friend S, George TC, Barteneva N & Lieberman J Numbers matter: quantitative and dynamic analysis of the formation of an immunological synapse using imaging flow cytometry. J Immunol Methods 347, 79–86, 10.1016/j.jim.2009.05.014 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett DM et al. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: a strategy for preclinical modeling. Blood 118, e112–117, 10.1182/blood-2011-04-346528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data 1 | CONSORT structure of clinical trials of CD22 CAR T cells in adults and children.
Extended Data 2 | Concentration of serum cytokines in patients treated with CAR22-long T cells. Adult patient 1 had grade 3 CRS with mean GCSF, IL-6 and MCP-1 greater than all other patients (denotated with an *), and significantly higher levels of IL-10, IFNγ, IL-1Ra, IL-2R, IL-8, MIP-1α and VEGF than at least 3 other patients.
Extended Data 4 | CART22 phenotypes over time. a, Cell growth during T cell engineering. Expression of b, PD-1, c, LAG3, and d, Tim3. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR22-short and long T cells. Statistics reflect differences between CAR22-short and long T cells.
Extended Data 3 | Re-expansion of CART19 cells in patients treated with CART22. a-d, CART19 and CART22 composition in apheresis and CART22 products in a, pediatric patient #2 and b, adult patient #2, and expansion of CART19 and CART22 in c, pediatric patient #2 and d, adult patients #2 over time after infusion of CART22. e-i, Evaluation of peripheral blood from adult patient 2 at peak CART19 expansion (day 21). CD3+ peripheral blood mononuclear cells (PBMCs) from a healthy donor were used as either e, negative, untransduced control or f, CAR19 and CAR22 dual-engineered positive control. g, CD3+ PBMCs, h, CD3+CD4+ PBMCs, and i, CD3+CD8+ PBMCs from adult patient 2.
Extended Data 6 | CART22-short cells are primed for activation. a, Difference in phospho-peptide quantity in stimulated CAR22-short compared to CAR22-long T cells. Proteins with >1.5-fold difference are shown. b, Upregulated transcriptional programs in CAR22-short compared to CAR22-long T cells than have been stimulated by CD22-coated beads for 18 hours. c, Volcano plot of differentially expressed transcripts in CAR22-short compared to CAR22-long T cells.
Extended Data 5 | a-b, Fluorescent microscopy of T cells engineered with GFP-linked a, CAR19-short or b, CAR19-long constructs. c-f, CAR22-short T cells form more effective immune synapses. c, % of cells forming CART:tumor cell immune conjugates after 5 and 15 minutes of co-culture. d, phosphorylated CD3ζ (three independent experiments, n=820 measurements per group) and e, phosphorylated Zap70 (n=180 measurements per group) in CAR T cells engaged with Nalm6. f, Quantification of CAR T cell mobility in mice engrafted with Nalm6, as measured by % of cells moving within an intravital imaging field (n=5 per group).
Extended Data 8 | Functional characterization of CAR33-enginereed T cells. a, Growth of CD33+ Molm14 acute myeloid leukemia cells and b, CAR33-bearing T cells during in vitro co-culture. Quantification of c, IFNγ, d, IL-2 and e, TNFα by CART33 cells during an in vitro co-culture with Molm14 cells. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR33-short and long T cells.
Extended Data 7 | a, Measurement of activation-induced cell death by Annexin-V expression. Anti-tumor activity of an in vitro b, treatment or c, stress model of T cells bearing either a short or long CAR composed of the m971 scFv with the CD28 co-stimulatory domain. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR22-short and long T cells.
Extended Data 10 | Anti-tumor activity of all four CD22 CAR T cells in a xenograft model of SEM ALL. n=5 mice per condition; see Supplementary Figure 7 for individual animal responses.
Supplementary Figure 1 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART22-long or CART22-short cells. Summary data represented in Figure 4g.
Supplementary Figure 2 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART22-long or CART22-short cells in three additional replicate experiments.
Supplementary Figure 3 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART19-long or CART19-short cells. Summary data represented in Extended Data 9c.
Supplementary Figure 4 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CART19-long or CART19-short cells in an additional replicate experiment.
Supplementary Figure 5 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CD22-f2-long or CD22-f2-short cells. Summary data represented in Extended Data 5e.
Supplementary Figure 6 | Measurement of radiance over time in individual mice bearing Nalm6 ALL receiving control, CD22-f2-long or CD22-f2-short cells in three additional replicate experiments.
Supplementary Figure 7 | Measurement of radiance over time in individual mice bearing SEM ALL receiving a, control, b, m971-long, c, m971-short, d, CD22-f2-long or e, CD22-f2-short cells. Summary data represented in Extended Data 10.
Supplementary Figure 8 | Flow cytometry gating strategy.
Extended Data 9 | Functional characteristics of CAR19-engineered T cells. Anti-tumor activity in an in vitro a, treatment or b, stress model of T cells bearing either a short or long CD19-targeted CAR. c, Anti-tumor effect of long or short CART19 cells in a xenograft model of Nalm6 ALL (representative of two individual experiments, n=5 mice per condition; see Supplementary Figure 3 for individual animal responses and Supplementary Figure 4 for replicate experiment). Quantification of d, IFNγ, e, IL-2 and f, TNFα by CART33 cells during an in vitro co-culture with Molm14 cells. *P<0.05, **P<0.001, ***P<0.0001, ****P<0.00001. Statistics reflect differences between CAR19-short and long T cells.
Supplementary Movie 1
NSG mice were engrafted with GFP+ Nalm6 (green), and injected with 1×106 CAR22-short (red) and 1×106 CAR22-long (blue) T cells 48h later.
Supplementary Table 1: Additional clinical characteristics of subjects enrolled on NCT0288456 and NCT02650414.
Supplementary Table 2: Full toxicity reporting for subjects enrolled on NCT0288456 and NCT02650414. Given the small size of each study, each independent event represents occurrence in one patient (no event is shown more than once per patient), and thus each number reflects both number of patients as well as overall incidence.
Data Availability Statement
All requests for raw and analyzed pre-clinical data and materials will be promptly reviewed by the University of Pennsylvania to determine if they are subject to intellectual property or confidentiality obligations. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement. Sequences for the m971 CAR are publicly available under patent PCT/US2013/060332. Sequences for the CD22-f2 CAR are private property of Novartis. Raw data for Figures 3–5, Extended Data 4–10 and Supplementary Figures 2a–c (located with data for Figure 4g), 3a–c (located with data for ED9c), 4a–c (located with data for Figure 5d) and 5 (located with data for ED10) are available in the submitted Source Data files; Raw data for Supplementary Figures 1, 2d–l, 3d–f and 4d–l are located in the Supplementary Dataset.