
Fig. 3 |. Generic data-dependent acquisition workflows in quantitative proteomics.
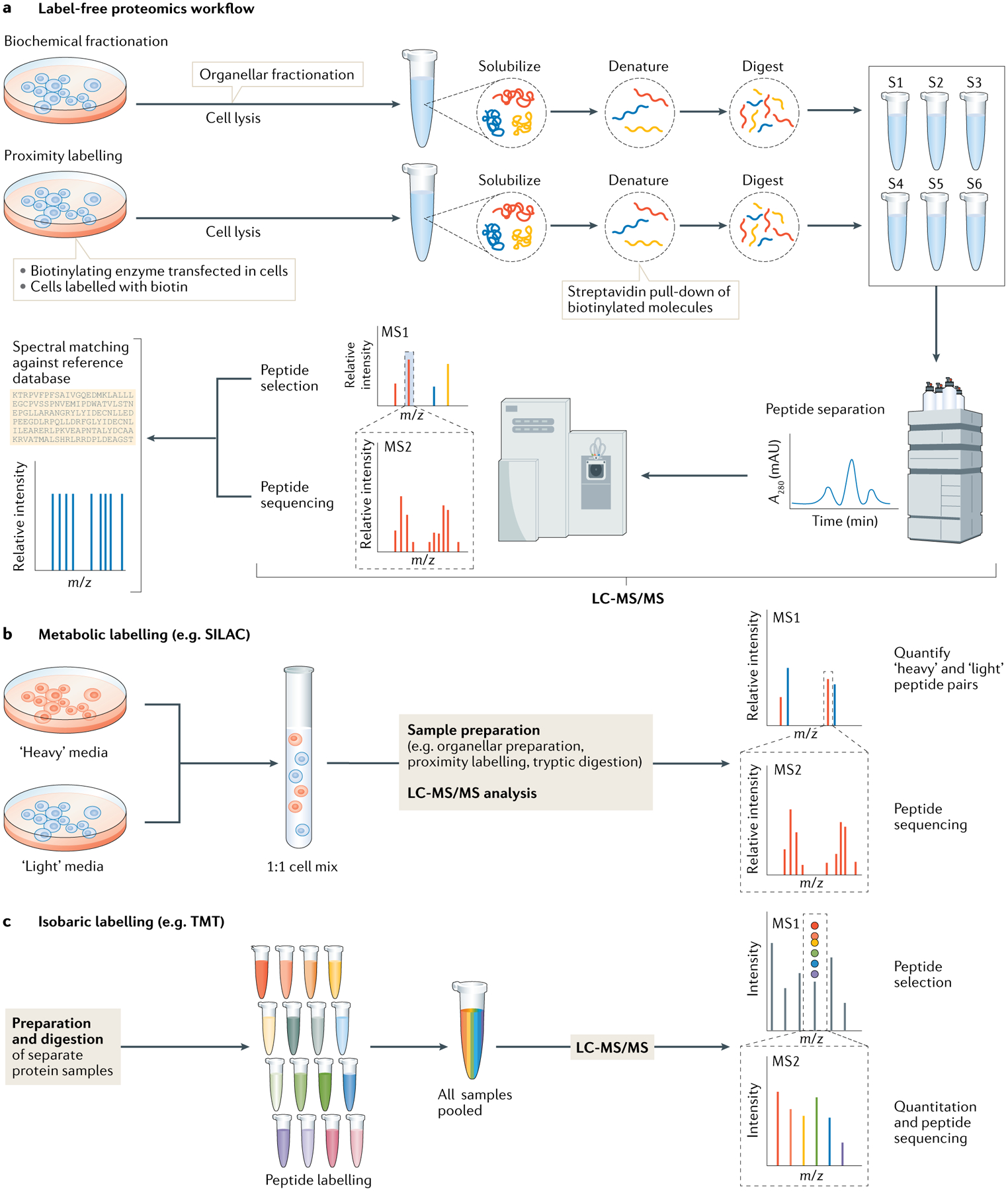
a | Two sample preparation workflows incorporating biochemical fractionation and proximity labelling techniques are shown in the context of a standard data-dependent acquisition (DDA) proteomics workflow. In DDA workflows, proteins are solubilized and denatured using buffers containing chaotropic agents and detergents, such as urea and SDS. Reduction of disulfide bonds and alkylation of free cysteine thiols allows for efficient digestion of the proteins to peptides using proteolytic enzymes, typically trypsin. Samples are then acidified and analysed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). MS/MS consists of an initial MS1 scan, which detects charged peptides (known as precursor ions), followed by isolation, fragmentation and detection of these ions in a subsequent MS2 scan to determine the amino acid sequence of the precursor ions. The complex spectra can then be deconvoluted using in silico reference databases, using algorithms that account for experimental parameters and sample preparations. b | Labelling-based proteomics methods use the above strategy, although additional steps are added to the workflow. For metabolic labelling methods such as stable isotope labelling by amino acid in culture (SILAC), a light and heavy isotopic version of an amino acid are added to the cell culture growth media, allowing metabolic incorporation of stable isotope-labelled amino acids into newly synthesized proteins. For complete incorporation, approximately six cell doublings are needed. Labelling enables sample pooling after cell harvest to minimize downstream technical variability. c | Isobaric labelling methods such as tandem mass tagging (TMT) also reduce technical variability. Labelling occurs post digestion and up to 16 samples can be multiplexed and measure in one MS run using TMT labelling. Although each tag has the same mass when bound to the peptides, upon fragmentation by higher-energy collisional dissociation during MS, their ion reporter components — which have distinguishing mass — are displaced from the peptide and can be observed in the low mass region of the MS2 spectra to determine the relative quantities of the same peptide across multiple samples. m/z, mass to charge ratio. S1–S6, sample 1–sample 6.