

ABSTRACT
DNA hypermethylation events occur frequently in human cancers, but less is known of the mechanisms leading to their initiation. Retinoblastoma, an intraocular cancer affecting young children, involves bi-allelic inactivation of the RB1 gene (RB−/-). RB1 encodes a tumour suppressing, cell cycle regulating transcription factor (pRB) that binds and regulates the RB1 core and other E2F responsive promoters with epigenetic functions that include recruitment of histone deacetylases (HDACs). Evidence suggests that bi-allelic epigenetic inactivation/hypermethylation of the RB1 core promoter (PrE-/E-), is specific to sporadic retinoblastomas (frequency~10%), whereas heritable RB1 promoter variants (Pr−/+, frequency~1-2%) are not associated with known epigenetic phenomena. We report heritable Pr−/- retinoblastomas with the expected loss of pRB expression, in which hypermethylation consistent with distal boundary displacement (BD) relative to normal peripheral blood DNAs was detected in 4/4 cases. In contrast, proximal BD was identified in 16/16 RB−/- retinoblastomas while multiple boundaries distal of the core promoter was further identified in PrE-/E-and PrE-/E+ retinoblastomas. However, weak or no DNA hypermethylation/BD in peripheral blood DNA was detected in 8/9 Pr−/+ patients, with the exception, a carrier of a microdeletion encompassing several RB1 promoter elements. These findings suggest that loss of boundary control may be a critical step leading to epigenetic inactivation of the RB1 gene and that novel DNA methylation boundaries/profiles identified in the RB1 promoter of Pr−/- retinoblastomas, may be the result of epigenetic phenomena associated with epimutation in conjunction with loss of pRB expression/binding and/or RB1 promoter interactions with boundary control elements.
KEYWORDS: Epimutations, DNA hypermethylation, loop extrusion, CTCF, RB1, promoter, retinoblastoma, cancer
Introduction
DNA hypermethylation in human cancers has been widely reported, but relatively little is known regarding the molecular signals that initiate epigenetic changes such as CpG hypermethylation, histone modification, and nucleosome packaging.
Retinoblastoma is an autosomal dominant retinal malignant tumour of the eye affecting young children with an estimated global incidence of ~1:18,000 live births. Of these, ~44% are heritable including ~27% bilateral disease and ~17% unilateral [1]. Retinoblastomas involve the tumour suppressor gene RB1 where tumour initiation requires pathogenic variants affecting both RB1 alleles. The second allele most commonly involves somatic loss of heterozygosity (LOH), which on average affects ~70% of retinoblastomas.
Pathogenic variants affecting the RB1 gene vary from deleterious high penetrance null variants including deletions and nonsense variants, to relatively low penetrance missense variants, cryptic splice variants and/or RB1 core promoter variants. The RB1 core promoter is a regulatory region containing elements sufficient for RB1 transcriptional expression (between nucleotides c.-220 to c.-170 upstream of the RB1 gene start codon) [2]. Known regulatory variants that occur within the RB1 core promoter are expected to affect RB1 gene expression by disrupting the binding of transcription factor(s). An alternative mechanism of RB1 gene regulatory inactivation observed in ~10% of retinoblastomas, involves bi-allelic epigenetic silencing of the RB1 promoter (PrE-/E-) [3]. Epigenetic silencing of the RB1 gene in retinoblastoma is generally accepted as a sporadic phenomenon [3] and is thought to involve DNA hypermethylation. In such cases, the primary DNA sequence of the RB1 gene and its promoter is otherwise unaffected.
While there has been generally no known association between promoter variants and DNA hypermethylation, a previous study identified hypermethylation of the RB1 core promoter in peripheral blood DNA and melanoma of a patient with a germline RB1 promoter variant [c.-188_-187delinsGG][4], an observation considered by the authors to be consistent with an ‘epimutation event’, where a genetic variant gives rise to epigenetic changes of adjacent DNA. No other previous studies have identified RB1 promoter variants in association with DNA hypermethylation while the requirements for epimutation(s) to exert epigenetic effects have not been explored.
RB1 gene transcription is ubiquitous with minimal expression variability between tissues (http://www.genecards.org/cgi-bin/carddisp.pl?gene=RB1). The retinoblastoma gene protein product (pRB) controls cell cycle G1/S transition by binding and inactivating E2F transcription factor. One of the several pocket proteins, pRB complexes directly with E2F-docking protein heterodimers (E2F-DP) where binding and masking the E2F transactivation domain can lead to transcriptional repression. Some promoters and transcription factors are inhibited by recruitment of histone deacetylase [5], a function related to possible epigenetic effects of pRB [6,7]. Other promoters, however, are resistant and are repressed by pRB binding directly to transcription factors [6]. The self-regulation of the RB1 gene may also involve activated expression of long non-coding RNAs, e.g., RB1-DT (aka LINC00441) which is transcribed in the opposite direction to the RB1 gene and which may repress RB1 gene expression by recruiting DNA methyltransferase [8,9] or alternative formation of a triple helix as previously described in other genes [10] might also be possible.
Evidence suggests that the organization of chromatin into distinct epigenetically defined regions involves cohesin, a protein which extrudes chromosomal DNA in a manner thought to involve the ‘loop extrusion’ model [11]. In this model, DNA is extruded through a handcuff-structured cohesin molecule generating loops which become anchored to cellular structures such as the nuclear lamina [12]. Certain DNA sequence motifs bind to CCCTC-Binding factors [13] (CTCF) which are needed to anchor looped DNA in order to regulate the 3D structure of chromatin [14] potentially forming boundaries between an active A domain (euchromatin) and an inactive B domain (heterochromatin). A single CTCF binding site has been recently experimentally verified immediately upstream of the RB1 gene core promoter with high homology to known CTCF binding sites [15]. The RB1 CTCF binding site consists of four separate DNA contact regions which are assumed to correspond to previously identified regions detected by DNAse I footprinting [16]. Interestingly, the CTCF binding site overlaps with the start of a long non-coding RNA transcript LINC00441 which evidence suggests down-regulates RB1 gene transcription in human cancers [8,9]. As the LINC00441 transcript overlaps with the CTCF binding site it is possible that CTCF binding and LINC00441 transcription are competing processes.
While much is known regarding CTCF and the biology of loop extrusion, the important subtle effects of how transcription factors and other factors modulate chromosomal boundary locations are less well characterized. In this study, we examine RB1 promoter methylation status of retinoblastomas and peripheral blood DNA affected by RB1 promoter variants to determine whether there may be a relationship between genetic/epigenetic changes and DNA methylation boundary control.
Materials and methods
Tissue specimens and bisulphite modification of DNA
All retinoblastomas (Germany, England and Toronto) were analysed anonymously following the guidelines of local Ethics committees. DNA was isolated by Roche High Pure DNA extraction kit. Genomic DNA (100 ng) extracted from frozen tumour specimens was treated with sodium bisulphite as previously described [17]. A summary of the DNA samples studied and their RB1 gene, LOH status, etc., is shown in Table 1.
Table 1.
Summary characteristics of retinoblastoma and peripheral blood DNA samples tested in this study CpGs counted as methylated includes any CpG with at least partial methylation (>10% per allele). (PB=peripheral blood). 1011 (homo c.1363C>T), 1012 (homo c.1814+1291T>G), 1013 (homo c.844G>T), 1014 (homo c.1215+1G>A), 1015 (c.2663+1G>A/c.-?1814+?del). *DNA methylation boundary location where CpGs are methylated up to and including boundary site with weak or no methylation downstream. Specimens PB=peripheral blood, T=tumour
| Sample ID | DNA sample origin |
13q14 locus zygosity |
RB1 promoter status |
CpGs (n=38) methylated (c.-1183-c.-259) |
CTCF CpGs (n=8) methylated (c.-240-c.-208) |
CP CpGs (n=7) methylated (c.-203-c.-170) |
CpGs (n=40) methylated (c.-167-c.+77) |
RB1 coding exon status |
DNA Methylation boundary CpGs* |
Heritable Status |
|---|---|---|---|---|---|---|---|---|---|---|
| M22508 | T | homo | c.-197_-196delinsTT | 21/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-516 | germline |
| 3208 | T | homo | c.-198G>A | 21/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-516 | germline |
| 3532 | T | homo | c.-198G>A | 23/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-481 | germline |
| 1706 | T | homo | c.-212G>T | 36/38 | 7/8 | 4/7 | 4/40 | RB+/+ | c.-190 | germline |
| 1323 | PB | hetero | Pr+/+ | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | normal |
| 1456 | PB | hetero | Pr+/+ | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | normal |
| 1789 | PB | hetero | Pr+/+ | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | normal |
| 1876 | PB | hetero | Pr+/+ | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | normal |
| 1987 | PB | hetero | Pr+/+ | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | normal |
| 1011 | T | Homo | Pr+/+ | 0/38 | 0/8 | 0/8 | 0/40 | RB−/- | c.-1365 | sporadic |
| 1012 | T | homo | Pr+/+ | 0/38 | 0/8 | 0/8 | 0/40 | RB−/- | c.-1365 | germline |
| 1013 | T | homo | Pr+/+ | 0/38 | 0/8 | 0/8 | 0/40 | RB−/- | c.-1365 | sporadic |
| 1014 | T | homo | Pr+/+ | 0/38 | 0/8 | 0/8 | 0/40 | RB−/- | c.-1365 | sporadic |
| 1015 | T | hetero | Pr+/+ | 0/38 | 0/8 | 0/8 | 0/40 | RB−/- | c.-1365 | sporadic |
| M22507 | PB | hetero | Pr+/c.-197_-196delinsTT | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 3208 | PB | hetero | Pr+/c.-198G>A | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 3532 | PB | hetero | Pr+/c.-198G>A | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 1706 | PB | hetero | Pr+/c.-212G>T | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 664 | PB | hetero | Pr+/c.-189G>T | 8/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 2149 | PB | hetero | Pr+/c.-197G>A | 10/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| M5616 | PB | hetero | Pr+/c.-198G>A | 8/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| M25851 | PB | hetero | Pr+/c.-239G>T | 8/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| D16.31062 | PB | hetero | Pr+/c.-198G>T | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
| 1342 | PB | hetero | Pr+/c.-206_-189del | 23/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-481 | germline |
| 4220 | PB | hetero | Pr+/c.-212G>T | 7/38 | 0/8 | 0/8 | 0/40 | RB+/+ | c.-978 | germline |
Primer design and PCR
Bisulphite treated DNA specific primers were designed for four amplicons spanning the RB1 promoter (summary of consensus sequences and CpG site locations in Figure 1, Amplicons in Fig. S1, Table S1) which were analysed by manual DNA sequencing and/or nextgen sequencing (BisNGS). Amplification primers for BisNGS (Amplicons 3 and 4 only) included a ten nucleotide sample-specific tag (barcode) which was added to the 5ʹ end of each forward and reverse primer sequence (Table S1). Nested PCR was performed for 30 cycles in a 25 μl reaction.
Figure 1.
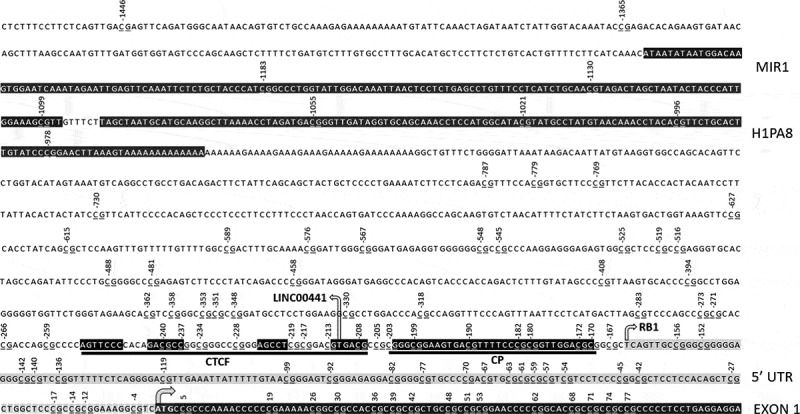
Known promoter elements and CTCF binding motif with DNA contact regions in black. Numbers indicate CpG location relative to translation start site (arrowed). RB1 gene and LINC00441 transcription start sites also arrowed. RB1 5ʹ untranslated region in grey. Core promoter (CP) and ALU repeat sequences (MIR1 and H1PA8) highlighted in black.
Nextgen sequencing of PCR products
For a single 454 sequencing run, equimolar amounts of 46 amplicons were pooled in a single tube. The emulsion PCR and subsequent sequencing reaction was performed as described in the GS FLX emPCR Method Manual (USM-00033.A, Roche, Mannheim, Germany). Using this configuration the expected yield was approx. ~1000 reads per amplicon and 17.5 Mbp sequence content in total.
A total of ~89,000 target reads were obtained from a single run (Summary of reads per tested sample can be seen in (Table S2)). As each single read corresponds to an individual DNA molecule, the fraction methylation was determined as the ratio Cn/(C + T)n at each CpG. To confirm the overall results of BisNGS analysis, Sanger sequencing (T-tracking) analysis was carried out for all amplicons obtained from amplification of bisulphite treated DNA as previously described [18]. In our hands, T-tracking reproducibility of replicate samples was within ± 10% margin of error of BisNGS results (not shown). Partial DNA methylation levels of amplicons 3 and 4 (Fig. S1), were corrected for bisulphite PCR amplification bias by the polynomial method (Fig. S2) as previously described [19]. Partially methylated CpGs were rare for Amplicons 1 and 2 (Figure S1). CpGs were either fully methylated or unmethylated and so no correction for amplification bias was carried out for these amplicons.
In addition to BisNGS, some peripheral blood DNA samples were analysed by DNA methylation microarray of bisulphite treated DNA (see Supplementary Figures for method summary). Statistical comparisons between methylation profiles in some cases were carried out by the standard Student’s t-test.
Results
RB1 Promoter DNA methylation of Pr−/- retinoblastomas
To determine whether DNA hypermethylation might be a recurrent feature of Pr−/- retinoblastomas, four available retinoblastomas homozygous for RB1 promoter variants (1706 3208, 3252, and M22507) were analysed. The first (1706) contained a variant c.-212 G > T affecting the most distal one of the four elements of CTCF. The remaining three were found to have variants within putative overlapping Sp1, RBF-1 and p53 transcription factor binding sites (SpRp53) within the RB1 core promoter (variants and promoter element motifs summarized in Figure 2). DNA methylation analysis was performed by BisNGS and the four retinoblastomas were initially compared to DNA extracted from normal peripheral blood and RB−/ – retinoblastomas. Of the four Pr−/- retinoblastomas, 1706 was found to be the most extensively methylated with 64/94 at least partially methylated CpGs (within RB1 promoter region extending from c.-1183 to c.+77) including 4/7 affecting the core promoter and 7/8 affecting the CTCF binding site region (Figure 3(a,b)) and summarized in Table 1). In contrast, peripheral blood DNA was not methylated in this region (Figure 3(c), Table 1). One of the eight core promoter CpGs of sample 1706 was abolished by the c.-212 G > T variant hence the core promoter of this sample, unlike other samples, contains 7 instead of 8 CpGs. In the remaining three SpRp53−/- retinoblastomas, however, DNA methylation was detected in only 20–29/94 CpGs which occur at a location proximal of the CTCF binding site (c.-1183 to ~c.-400-500, Figure 4(a–c) and Table 1) including four upstream CpGs (c.-787, c.-779, c.-768, and c.-730) (Fig. S3(a)). The methylation profile of the four Pr−/- retinoblastomas contrasted with all analysed normal peripheral blood DNAs and RB1−/- retinoblastomas (Figure 3(c) and Table 1). In the former, a single boundary where CpGs proximal and including c.-978 were fully methylated in the peripheral blood DNA of analysed normal individuals (summarized Table 1), a result which was further confirmed by DNA methylation microarray analysis (see below). The DNA methylation profiles of five RB1−/- retinoblastomas analysed by manual Sanger sequencing only were found to have weak or no methylation of CpGs proximal to c.978 such that CpGs distal to c.-1365 were unmethylated (example shown in Fig. S3(b)), consistent with a boundary more proximal to that identified in normal peripheral blood DNA (summarized Table 1). A further 11 RB1−/- retinoblastomas showed similar results (not shown). Taken together these results suggest that relative to peripheral blood DNA, DNA hypermethylation/distal boundary displacement may be a recurrent feature of Pr−/- retinoblastomas, whereas methylation profiles suggestive of proximal boundary displacement were consistently observed in RB1−/- retinoblastomas.
Figure 2.
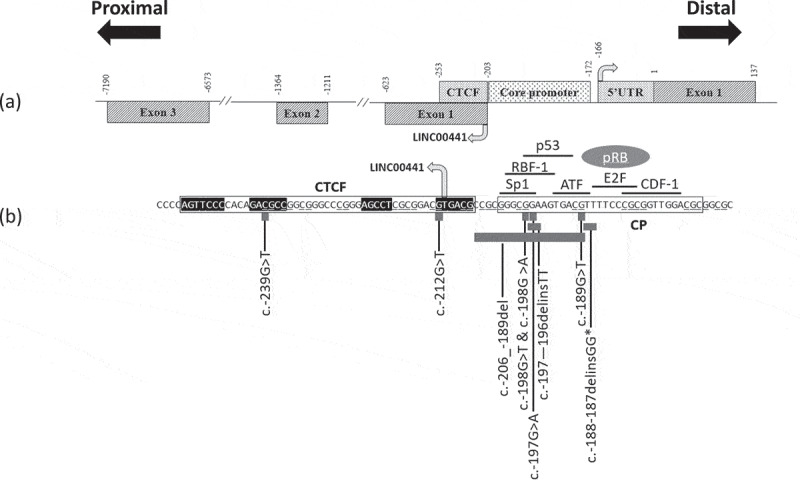
(a) Exons, 5ʹUTR, CTCF binding site, Core promoter region and long non-coding RNA exons. (b) CTCF binding site (boxed, contact regions in black), RB1 gene core promoter (CP) (boxed) with variants reported here (spanning nucleotides −239 to −187, see text) and elsewhere (*) [4], CTCF (boxed), and putative transcription factor binding elements. RB1 and LINC00441 transcription start sites (arrowed). Transcription factor pRB (grey ellipse) known to bind E2F directly.
Figure 3.
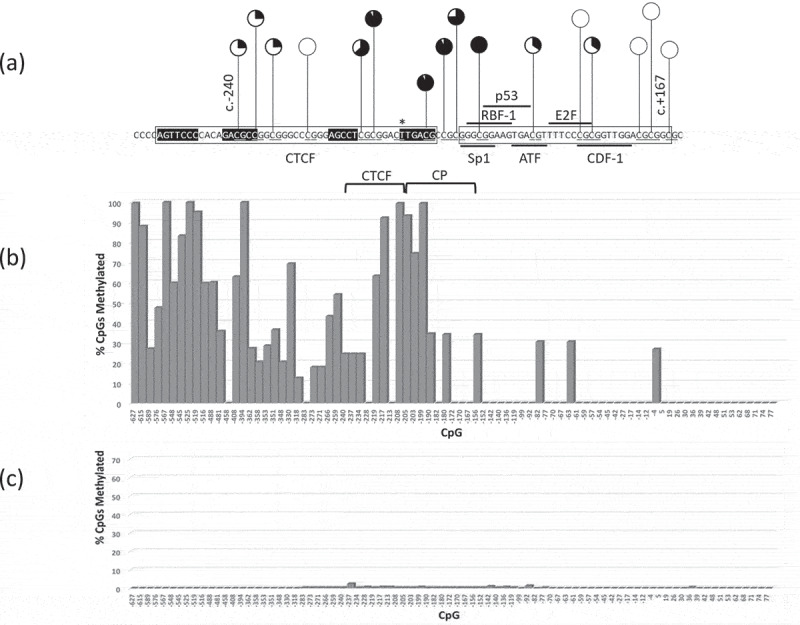
RB1 promoter CpG methylation profile of a Pr−/- retinoblastoma (1706) homozygous for the promoter variant c.-212 G > T spanning CpGs −627-+77 (underlined). (a) pie chart summary of RB1 promoter DNA methylation of CpGs that include putative CTCF binding site, core promoter (CP), coding and untranslated exons; (b) bar chart summary of 1706 CpG methylation of RB1 upstream region −627 to+77; (c) bar chart summary of normal peripheral blood DNA.
Figure 4.
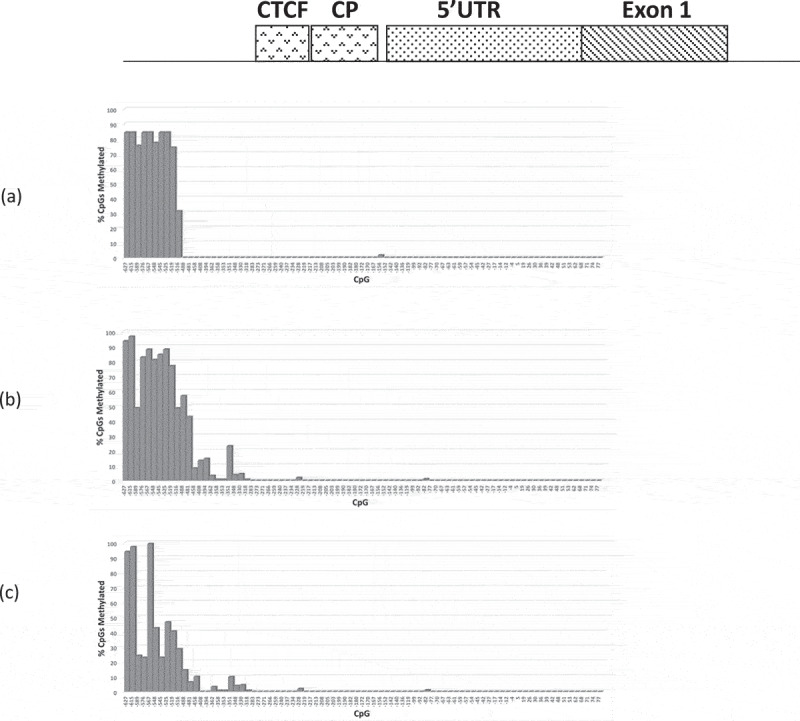
Bar chart summary of RB1 promoter CpG methylation profiles (CpGs c.-627-+77) of (a) 3208 (b) 3532 and (c) M22507; (a) and (b) are retinoblastomas homozygous for the RB1 variant c.-198 G > A, (c) retinoblastoma homozygous for c.-197_-196delinsTT variant.
As the RB1 promoter hypermethylation identified in Pr−/- retinoblastomas was associated with homozygous alleles, it was unclear whether heterozygous RB1 promoter variants in isolation might be sufficient to initiate hypermethylation in cis. If so then DNA hypermethylation of the RB1 promoter region in the peripheral blood DNA of patients carrying heterozygous promoter variants would also be expected.
RB1 promoter DNA methylation in heterozygous Pr−/+ peripheral blood DNA
To determine whether heterozygous promoter variants (Pr−/+) might be sufficient to initiate RB1 promoter DNA hypermethylation, we analysed corresponding peripheral blood DNAs of retinoblastomas M22507, 3208, 3532, and 1706 as well as five other patients carrying RB1 promoter variants for whom no tumour was available. The RB1 promoter variants of these patients are shown in Table 1. Relative to normal peripheral blood DNA (Fig. S4(a)) and RB−/- retinoblastomas (Fig. S4(b)), weak or no hypermethylation was detected in the peripheral blood DNA of 8/9 carriers of RB1 core promoter variants (Fig. S4(c)-(i), Table 1). To further confirm this result, we performed in-depth microarray DNA methylation analysis of the RB1 promoter of peripheral blood DNA (Region: hg19 13:48,876,603–13:48,892,244, Fig. S5). DNA of sufficient quantity and quality was available from three patients (664, D16.31062, and 3531, Table 1) which were analysed by methylation microarray, and compared with 20 controls 19 of which were derived from the Gene Expression Omnibus (GEO) database. No significant hypermethylation throughout the RB1 promoter region downstream of CpG at c.-978 was detected. These findings further support the results obtained by manual sequencing and BisNGS of heterozygous carriers of RB1 promoter variants indicating that there is little or no hypermethylation/boundary displacement in the peripheral blood DNA of carriers heterozygous which was comparable to normal peripheral blood DNA.
Only 1/9 carriers of heterozygous RB1 promoter variants (patient 1342, c.-206_-189del) were identified with significant RB1 promoter DNA hypermethylation (CpGs between c.-627 to c.-458, Fig. S4(j)) consistent with distal BD of a single allele. The variant identified in patient 1342 was an 18 bp deletion resulting in the loss of several putative RB1 core promoter transcription factor binding sites including Sp1, RBF-1, p53, and ATF while adjacent CTCF binding site and putative E2F element were unaffected. It is not clear why monoallelic DNA hypermethylation/boundary displacement was observed in 1342 but not other Pr−/+ carriers of promoter variants. One possibility is that the deletion of multiple transcription factor binding sites might affect the loop extrusion process and/or influence the location of DNA anchoring points.
The RB1 promoter DNA hypermethylation detected in retinoblastomas 3208, 3532, M22507, and peripheral blood DNA of 1342 (~c.-500) (Figs. S3, S4(a)-(c), and S4(j)) are each consistent with distal BD to a new location at ~c.-500. A DNA methylation boundary at this location occurs ~250 base pairs upstream from a CTCF binding element as shown in Figure 1. This result suggests that the variants affecting core promoter transcription factors are able to exert epigenetic changes ~300 bp upstream despite a single CTCF insulating barrier separating the two regions.
DNA methylation analysis of RB1 promoter in PrE-/E- and PrE-/E+ retinoblastomas
One explanation for the distal BD observed in Pr−/- retinoblastomas, is the possibility of coincidental epigenetic inactivation as a separate somatic event. If DNA hypermethylation was coincidental and in addition to the presence of promoter variants then the obtained methylation profiles might be expected to resemble typical PrE-/E- and/or PrE-/E+ retinoblastomas. This possibility was assessed by comparing the DNA methylation profiles across 94 CpGs (c.-1183 to c.+77) of Pr−/- retinoblastomas and comparing with epigenetically silenced retinoblastomas including 22 available PrE-/E- and 8 PrE-/E+ sporadic retinoblastomas (examples shown in Figure 5(a–f), and Table S3). In all 22 PrE-/E- and 8 PrE-/+ retinoblastomas examined, hypermethylated CpGs were detected in most or all upstream CpGs including the core promoter (c.-1183-c.-180) and in some cases further distal (e.g. Figure 5(a,b,e)). Interestingly, hypermethylation extended to a relatively concise location just beyond the core promoter (~c.-100) in 4/22 PrE-/E- and 4/8 PrE-/+ retinoblastomas (Figure 5(d,f), Table S3). In some PrE-/E- and PrE-/+ cases, however, boundary displacement was not a clear mechanism, e.g., the retinoblastoma DNA of patient 4927 (Figure 5(c)), where significant regions of hypomethylation were observed mainly distal of the core promoter; however, a clear methylation boundary was not identified despite significant regions of hypomethylation. A distinct boundary for 4927 could not be ruled out as CpGs further downstream of the RB1 promoter region were not analysed. Two other PrE-/E- retinoblastomas were found with DNA hypermethylation beginning to subside at CpGs approaching c.+77 (Fig. S6), suggesting a possible further distally placed boundary, but we did not confirm this result by further analysis of downstream CpGs. Collectively these results indicate that of 22 PrE-/E- and 8 PrE-/E+ retinoblastomas, a variety of displaced methylation boundaries distal of the core promoter were detected that included 6/30 retinoblastomas consistent with a common boundary location at ~c.-100. Also observed was that the boundaries of PrE-/E- and PrE-/E+ retinoblastomas were often associated with hypervariable CpGs methylation and weakly methylated CpGs frequently detected at or near the new boundary, but no clear consensus pattern observed between samples (e.g. Figure 5(d,f)).
Figure 5.
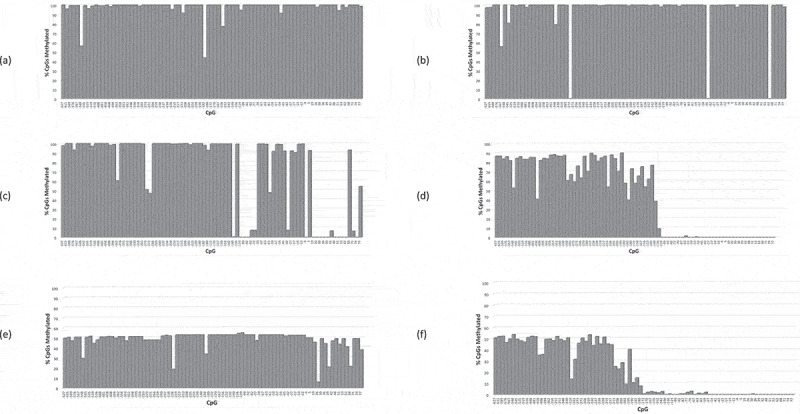
RB1 promoter CpG methylation profiles (CpGs −627 – +77) of PrE-/E- retinoblastomas, patients (a) 5492, (b) 3093, (c) 4927, and (d) 2504; and PrE-/E+ retinoblastomas, patients (e) 17,921 and (f) 2958.
To determine whether the RB1 promoter DNA methylation profile of Pr−/- retinoblastomas was comparable to PrE-/E- and/or PrE-/E+ retinoblastomas, the CpG methylation profiles were assessed and compared on the basis of three criteria, (a) boundary location, (b) the CpG methylation profile of putative functional elements within CTCF and core promoter elements (c.-240 to c.-170) and (c) the DNA methylation profile of 38 CpGs upstream of the core promoter (c.-627 to c.-259). Of 3/4 Pr−/- retinoblastomas (3532, 3208 and M22507), the CpG methylation profiles did not match PrE-/E- and/or PrE-/E+ retinoblastomas in any of the above three criteria, since no methylation affecting putative functional elements were detected and the displaced boundary was clearly considerably 5ʹ of the core promoter. As the RB1 promoter DNA methylation profile of retinoblastoma 1706 was distinct from the other three Pr−/- retinoblastomas, DNA from this tumour sample was evaluated separately.
To determine whether the RB1 promoter methylation profile of retinoblastoma 1706 (c.-212 G > T) might be consistent with coincidental hypermethylation matching PrE-/E- and PrE-/E+ retinoblastomas, CpG methylation was again compared in relation to the three criteria outlined above. While the DNA methylation boundary of 1706, which was the most extensively hypermethylated Pr−/- retinoblastoma, was not sharply defined, DNA hypermethylation had clearly ceased at a location distal of the core promoter (Figure 3) and was hence potentially similar to some PrE-/E- and/or PrE-/E+ retinoblastomas (Fig. S7, S8). With respect to the second criteria, CpGs spanning CTCF and putative RB1 core promoter functional elements (CpGs in the region c.-240 to c.-170, n = 16) were quantitatively compared based on the difference of their mean and standard deviation between and individual PrE-/E- (~40.5% per allele) and PrE-/E+ (~39.3% per allele) retinoblastomas where all the former (Fig.S7(a)) reached statistical significance (p < 0.001) whereas 2/7 of the latter (Fig.S7(b)) did not reach significance. In addition, CpGs in the CTCF region were significantly undermethylated compared to all PrE-/E- and PrE-/E+ retinoblastomas whereas the core promoter was only marginally less methylated (Fig. S8). These results suggest that while the 1706 methylation profile appears unusual, a subset of PrE-/E+ retinoblastomas was identified which were relatively closely matched at least with respect to CpGs spanning CTCF and the core promoter.
A comparison between 1706 and PrE-/E- as well as PrE-/E+ retinoblastomas regarding the third of three criteria was carried out in which CpGs upstream of the RB1 core promoter (n = 38 CpGs c.-627 to −259) was analysed. The relative mean and overall variance of CpG methylation across these upstream CpGs was compared. The mean CpG methylation was significantly reduced in comparison to all PrE-/E- as well as PrE-/E+ (Box plot and p value summaries of the CpG DNA methylation status shown in (Fig. S9(a) and 9(b)). These results indicate that while core promoter CpG methylation and the boundary location identified in 1706 were potentially similar to two PrE-/E+ retinoblastomas, there was a significant difference (p < 0.001) in methylation of the 38 CpGs upstream of the RB1 core promoter (c.-627 to c.-259), indicating that the overall DNA methylation profile of 1706 was not matched to any of our cohort of PrE-/E- or PrE-/E+ retinoblastomas and most likely has a different mechanism of origin. It is possible that the c.-212 G > T variant may either weaken the binding of CTCF or alternatively distort its conformation at a critical region immediately upstream of core promoter transcription factors. With the expected loss of pRB expression after homozygosity of the CTCF variant, a cooperative effect of these two events may be sufficient for the observed DNA hypermethylation/BD affecting the RB1 promoter.
Finally, the critical nature of CTCF-DNA contact regions is suggested by the placement of known SNP variants (summarized Fig.S10). Variants identified in this CTCF binding region are rare, particularly within the CTCF DNA contact regions (Figure 1). Variants associated with retinoblastoma are not otherwise known to dbSNP (c.-212 G > T c.-198 G > A and c.-189 G > T, Fig.S10). The c.-239 G > T variant reported here (patient M25851, Table 1) has not been seen in dbSNP although c.-239 G > A has been seen multiple times (Fig.S10). Unfortunately, no retinoblastoma tissue is available for M25851 which would have been of considerable interest to determine whether this variant might also lead to hypermethylation/boundary displacement given that a DNA contact region of the CTCF binding site is potentially affected. In the absence of such data c.-239 G > T remains a variant of uncertain significance.
Discussion
We have identified novel and unique DNA methylation profiles in retinoblastomas homozygous for RB1 promoter variants and in the peripheral blood DNA of one patient heterozygous for a deleterious germline 18 bp deletion. The DNA methylation profiles observed in Pr−/- retinoblastomas were clearly distinguishable from all other PrE-/E-, PrE-/E+, and RB−/- retinoblastomas that we have analysed. One trivial possibility that might have explained the hypermethylation detected in Pr−/- retinoblastomas was coincidental iso-allelic epigenetic silencing. Independent epigenetic inactivation as a separate coincidental sporadic event might only have been expected if the promoter variants per se were insufficient to initiate a clonal tumour expansion and therefore requiring a more damaging event in cis for the allele to be pathogenic. If true, then an independent promoter hypermethylation event would be no more effective or probable than a nonsense/missense variant or deletion affecting the RB1 gene in cis. Given that PrE-/E- retinoblastomas account for ~10% of retinoblastomas, then coincidental iso-allelic DNA methylation, and not other pathogenic variants in 6/6 Pr−/- retinoblastomas (five here and one reported elsewhere [4]) is extremely unlikely (χ2~48.6, p < 0.00001, 5 degrees of freedom). However, all available molecular and familial segregation evidence suggests that promoter variants affecting transcription factor binding sites are pathogenic and sufficient to cause disease, but with no evidence of additional iso-allelic damaging events necessary to inactivate RB1. In addition, as discussed below the DNA methylation profiles of all four Pr−/- retinoblastomas were dissimilar to all other analysed retinoblastomas, suggesting a novel mechanism giving rise to DNA hypermethylation/boundary displacement.
Of the four Pr−/- retinoblastomas, the most distinctly unique RB1 promoter methylation profiles were found in 3/3 SpRp53−/- retinoblastomas where limited RB1 promoter DNA methylation/distal BD was identified. In this subset of Pr−/- retinoblastomas, CpG hypermethylation/boundary displacement (CpGs ~c.-627 to ~c.-450) did not affect CTCF or the RB1 core promoter itself. To our knowledge, no known functional elements or pathogenic variants or deletions affecting this upstream hypermethylated region have been reported, which suggests that methylation of these upstream CpGs in isolation is unlikely to lead to inactivation of RB1 gene expression. Moreover, if functional elements critical for gene expression were indeed present in the c.-627 and ~c.-450 region and/or methylation of this region alone was sufficient to disrupt RB1 gene expression, then a subset of PrE-/E- and/or PrE-/E+ retinoblastomas with matching hypermethylation profiles not affecting the core promoter would be expected. Among our cohort of retinoblastomas without RB1 promoter variants none matched such a methylation profile, and to our knowledge, none have been previously reported. These observations suggest, therefore, that the unique hypermethylation profiles of 3/3 SpRp53−/- retinoblastomas are not coincidental events but rather epimutation driven phenomena.
Other evidence suggesting that the hypermethylation of upstream CpGs (~c.-627 to~c.-450, Figure 4) in Pr−/- retinoblastomas is not a coincidental somatic event, can be inferred from the analysis of the peripheral blood DNA of 1342 (heterozygous for c.-206_-189del, no tumour available), where a germline deletion encompassing multiple transcription factor binding sites including putative Sp1, RBF-1, p53, and ATF but not CTCF and putative E2F-DP elements (Figure 2) was identified. Comparable to the three homozygous SpRp53−/- retinoblastomas, the single allele DNA hypermethylation profile of 1342 (Figure S4(j)) is consistent with a significant distal displacement of the 5ʹ DNA methylation boundary of the affected allele but not extending as far as the RB1 core promoter. However, in contrast to the bi-allelic RB1 gene inactivation of clonal retinoblastoma cells, the presence of methylation in peripheral blood lymphocytes indicates a germline and not a somatically acquired event. To our knowledge, the only other known example of germline hypermethylation at the RB1 locus was a previously reported translocation of chromosome 13 to an inactive X chromosome [20]. We have identified one similar case (not shown) but found that the entire RB1 locus was hypermethylated, which was in sharp contrast to 1342 in which detected hypermethylation was restricted to a relatively small region ~2-400 bases upstream of the RB1 core promoter. These observations support hypermethylation observed in 1342 occurring in response to a deleterious germline promoter epimutation rather than a coincidental independent somatic event.
Only 1/4 Pr−/ – retinoblastomas analysed was found to have extensive DNA methylation that included the RB1 core promoter region (1706), where hypermethylation of 11/15 CTCF/core promoter CpGs (Figure 3(a)) was identified. While the RB1 core promoter methylation profile, and overall boundary displacement of 1706 was potentially similar to PrE-/E- and/or PrE-/+- retinoblastomas, the overall DNA methylation profile, in particular, CpGs upstream of the core promoter (c.-627 to −259) was qualitatively and quantitatively different to all other retinoblastomas that we have analysed. Specifically, we observed a reduced mean and increased variance of DNA methylation across the 38 CpGs in this region (further discussion below) when compared with our cohort of PrE-/E- and PrE-/E+ retinoblastomas. We interpret this result to support a novel mechanism of hypermethylation/BD which is most likely epimutation driven.
The mechanism of RB1 gene inactivation caused by core promoter or CTCF variants might be due to one of the two possibilities. One is that the variants simply inactivate the RB1 core promoter by the disruption of transcription factor binding. The second possibility is that the variants tip the balance favouring activation of transcription of LINC00441 transcription a negative feedback process which may down-regulate RB1 gene transcriptional repression. Evidence suggests that LINC00441 transcription may be linked to the recruitment of DNA methyltransferase [8,9]. While we found no significant hypermethylation in the peripheral blood DNA of promoter variant carriers, activation of LINC00441 leading to transcriptional inactivation of the RB1 gene in retinoblastomas via a negative feedback pathway involving triple helix formation as previously described [10], remains a possibility.
As the transcription start site of LINC00441 is immediately upstream (1 bp) of the c.-212 G > T variant identified in 1706, the non-coding transcript needs to be considered as a possible factor in DNA hypermethylation identified in this retinoblastoma. Our DNA methylation data, however, suggest that an involvement of LINC00441 in RB1 gene inactivation is less likely in case 1706. The core promoter in 1706 was clearly hypermethylated, a result strongly suggesting that it is no longer transcriptionally active either forwards or backwards rendering any effect of LINC00441 unlikely. Furthermore, in the peripheral blood DNA of this patient significant hypermethylation of the core promoter region was not detected suggesting an initial non-epigenetic mechanism of RB1 gene inactivation of the heterozygous allele. Such single allele gene inactivation is possible if CTCF is indeed critical for a fully functional RB1 promoter as previously suggested [15]. However, after c.-212 G > T homozygosity in retinoblastoma, hypermethylation is best explained by both alleles initially being non-epigenetically inactivated and only after the expected loss of pRB expression does hypermethylation/boundary displacement in 1706 become manifest.
One of CTCF’s essential functions is as an insulator and to anchor DNA to nuclear lamina in order to separate gene domains; hence, it is of considerable interest as to how variants affecting the SpRp53 region, which occur on the 3ʹ side of the RB1 CTCF binding site might influence a shift in the chromatin/heterochromatin boundary several hundred bases away on the 5ʹ side (Figure 4). One possibility is that the cohesin associated loop extrusion process is partially disrupted by genetic changes affecting the RB1 promoter. Alternatively, loop extrusion may occur normally, but the location of DNA anchoring points may be affected, for example, by the combination of promoter variants and/or expected loss of pRB expression.
Hypomethylation of CpGs upstream of the RB1 core promoter (~c.-1300 and c.-900) found in all RB−/- retinoblastomas with wild type RB1 promoter that we have analysed, is consistent with a proximal relocation of the DNA methylation boundary relative to normal peripheral blood DNA. This result may be explained by the functional loss or absent expression of pRB leading to reduced binding of HDACs to the RB1 promoter. Reduced HDAC binding is expected to increase histone acetylation and reduce the binding affinity of adjacent nucleosomes followed by CpG hypomethylation. Alternatively, it is possible that the presence of pRB may be involved in limiting loop extrusion, a process which albeit may still be dependent on pRB’s HDAC recruiting function. We have observed similar hypomethylation in the DNA of mainly primordial RB+/+ cells (e.g. embryonic stem cells and germ cells) (not shown) where although the RB1 gene is intact, a role associated with loss of pRB function is still possible since cell cycle-dependent inactivation of pRB is known to occur indirectly by other mechanisms such as phosphorylation [21].
Although the loss of pRB binding in conjunction with other transcription factors offers the simplest explanation for the observed hypermethylation/BD of Pr−/- retinoblastomas, given that pRB is a transcription factor involved in the regulation of a multitude of other E2F responsive genes, we cannot rule out that loss or activation of other transcription factors regulated by pRB might also have a role. Furthermore, it is possible that there are other expressed factors specific to retinoblastomas but not peripheral blood that might contribute to an epimutation effect of RB1 promoter variants; however, available evidence suggests that this is unlikely. Firstly, RB1 transcription/expression is ubiquitous with relatively little variation between cell types (http://www.genecards.org/cgi-bin/carddisp.pl?gene=RB1). Secondly, the DNA methylation boundary identified in peripheral blood DNA is highly conserved since it is also found in at least several other adult cell types that we have analysed, e.g., cell lines HCT116 (colon), RK0 (colon), and IPSc (foreskin fibroblasts) (not shown). As RB1 expression levels and DNA methylation boundaries in different adult tissue types are both highly conserved, any observed methylation differences between peripheral blood and retinoblastoma DNA are unlikely due to tissue-specific differences.
To our knowledge, the intriguing finding of the relocation of DNA methylation boundaries to a relatively concise location identified in 4/22 PrE-/E- and 4/8 PrE-/E+ retinoblastomas (~c.+5 to -c.-140) (Fig. S5(d), S5(f) respectively, and Table S3) has not been previously reported. It is currently not known what factors might lead to such a defined and relatively stable location just distal of the RB1 core promoter in some PrE-/E- and/or PrE-/E+ retinoblastomas but not others (Figs.6(a) and 6(b), and Table S3). Of the remaining PrE-/E- and PrE-/E+ retinoblastomas, we did not explore the full extent of methylation boundary displacement as we have analysed CpGs only as far as c.+77. The displacement of the DNA methylation boundary to variable locations in epigenetically silenced retinoblastomas despite an in tact CTCF binding site, is perhaps an indication of a malfunctioning loop extrusion and/or anchoring process.
A recurrent feature of PrE-/E-, PrE-/E+ and Pr−/- retinoblastomas was the high degree of CpG hypermethylation variation surrounding the new boundary location, whereas little variation was detected in normal cells such as peripheral blood DNA. In the latter, the boundary was consistently very sharply defined (Fig. S5) with CpG methylation at or near 100% for CpGs upstream of c.-978 and ~0% for CpGs downstream of this boundary location. It is currently not clear what determines the relatively high degree of CpG methylation variation at or near the displaced boundaries identified in epimutation and epigenetically silenced retinoblastomas.
The displacement of DNA methylation boundaries reported here is consistent with the compaction and/or relaxation of nucleosomes. For example, the most 5ʹ DNA methylation boundary was observed in RB−/- retinoblastomas (CpGs up to and including c.-1365) potentially ~2-3 nucleosomes from the methylation boundary of normal peripheral blood DNA (CpGs at c.-978). Also, the DNA methylation boundary of SpRp53−/- retinoblastomas (ca. ~c.-500) corresponds to a distal displacement ~4-500 bp from the DNA methylation boundary found in normal peripheral blood DNA a genetic distance equivalent to ~3 nucleosomes. Furthermore, the DNA boundaries at ~c.140 in four PrE-/E- and four PrE-/E+ retinoblastomas (Table S3) correspond to a possible further compaction of 1–2 nucleosomes relative to SpRp53−/- retinoblastomas. Of at least four different DNA methylation boundaries (RB−/- retinoblastomas, normal peripheral blood, three SpRp53−/-, four PrE-/E- and four PrE-/E+ retinoblastomas), the relative distance between boundaries is consistent with defined nucleosomal units in varying degrees of compaction.
Interestingly, the only other known example of promoter epimutation driven single allele DNA hypermethylation in peripheral blood DNA was the previously identified +/c.-188_-187delinsGG variant [4] a deleterious variant affecting both the last and first base of putative ATF and E2F elements, respectively. Compared to heterozygous RB1 promoter variants reported here, the previously reported epimutation [4] (Figure 2) is expected to be highly deleterious resulting in the disruption of pRB binding, since pRB directly interacts the E2F-DP heterodimer [22]. Of interest is that the epimutation leads to significant single allele hypermethylation/boundary displacement even though the CTCF binding sequence remains intact and pRB is still produced from the unaffected allele. This previously reported case [4] in conjunction with 1342 represent the only known examples of epimutation induced single allele hypermethylation associated with RB1. One explanation for these observed cases is that RB1 core promoter variants, particularly those expected to affect the binding of the transcription factor pRB directly may be more likely to result in significant single allele hypermethylation/BD of the RB1 promoter. In contrast, other variants such as c.-198 G > A having less expected direct effect with regard pRB binding to the RB1 promoter may be more dependent on loss of pRB expression as found in Pr−/- retinoblastomas, before more significant hypermethylation/BD is observed.
A question arises from the methylation profiles of analysed PrE-/E- and PrE-/E+ retinoblastomas as to whether RB1 core promoter and/or putative CTCF binding site hypermethylation or as yet some other unidentified process in some way perhaps disrupts loop extrusion/anchoring leading to the variable and erratic DNA methylation boundaries observed in these retinoblastomas. Of interest was that the boundaries of 4/8 (50%) of available PrE-/E+ retinoblastomas were just 3ʹ of the core promoter, whereas only 3/22 (14%) of PrE-/E- retinoblastomas had such a relatively upstream boundary (p < 0.001) (Table S3), while the remainder (86%) were post c.+77. The significance of these results in explaining the hypervariable boundary locations of epigenetically silenced retinoblastomas is not known but may suggest that boundary displacement is not a static event but rather may be associated with gradual deterioration of the loop extrusion process occurring during tumour evolution.
Obtaining a more detailed understanding of the molecular mechanisms associated with hypermethylation/BD in retinoblastoma has proven extremely difficult given that there are no known methods of stably hypermethylating small chromosomal regions. Studies involving RB1 promoter epimutations, however, are feasible and could potentially allow a useful approach towards further delineating factors influencing RB1 gene boundary control.
As shown in Fig.S10, rare SNPs are prevalent throughout the CTCF binding site, but very few are known to be pathogenic, other than those which have been reported associated with retinoblastoma although c.-239 G > A remains a variant of uncertain significance. Our findings suggest that the functional effect of RB1 promoter variants such as c.-239 G > A could potentially be explored through their analysis in a cell line model preferably by introducing CrispR/cas9 knock in gene edits. Ideally, homozygous Pr−/- or Pr−/RB− compound heterozygous variants at the RB1 locus would be introduced such that pRB inactivation in combination with variants of interest could be analysed. However, an alternative simpler approach might be to introduce single allele variant edits, and silence pRB expression by the addition of antisense oligos targeting RB1 gene mRNA. Through experimental analyses such as these genuine pathogenic variants affecting the RB1 promoter may result in DNA hypermethylation/boundary displacement of the affected allele potentially offering important functional evidence of their pathogenicity. Introduction of such edits in plasmids for the purpose of stable transfection assays may not be as informative due to unpredictable position effects. Transient transfection assays would not be informative since free plasmid DNA would not be subject to the epigenetic forces present in chromosomal DNA.
In summary, the detection of hypermethylation and/or the displacement of DNA methylation boundaries in retinoblastomas suggests that components of the RB1 promoter complex including pRB may be directly or indirectly interacting with boundary control/loop extrusion elements. Further study of epimutation events potentially offers a powerful approach to help refine our understanding of the early molecular events leading to promoter hypermethylation in cancer cells and in future facilitate the identification of novel factors involved in epigenetic silencing that may serve as more specific targets for future drug therapies.
Supplementary Material
Acknowledgments
We are extremely grateful to Deniz Kanber, Dietmar Lohmann, Diane Rushlow and Trevor Cole for consented patient DNA specimens, the Cancer Society (Canterbury/Westland branch) for partial funding of this study, Anthony Thrush for 454 nextgen sequence analysis and Darren Walmsley for his assistance in Linux-based DNA methylation software.
Acronyms
Pr−/+ =genetic silencing of single allele; PrE-/E+ = epigenetic silencing of single allele; SpRp53 = overlapping Sp1, RBF-1 and p53 binding motifs; CTCF = CCCTC-Binding factor; BisNGS = nextgen sequencing of bisulphite treated DNA; BD = Boundary displacement; HDACS=histone deacetylases SNPs=single nucleotide polymorphisms.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Broaddus E1, Topham A, Singh AD.. Incidence of retinoblastoma in the USA, 1975–2004. Br J Ophthalmol. 2009;93:21–23. [DOI] [PubMed] [Google Scholar]
- [2].Fujita T, Ohtani-Fujita N, Sakai T, et al. Low frequency of oncogenic mutations in the core promoter region of the RB1 gene. Hum Mutat. 1999;13:410–411. [DOI] [PubMed] [Google Scholar]
- [3].Ohtani-Fujita N, Dryja TP, Rapaport JM, et al. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet. 1993;98:43–49. [DOI] [PubMed] [Google Scholar]
- [4].Quiñonez-Silva G, Dávalos-Salas M, Recillas-Targa F, et al. Monoallelic germline methylation and sequence variant in the promoter of the RB1 gene, a possible constitutive epimutation in hereditary retinoblastoma. Clin Epigenetics. 2016;8(8):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brehm A, Miska EA, McCance DJ, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391(6667):597–601. [DOI] [PubMed] [Google Scholar]
- [6].Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92(4):463–473. [DOI] [PubMed] [Google Scholar]
- [7].Ferreira R, Magnaghi-Jaulin L, Robin P, et al. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci USA. 1998;95(18):10493–10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tang J, Xie Y, Xu X, et al. Bidirectional transcription of Linc00441 and RB1 via H3K27 modification-dependent way promotes hepatocellular carcinoma Cell. Cell Death Dis. 2017;8(issue 3):page e 2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou J, Shi J, Fu X, et al. Linc00441 interacts with DNMT1 to regulate RB1 gene methylation and expression in gastric cancer. Oncotarget. 2018;9:pp: 37471–37479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou Z, Giles KE. Felsenfeld G DNA·RNA triple helix formation can function as a cis-acting regulatory mechanism at the human β-globin locus PNAS March 26. Proc Nat Acad Sci. 2019;116:6130–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barrington C, Finn R, Hadjur S. Cohesin biology meets the loop extrusion model. Chromosome Res. 2017;25:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Phillips JE, Corces VG. “CTCF, master weaver of the genome”. Cell. 2009;137:1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cuddapah S, Jothi R, Schones DE, et al. “Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains”. Genome Res. 2009;19:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rubio ED, Reiss DJ, Welcsh PL, et al. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci USA. 2008;105:8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].De La Rosa-Velázquez A, Rincón-Arano H, Benítez-Bribiesca L, et al. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. 2007;67(6):2577–2585. [DOI] [PubMed] [Google Scholar]
- [16].Temple MD, Murray V. Footprinting the ‘essential regulatory region’ of the retinoblastoma gene promoter in intact human cells. Int J Biochem Cell Biol. 2005;37(3):665–678. [DOI] [PubMed] [Google Scholar]
- [17].Raizis AM, Schmitt F, Jost JP. A bisulfite method of 5-methylcytosine mapping that minimizes template degradation. Anal Biochem. 1995;226(1):161–166. [DOI] [PubMed] [Google Scholar]
- [18].Leakey TI, Zielinski J, Siegfried RN, et al. A simple algorithm for quantifying DNA methylation levels on multiple independent CpG sites in bisulfite genomic sequencing electropherograms. Nucleic Acids Res. 2008;36:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moskalev EA, Zavgorodnij MG, Majorova SP, et al. Correction of PCR-bias in quantitative DNA methylation studies by means of cubic polynomial regression. Nucleic Acids Res. 2011;39:e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dries D1, Baca K, Truss L, et al. Interstitial deletion of 13q and a 13; X chromosome translocation results in partial trisomy 13 and bilateral retinoblastoma. Ophthalmic Genet. 2003;24(3):175–180. [DOI] [PubMed] [Google Scholar]
- [21].Ewens KG, Bhatti TR, Moran KA, et al. Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med. 2017;6(3):619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rogers KT, Higgins PD, Milla MM, et al. DP-2, a heterodimeric partner of E2F: identification and characterization of DP-2 proteins expressed in vivo. Proc Natl Acad Sci U S A. 1996;93:7594–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.