

SUMMARY
Metabolic reprogramming is a common feature of many human cancers, including acute myeloid leukemia (AML). However, the upstream regulators that promote AML metabolic reprogramming and the benefits conferred to leukemia cells by these metabolic changes remain largely unknown. We report that the transcription factor ATF3 coordinates serine and nucleotide metabolism to maintain cell cycling, survival, and the differentiation blockade in AML. Analysis of mouse and human AML models demonstrate that ATF3 directly activates the transcription of genes encoding key enzymatic regulators of serine synthesis, one-carbon metabolism, and de novo purine and pyrimidine synthesis. Total steady-state polar metabolite and heavy isotope tracing analyses show that ATF3 inhibition reduces de novo serine synthesis, impedes the incorporation of serine-derived carbons into newly synthesized purines and disrupts pyrimidine metabolism. Importantly, exogenous nucleotide supplementation mitigates the anti-leukemia effects of ATF3 inhibition. Together, these findings reveal the dependence of AML on ATF3-regulated serine and nucleotide metabolism.
Graphical Abstract
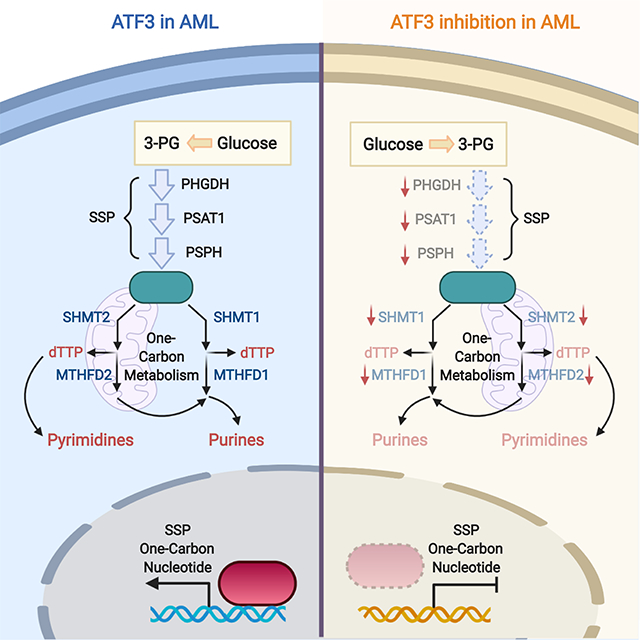
eTOC Blurb
Metabolic reprogramming is a hallmark of human cancer including acute myeloid leukemia (AML), however, many of its upstream drivers remain largely unknown. Di Marcantonio et al. have discovered that ATF3 is a previously unrecognized transcriptional regulator of serine and nucleotide metabolism and through this function supports AML cell biology.
INTRODUCTION
Acute myeloid leukemia (AML) is a hematopoietic malignancy that is characterized by the accumulation of undifferentiated myeloid blasts in the bone marrow (BM) and periphery. Although the genetic abnormalities that drive AML are well-annotated (Cancer Genome Atlas Research Network, 2013; Papaemmanuil et al., 2016), the non-mutated molecular processes that support this disease remain largely unknown. De-regulation of cellular metabolism has emerged as a focal point of investigation and therapeutic intervention in AML (Castro et al., 2019; Kreitz et al., 2019; Stuani et al., 2019). Recent studies have exposed the targeting potential of certain amino acid metabolic pathways in AML. Specifically, multiple experimental models of AML, including those bearing internal tandem duplications of the FLT3 gene (FLT3ITD), display a unique dependency on and are sensitive to inhibitors of glutaminolysis (Gallipoli et al., 2018; Gregory et al., 2019). Blockade of amino acid uptake by the combination of venetoclax and azacitidine has been shown to selectively target leukemia stem cells (Jones et al., 2018). Furthermore, disruption of branched-chain amino acid metabolism antagonizes the leukemia initiating potential of human AML in vivo (Raffel et al., 2017). However, the synthesis and catabolism of many non-essential amino acids in AML have yet to be investigated.
Many solid cancers rely on the non-essential amino acid, serine, to maintain tumor cell proliferation and survival. Cells maintain serine pools through two processes, de novo synthesis and import from the extracellular environment, both of which are coopted in human cancer (Mattaini et al., 2016). Certain colon and lung cancer cell lines rely on exogenous serine to maintain proliferation, however, in the absence of extracellular serine, cells are also able to engage the serine synthesis pathway (SSP) via a p53-dependent mechanism (Labuschagne et al., 2014; Maddocks et al., 2013; Ou et al., 2015). The SSP is also activated in various cancer settings by ATF4, NRF2 and mTORC1 (Adams et al., 2007; DeNicola et al., 2015, Ben-Sahra et al., 2016; Bjelosevic et al., 2021), and the PHGDH gene, which encodes the rate limiting enzyme of the SSP, is amplified in both melanoma and breast cancer (Possemato et al., 2011; Locasale et al., 2011).
The high demand for serine has prompted efforts to understand the catabolic fates of serine in human cancer. Beyond the role of serine as a precursor for the generation of other non-essential amino acids, such as glycine and cysteine, serine also plays a critical role in redox homeostasis through the synthesis of glutathione and the maintenance of the NAD+/NADH ratio (Ducker et al., 2016; Diehl et al., 2019). Certain tumor cells also rely on serine for the de novo synthesis of purines as well as the pyrimidine, deoxythymidine triphosphate (dTTP) (Mattaini et al., 2016; Newman and Maddocks 2017a; Newman and Maddocks 2017b; DeNicola et al., 2015). Moreover, under limiting exogenous serine conditions, metastatic brain tumors drive serine synthesis to sustain nucleotide production (Ngo et al., 2020). While serine synthesis and catabolism have been well-defined in many human cancers, the upstream regulators of these processes as well as the role of serine metabolism in acute leukemia remain largely unknown.
Using a combination of transcriptomics and metabolomics, we have identified that the transcription factor (TF) ATF3 coordinates serine synthesis and catabolism as well as purine and pyrimidine synthesis to support AML cell cycling, survival and the differentiation blockade. These data reveal ATF3 as a previously unrecognized regulator of serine and nucleotide metabolism and potentially uncover new therapeutic targets in AML.
RESULTS
ATF3 supports KMT2A-rearranged and FLT3ITD-positive AML
We previously reported that the oncogenic TF, JUN supports AML pathogenesis (Zhou et al., 2017). To identify potential downstream target genes of JUN that also support AML, we utilized two gene query databases (Bagger et al., 2016; Hebestreit et al., 2012) to examine whether the expression of any JUN-targets correlated with AML prognosis. From this analysis, we found that elevated expression of ATF3 correlates with poorer outcomes in the AML-TGCA dataset (p=0.0248, n=172, Fig. S1A) and the German AMLCG 1999 trial (p=0.0049, n=163, Fig. S1B) (Cancer Genome Atlas Research Network, 2013; Metzeler et al., 2008). In a third AML patient dataset published by Verhaak et al. (Verhaak et al., 2009), we found that high levels of ATF3 are associated with poorer outcomes in AML patients that are wild type for NPM1 but FLT3ITD+ (p=0.0038, n=21, Fig. S1C). An examination of the frequency of various AML genetic lesions in “high” versus “low” ATF3 expressers showed that high ATF3 expressers display more cases of FLT3ITD+ AML compared to low ATF3 expressers in the TCGA dataset (Χ2 statistic with Yates correction=12.57, p=0.000392) but not in the Verhaak et al. dataset (Fig. S1D).
To validate whether ATF3 is a direct transcriptional target of JUN, we performed quantitative PCR (qPCR) analysis of human AML cells expressing JUN-targeting shRNAs and found that steady-state levels of ATF3 are decreased upon JUN inhibition in the human AML cell lines, THP-1, NOMO-1, OCI-AML3 and HL-60 (Fig. S1E). Analysis of JUN chromatin immunoprecipitation sequencing (ChIP-seq) data from the UCSC genome browser (Wang et al., 2013; Davis et al., 2018) suggested that JUN localizes to the transcriptional start site (TSS) of the ATF3 promoter, which we confirmed by ChIP analysis in the human AML cell lines, THP-1 and OCI-AML3 (Fig. S1F). A comparison of ATF3 and JUN expression in both the AML-TCGA and Verhaak et al. datasets show that the expression of ATF3 and JUN positively correlate in a variety of AML genetic sub-types and lesions (Fig. S1G).
To determine whether ATF3 is functionally relevant in AML, we assessed how ATF3 inhibition impacted leukemia cell expansion both in vitro and in vivo. First, we transduced several human acute leukemia cell lines (THP-1, NOMO1, MOLM-14, HL-60, MV4–11, OCI-AML3, OCI-AML2 and K562) with recombinant lentiviruses that co-express GFP and either non-targeting control shRNAs (shNT) or human ATF3 targeting shRNAs (shATF3–1 and shATF3–2) (Fig. 1A and S1H–N). We then monitored each cell line and shRNA condition for changes in the percentage of GFP+ (i.e. shRNA-expressing) cells over time. With the exception of K562, all of the cell lines expressing shATF3 displayed a substantial decline in the percentage of GFP+ cells compared to shNT-expressing cells (Fig. 1A and S1H–N). We also found that shRNA-mediated inhibition of Atf3 significantly reduced the colony forming capacity (CFC) of two mouse AML cell lines that we generated by transducing healthy murine BM progenitor cells with retroviruses expressing either of the KMT2A-rerrangements, MLL-AF9 or MLL-ENL (Fig. 1B). However, shRNA-mediated inhibition of Atf3 did not significantly impact the CFC of lineage-depleted healthy mouse BM (Fig. 1B). To assess whether ATF3 inhibition impacted leukemia biology in vivo, we employed two mouse models of AML; one driven by MLL-AF9 and the other by a combination of FLT3ITD expression with compound deletion of Dnmt3a and Tet2 (FLT3ITD;Dnmt3a−/−;Tet2−/−, hereafter referred to as FDT cells). Briefly, leukemia cells recovered from the spleens of mice displaying frank leukemia from either model were transduced with control or Atf3-targeting shRNAs and subsequently transplanted into sub-lethally irradiated syngenic mice. Recipient mice from each condition were then monitored for the development of disease. In both models, the time of leukemia onset for recipient mice transplanted with cells expressing Atf3-targeting shRNAs was significantly longer than mice transplanted with shNT-expressing cells (Fig. 1C, 1D & S1O). Having observed that ATF3 expression was associated with patient outcome in normal-karyotype AML (NK-AML), we evaluated how ATF3 inhibition impacted the ability of NK-AML patient-derived cells (PD-AML – mutational spectrum listed in Supplemental Table 1) to persist in co-culture with HS-27 stromal cells (Klco et al., 2013; Klco et al., 2014; Di Marcantonio et al., 2018). PD-AML cells were transduced with shRNA-expressing recombinant lentiviruses and subsequently analyzed for the percentage of live cells positive for both GFP and human hematopoietic surface marker CD45 (hCD45) by flow cytometry. In the three PD-AML samples evaluated – one that is FLT3 wild type (TUHS-AML-014) and two that carry FLT3ITD (TUHS-AML-002 & −015) – the percentage of live GFP/hCD45+ PD-AML cells expressing shATF3–1 significantly decreased over time compared to shNT-expressing cells (Fig. 1E).
Figure 1. ATF3 inhibition supports AML in vitro and in vivo.
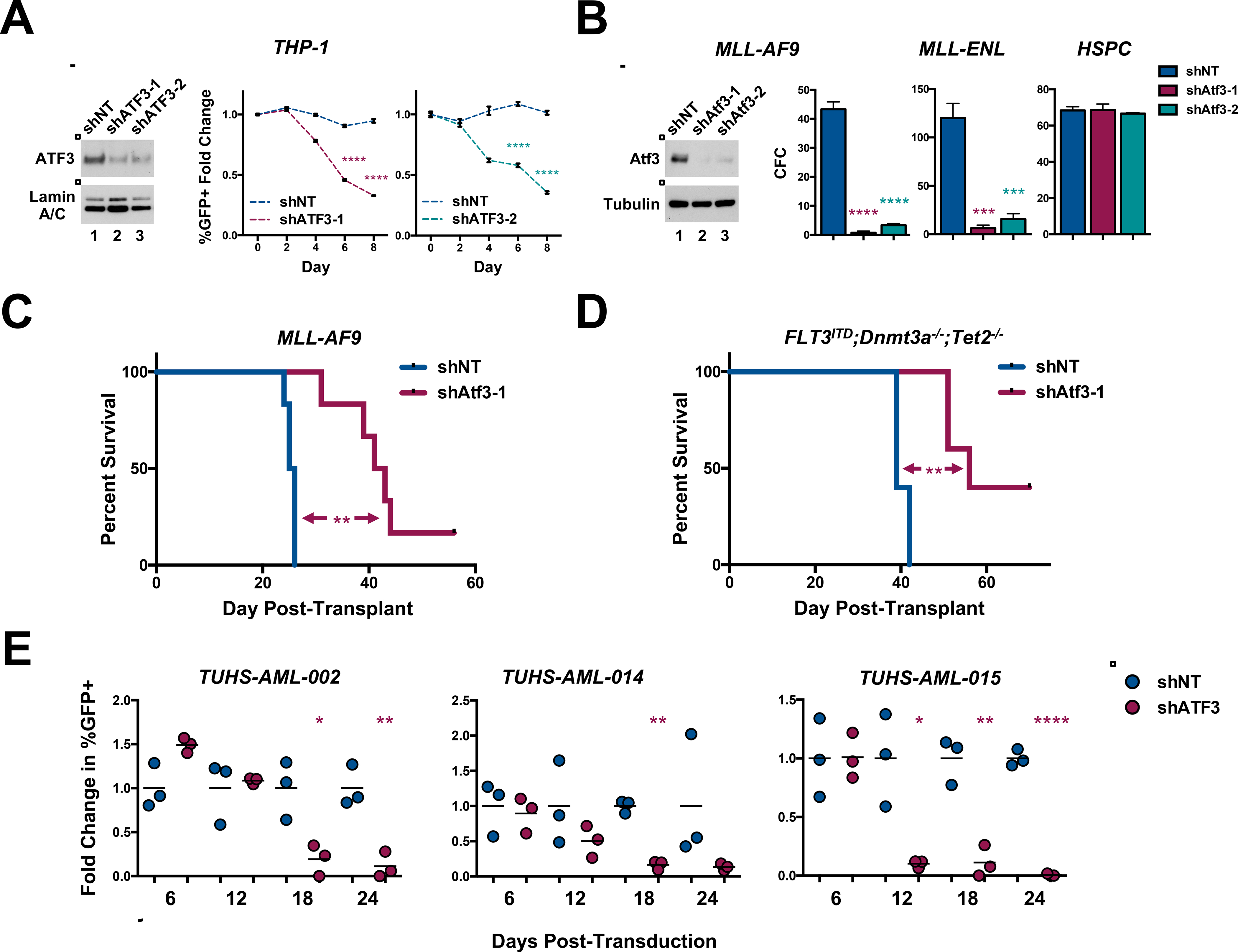
(A)Left, western blot analysis of the nuclear fraction of THP-1 cells expressing control shRNA (shNT) or human ATF3 targeting (shATF3–1 and −2) shRNAs. Middle and right panels, in vitro growth curve of THP-1 cells co-expressing GFP and shNT or shATF3–1 (middle) or shATF3–2 (right). %GFP+ cells were evaluated every 2 days by flow cytometry and plotted as the fold change in %GFP+ compared to that at day 2 post-transduction (day 0). Data represent the mean ± SD of three technical replicates. (B) Left, western blot analysis of fluorescence-activated cell-sorting (FACS)-purified GFP+ mouse MLL-AF9 cells expressing shNT, Atf3–1 or Atf3–2 shRNAs. 200 GFP+ MLL-AF9 cells (left, middle), 500 GFP+ MLL-ENL cells (right, middle) or 1,000 GFP+ lineage low healthy HSPCs from each shRNA condition were cultured in cytokine-enriched methylcellulose for 5 days. Data represent the mean ± SD of three replicates. (C & D) Kaplan–Meier survival curve of mice transplanted with shNT- or Atf3–1-expressing: (C) mouse MLL-AF9 cells (Log-rank (mantel-cox) test; n=6) or (D) or FLT3ITD;Dnmt3a−/−; Tet2−/− cells (Log-rank (mantel-cox) test (n=5). (E) Patient samples co-expressing GFP and shNT or Atf3–1 shRNAs were co-cultured with irradiated HS-27 fibroblasts and analyzed by flow cytometry for the expression of GFP and human CD45. Cells were analyzed 27 days post-transduction and plotted as fold change in %GFP+. Data represent the mean ± SD of three wells from a single experiment. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S1 and T1.
ATF3 sustains AML cell cycling, survival and the differentiation blockade
We next investigated the impact of ATF3 inhibition on human and mouse AML cell cycling, survival and differentiation. Mouse MLL-AF9 cells expressing control or Atf3-targeting shRNAs were fixed and stained with propidium iodide (PI) at various times post-introduction of the shRNAs. Flow cytometric analysis revealed that Atf3 inhibition leads to an accumulation of MLL-AF9 cells in the G1-phase of the cell cycle, beginning at 78 hours and continuing to 96 hours post-transduction (Fig. S2A). ATF3 inhibition also led to G1 accumulation of THP-1 and OCI-AML3 (Fig. S2B). MLL-AF9, THP-1 and OCI-AML3 also each displayed significantly reduced BrdU incorporation upon knockdown of ATF3 (Fig. 2A and 2B). Flow cytometric analysis of Annexin V-stained control or Atf3-targeting shRNA-expressing MLL-AF9 showed that inhibition of Atf3 expression significantly diminished cell survival starting at 96 hours and maximizing at 120 hours post-transduction (Fig. 2C and S2C). Reducing ATF3 expression also significantly decreased the survival of THP-1, MV4–11, MOLM-14, NOMO1, OCI-AML3 cells as well as FDT cells (Fig. 2D, S2D and S2E).
Figure 2. ATF3 inhibition promotes AML cell cycle arrest, death and differentiation.
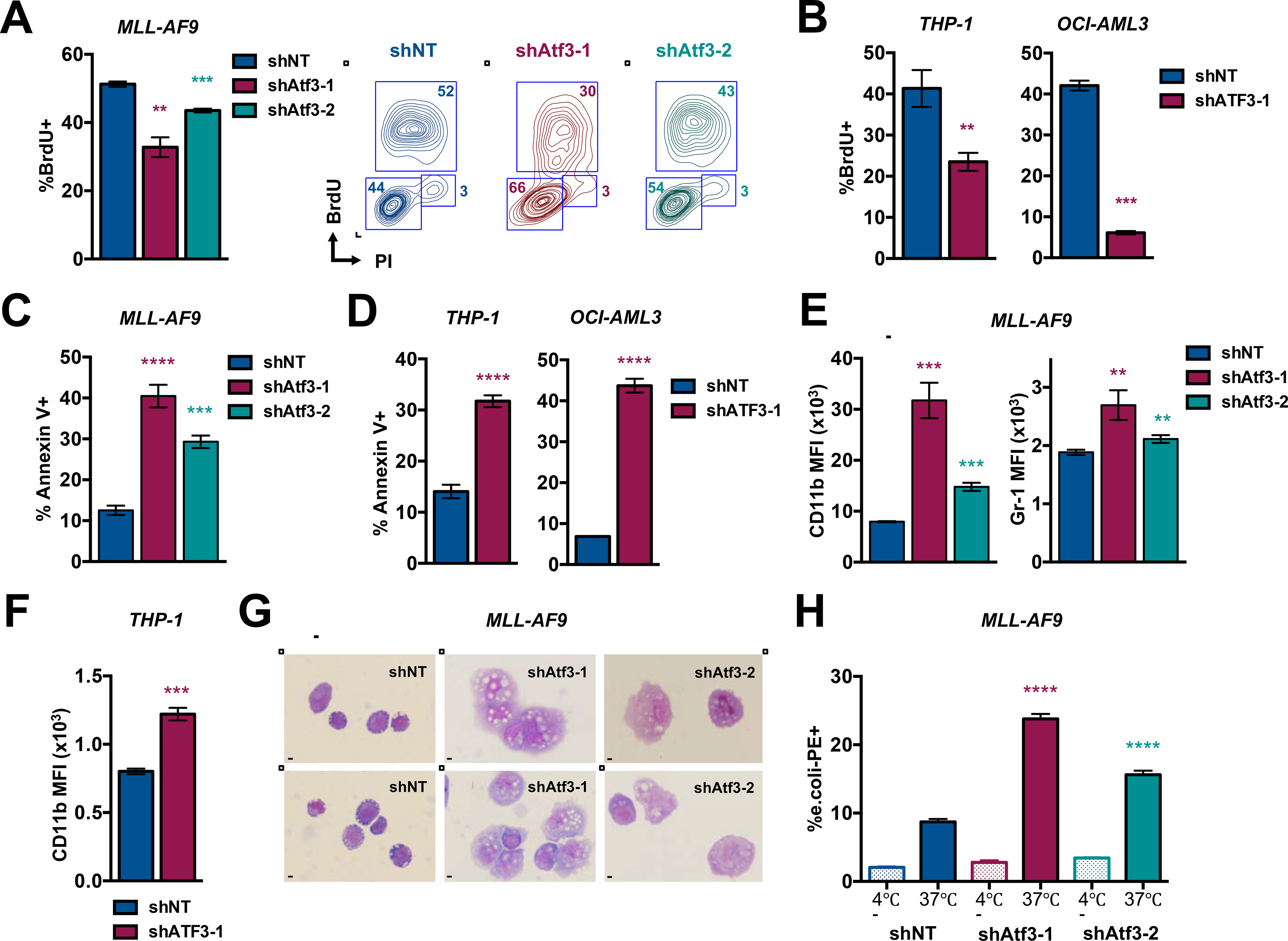
(A) Sorted GFP+ MLL-AF9 expressing shNT, shAtf3–1 or shAtf3–2 were treated with 10uM of BrdU, stained with anti-BrdU and PI and assessed by flow cytometry (left). Contour plot analysis depicts gate definition for G0-G1, S and G2-M phases of the cell cycle (right). (B) BrdU analysis of human THP-1 and OCI-AML3 cell lines as described in 2A. (C) MLL-AF9 cells expressing shNT, shAtf3–1 or shAtf3–2 were analyzed by flow cytometry for the %Annexin-V+ cells at 5 days post-transduction. (D) THP-1 (left) and OCI-AML3 (right) cells expressing shNT or shATF3–1 were analyzed by flow cytometry for the %Annexin-V+ cells at 8 days post-transduction. (E) MLL-AF9 cells expressing shNT, shAtf3–1 or shAtf3–2 were analyzed by flow cytometry for CD11b and Gr1 expression (MFI = median fluorescence intensity). (F) THP-1 cells expressing shNT or shATF3–1 were analyzed by flow cytometry for CD11b MFI and %Annexin-V+ at 6 days post-transduction. (G) Wright-Giemsa staining of MLL-AF9 expressing shNT, shAtf3–1 or shAtf3–2 shRNAs at 5 days post-transduction (40X magnification - bar = 20μm). (H) Internalization of fluorescently labeled (PE) E. coli peptides by MLL-AF9 cells expressing shNT, shAtf3–1or shAtf3–2 was assessed by flow cytometry at day 5 post-transduction. All graphed data represents the mean ± SD of three technical replicates. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S2.
To explore a possible role of ATF3 in the differentiation blockade, we first assessed the expression of the mature myeloid markers, CD11b and Gr-1, on the surface MLL-AF9 and FDT leukemia cells expressing control or Atf3-targeting shRNAs. Flow cytometric analysis showed that Atf3 inhibition led to increased CD11b and Gr-1 expression on MLL-AF9 cells as well as increased Gr-1 levels on FDT cells (Fig. 2E and S2F). ATF3 inhibition also led to increased CD11b expression on several human AML cell lines (Fig. 2F and S2G). Myeloid leukemia cells undergoing maturation display morphological changes such as increased size, cytoplasmic to nuclear ratio and asymmetry. Wright-Giemsa staining of MLL-AF9 cells showed that, compared to controls, cells expressing shAtf3–1 or shAtf3–2 were asymmetrical and displayed a substantial increase in cell size and cytoplasmic volume (Fig. 2G). We also observed similar results in THP-1 cells expressing shATF3–1 (Fig. S2H). Lastly, we examined whether ATF3 inhibition altered the ability of AML cells to phagocytose fluorescently-labeled E. coli peptides. From this analysis, we observed that MLL-AF9 cells expressing shAtf3–1 or shAtf3–2 internalized E. coli peptides at a significantly higher percentage compared to the shNT controls (Fig. 2H and S2I). Collectively, these data suggest that ATF3 inhibition promotes the terminal maturation of AML cells.
ATF3 initiates transcriptional programs associated with serine and nucleotide metabolism
To identify downstream molecular effectors of ATF3 in AML, we performed RNA sequencing (RNA-seq) of MLL-AF9 cells expressing shNT or shAtf3–1. A comparison of the transcriptomes using a statistical cut-off of p<0.001 and fold change (log2) > 0.5 and < −0.5 revealed that inhibition of Atf3 led to a significant decrease in the expression of 1164 transcripts while increasing the expression of 772 (Fig. 3A, 3B and data not shown). To identify molecular pathways impacted by Atf3 inhibition, we carried out a pathway enrichment analysis using the DAVID bioinformatic resource v6.8 (Huang da et al., 2009). An analysis (FDR p<0.05) of genes down-regulated by Atf3 inhibition identified 49 gene sets from the Gene Ontology Biological Processes (GO_BP) and 19 from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (File S1). Many of the highly enriched gene sets segregated into two themes; serine metabolism and DNA replication (Fig. 3C and 3D). Furthermore, many of the DNA replication-associated genes are directly involved in purine and pyrimidine metabolism (Fig. 4A and 4B).
Figure 3. ATF3 regulates transcriptional programs associated with serine and nucleotide metabolism.
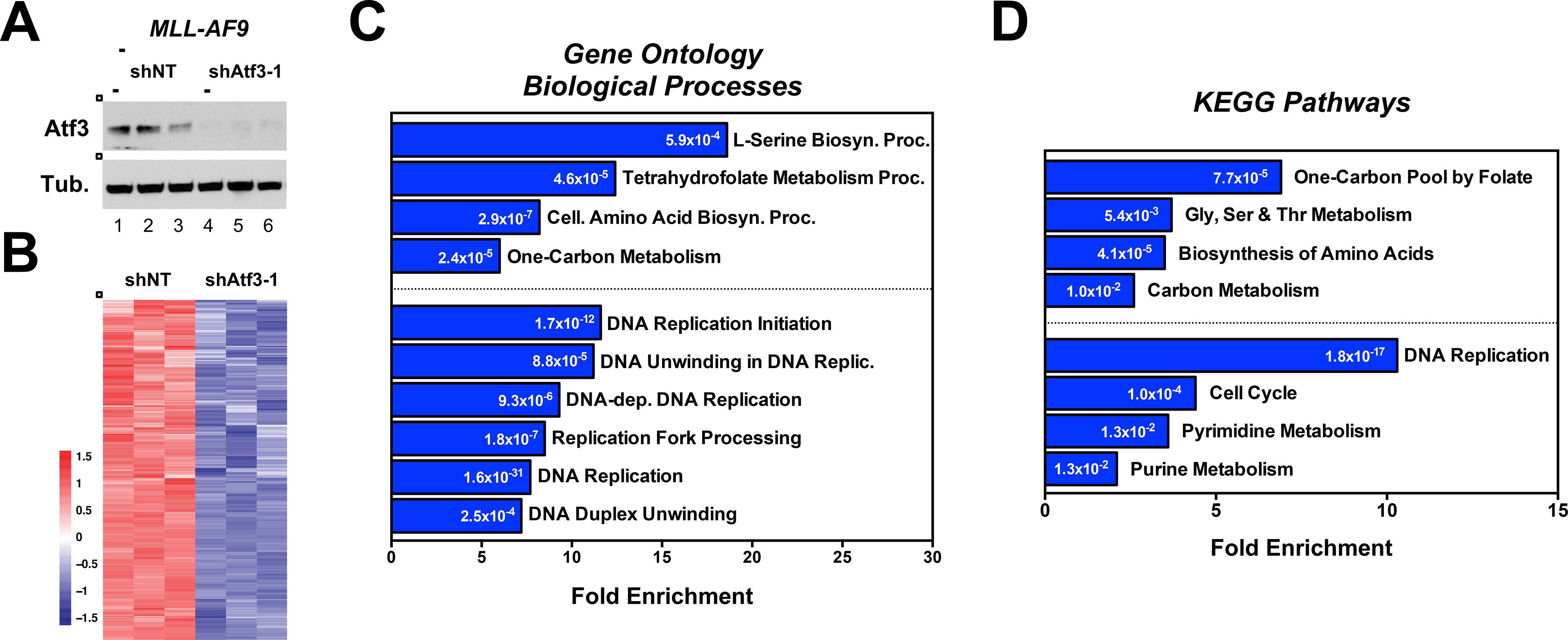
(A) Western blot analysis of MLL-AF9 cells expressing shNT or shAtf3–1 used for RNA-seq analysis. (B) Heatmap of genes that are downregulated in Atf3-shRNA expressing MLL-AF9 cells compared with shNT controls. (C & D) Pathway enrichment analysis using the DAVID bioinformatic resource v6.8. The analysis was performed using: (C) GO_BP and (D) KEGG. Numbers within the bars represent p-values. See also File S1.
Figure 4. ATF3 maintains pools of purine and pyrimidine metabolites.
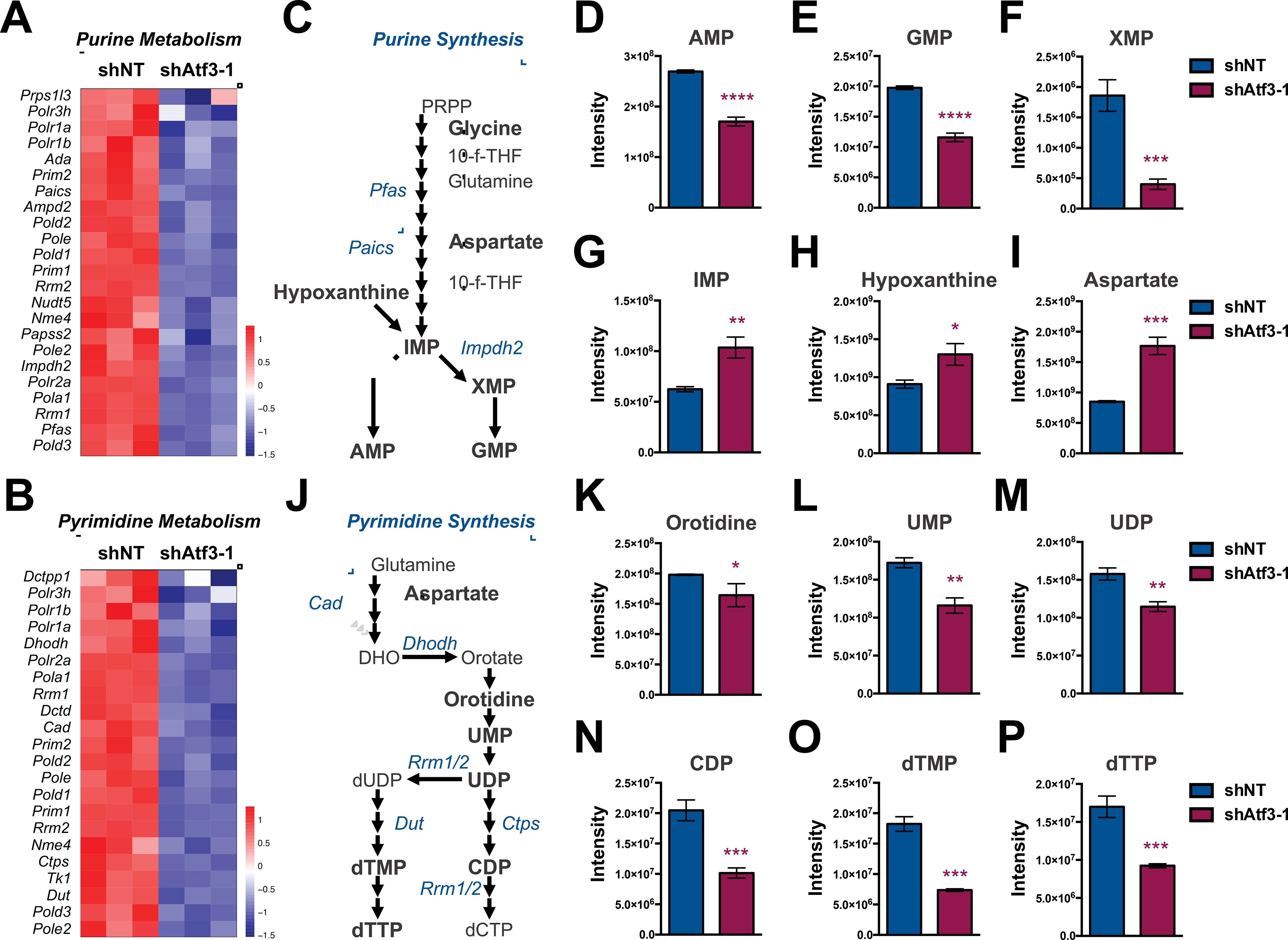
(A & B) Heatmaps of genes associated with (A) purine and (B) pyrimidine metabolism that are down-regulated in Atf3–1 shRNA-expressing MLL-AF9 cells compared to controls. (C) Schematic of the purine synthesis pathway. (D-I) Steady-state levels of polar metabolites extracted from shNT- or shAtf3–1-expressing MLL-AF9 at 72-hours post-transduction: (D) AMP, (E) GMP, (F) XMP, (G) IMP, (H) Hypoxanthine, (I) Aspartate. (J) Schematic of the pyrimidine synthesis pathway. (K-P) Steady-state levels of polar metabolites extracted from shNT- or shAtf3–1-expressing MLL-AF9: (K) Oritidine, (L) UMP, (M) UDP, (N) CDP, (O) dTMP and (P) dTTP. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S3.
ATF3 maintains pools of purine and pyrimidine metabolites
Based on our enrichment analysis, we next utilized hydrophilic interaction liquid chromatography mass spectrometry (HILIC-MS) to compare the steady-state polar metabolite profiles of shNT- and shAtf3–1-expressing MLL-AF9 cells. To ensure that polar metabolite profiles reflected a reduction in Atf3 expression and not alterations in cell cycle, survival or differentiation mediated by Atf3 inhibition, we collected metabolites extracts from each shRNA condition at 72 hours post-transduction. The initial steps of de novo purine synthesis is the conversion of phosphoribosyl pyrophosphate (PRPP) into inosinic acid (IMP), which is then converted into either adenosine monophosphate (AMP) or guanosine monophosphate (GMP), via the intermediate xanthine monophosphate (XMP) (Fig. 4C). Atf3 inhibition resulted in a significant reduction in steady-state levels of AMP, GMP and XMP but an increase in inosinic acid (IMP) levels (Fig. 4D–G). We also found that Atf3 inhibition leads to significantly higher levels of hypoxanthine, which is a metabolite generated by the nucleotide salvage pathway to resupply IMP levels under insufficient purine conditions (Fig. 4H). Atf3 inhibition also resulted in increased levels of adenosine triphosphate (ATP) and guanosine triphosphate (GTP), which are influenced by several cellular processes including purine synthesis, energy metabolism and signal transduction (Fig. S3A and S3B). While we did observe an increase in levels of aspartate upon Atf3 inhibition (Fig. 4I), we did not observe a difference in levels of glycine between shRNA conditions (Fig. S3C). The early steps of de novo pyrimidine synthesis utilize glutamine, aspartate and ribose sugars to generate uridine diphosphate (UDP), which is subsequently used to generate pyrimidines (Fig. 4J). In addition to elevated aspartate, we also observed that glutamine levels were significantly higher in shAtf3–1-expressing MLL-AF9 cells compared to shNT controls (Fig. S3D). However, steady-state levels of oritidine, uridine monophosphate (UMP), UDP, cytidine monophosphate (CMP) and cytidine diphosphate (CDP) were all significantly lower in shAtf3–1-expressing MLL-AF9 cells compared to controls (Fig. 4K–N and S3E). Additionally, steady-state levels of deoxythymidine mono- (dTMP), di- (dTDP) and triphosphate (dTTP) were all significantly decreased in MLL-AF9 cells expressing Atf3-targeting shRNAs (Fig. 4O, 4P and S3F).
ATF3 supports de novo serine synthesis
The most enriched pathway in our RNA-seq analysis showed that Atf3 inhibition decreased the expression of the genes that encode the three SSP enzymes, Phgdh, Psat1 and Psph (Fig. 5A). qPCR validation confirmed that reducing ATF3 in MLL-AF9, NOMO-1, THP-1 or HL-60 cells significantly decreased the expression of all three genes and a significant decrease in steady-state levels of PHGDH and PSPH in OCI-AML3 cells expressing shATF3 (Fig. 5B, S4A–D). ATF3 inhibition also resulted in decreased steady-state protein levels of PHGDH and PSAT1 in both mouse MLL-AF9 and NOMO1 cells (Fig. 5C and S4E). ChIP assays with anti-ATF3 antibodies showed that ATF3 localizes to a promoter region embedded within intron 1 (+600bp) of the mouse Phgdh gene, a promoter region of Psat1 over-lapping the TSS and an enhancer element located within intron 2 (+9500bp) of the Psph gene (Fig. 5D). We also observed that ATF3 binds to analogous regions within the human PHGDH, PSAT1 and PSPH genes (Fig. S4F–H). We found that ATF4, which is a known transcriptional regulator of SSP enzymes (Adams et al., 2007), localizes to similar SSP promoter regions as ATF3 in both mouse and human AML cell lines (Fig. 5D, S4F and S4G). shRNA-mediated inhibition of ATF4 also led to reduced mRNA levels of Phgdh, Psat1 and Psph (Fig. S4I and S4J). We also observed that shRNA-mediated inhibition of Atf3 resulted in decreased ATF4 mRNA and protein levels (Fig. S4K). Consistent with previous studies (Zhao et al., 2016), we observed that depletion of extracellular serine and glycine increased localization of ATF4, but not ATF3, to the promoters of SSP genes (Fig. S4L).
Figure 5. ATF3 inhibition disrupts de novo serine synthesis.
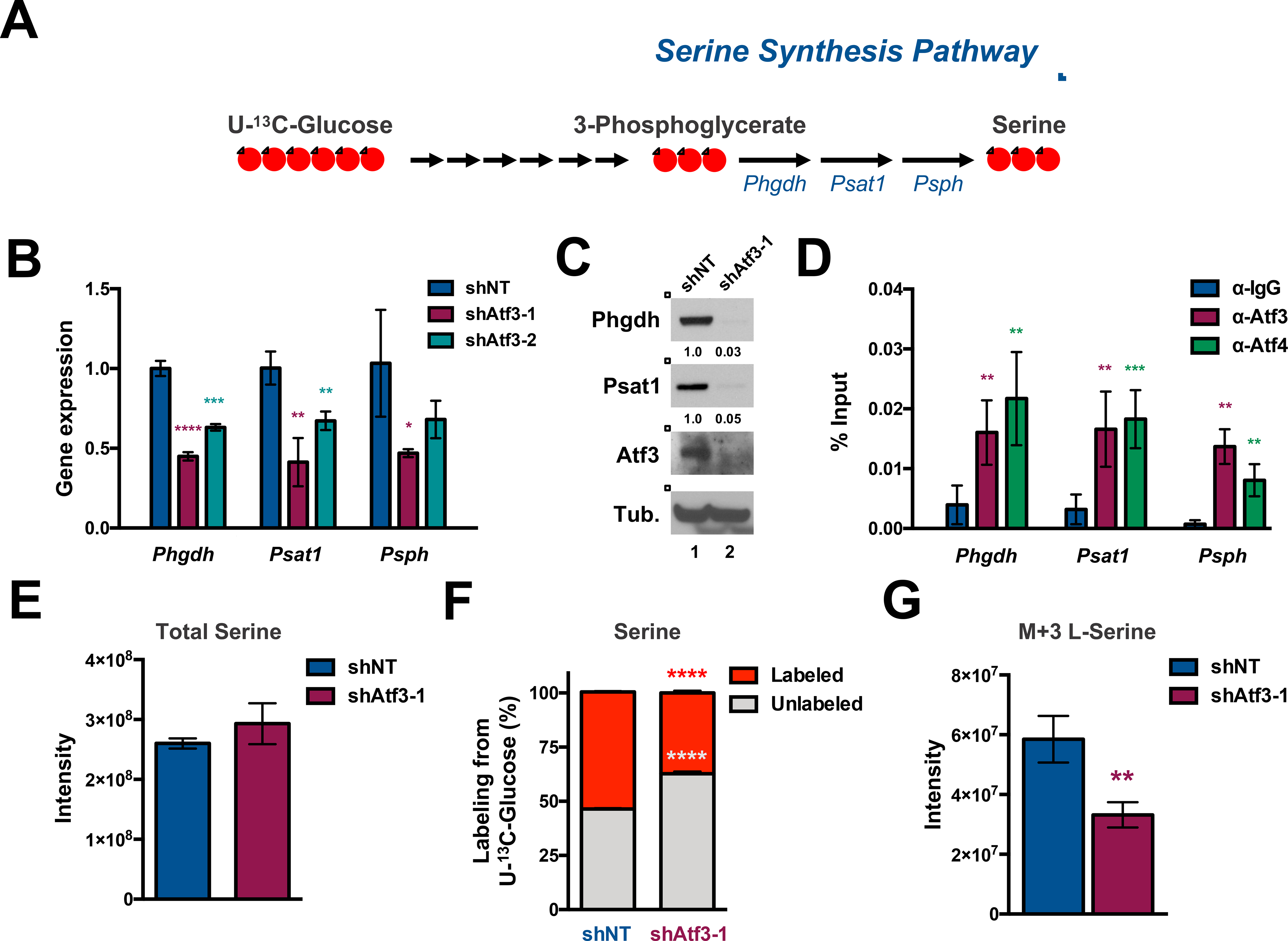
(A) Schematic of the SSP. Red circles represent 13C atoms derived from U-13C-Glucose. (B) qPCR analysis of Phgdh, Psat1 and Psph in shNT-, shAtf3–1- and shAtf3–2-expressing MLL-AF9 cells at 2 days post-transduction. (C) Western blot analysis of FACS-sorted GFP+ MLL-AF9 cells expressing shNT or shAtf3–1 at 3 days post-transduction with the indicated antibodies. Quantified proteins levels were normalized to tubulin. (D) qPCR quantification of anti-IgG, -ATF3 or -ATF4 ChIP samples with primers amplifying regions of the Phgdh, Psat1, and Psph genes. The approximate promoter regions are: Phgdh (0 - +1000); Psat1 (−500 - +500); Psph (+9000 - +10000). Data are presented as the % of input. (E) Quantification of steady-state levels of serine from shNT- or shAtf3–1-expressing MLL-AF9 at 3 days post-transduction. (F & G) shNT or shAtf3–1–expressing MLL-AF9 at 48 hours post-transduction were cultured in media supplemented with U-13C-Glucose for 24 hours after which metabolites were extracted and analyzed by HILIC-MS. (F) Percentage of unlabeled (M+0, grey) or labeled (the sum of M+1, M+2 and M+3, red) serine levels. (G) Quantification of the steady-state levels of M+3 isotopologue fraction of serine. All graphed data represents the mean ± SD of three technical replicates. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S4.
We next evaluated whether Atf3 inhibition impacted serine metabolism in AML. Total levels of serine, which are influenced by serine synthesis, import and catabolism, were not significantly different between MLL-AF9 cells expressing shNT or shAtf3–1 (Fig. 5E). To assess the impact of Atf3 inhibition on de novo serine synthesis, we performed isotope tracing experiments where MLL-AF9-GFP+ cells expressing either shNT or shAtf3–1 were placed in glucose-free RPMI-1640 media supplemented with a glucose isotopologue containing six heavy carbons (U-13C- glucose) at a concentration of 11.1mM for 24 hours – the time at which we observed isotopic steady-state (data not shown). Polar metabolite extracts from each condition were then subjected to HILIC-MS to quantify polar metabolites. MLL-AF9-shAtf3–1 expressing cells displayed significantly lower proportions of labeled (the sum of M+1, M+2 and M+3, red) serine (37.29% vs. 54.03%, p < 0.0001) with a concomitant increase in unlabeled (M+0, grey) serine compared to control cells (Fig. 5F). Additionally, the proportion of fully isotope-labeled serine in shAtf3-expressing cells was significantly less than shNT-expressing cells (Fig. 5G). Previous studies have shown that inhibition of the SSP not only blocks serine synthesis but also significantly alters steady-state levels of multiple glycolytic, tricarboxylic acid and pentose phosphate intermediates as well as leads to decreased labeling of UMP with glucose-derived carbons (Reid et al. 2018). Consistent with that study, inhibition of Atf3 resulted in higher steady-state levels of glucose-6-phosphate (G-6-P) and decreased levels of 3-phosphoglycerate (3-PG), phosphoenolpyruvate (PEP), malate, sedoheptulose-7-phosphate as well as decreased labeling of UMP with glucose-derived carbons (Fig. S4M and S4N). Similar to Atf3 inhibition, we found that shRNA-mediated inhibition of Phgdh significantly impeded MLL-AF9 cell expansion in vitro (Fig. S4O).
ATF3 is required for the incorporation of serine-derived carbons into de novo synthesized purines
Our RNA-seq data indicated that ATF3 may be activating several genes within one-carbon metabolism, which supplies serine-derived carbons into multiple metabolic pathways (Fig. 6A). qPCR analysis of MLL-AF9 cells validated that Atf3 inhibition significantly decreases the expression of Shmt1, Shmt2, Mthfd1, Mthfd2 and Mthfd1l (Fig. 6B). ChIP assays confirmed that Shmt1, Shmt2, Mthfd1 and Mthfd1l are direct transcriptional targets of Atf3 in MLL-AF9 cells (Fig. 6C). We also observed that ATF3 inhibition leads to decreased mRNA expression of SHMT1, SHMT2, MTHFD1 and MTHFD2 in the human AML cell lines, THP-1, HL-60, NOMO-1 and OCI-AML3 cells (Fig. S5A–D). Furthermore, we observed that ATF3, as well as ATF4, localizes to the promoters of SHMT1, SHMT2, MTHFD1 and MTHFD2 in NOMO-1 and OCI-AML3 cells (Fig. S5E and S5F). Atf3 inhibition in mouse MLL-AF9 resulted in decreased steady-state protein levels of Shmt1, Shmt2 and Mthfd2 but not Mthfd1 (Fig. 6D). Protein levels of SHMT1 and SHMT2 were decreased in shATF3-expressing NOMO-1 cells, however, MTHFD1 and MTHFD2 protein levels were largely unaffected (Fig. S4E).
Figure 6. ATF3 inhibition abrogates serine-derived purine synthesis.
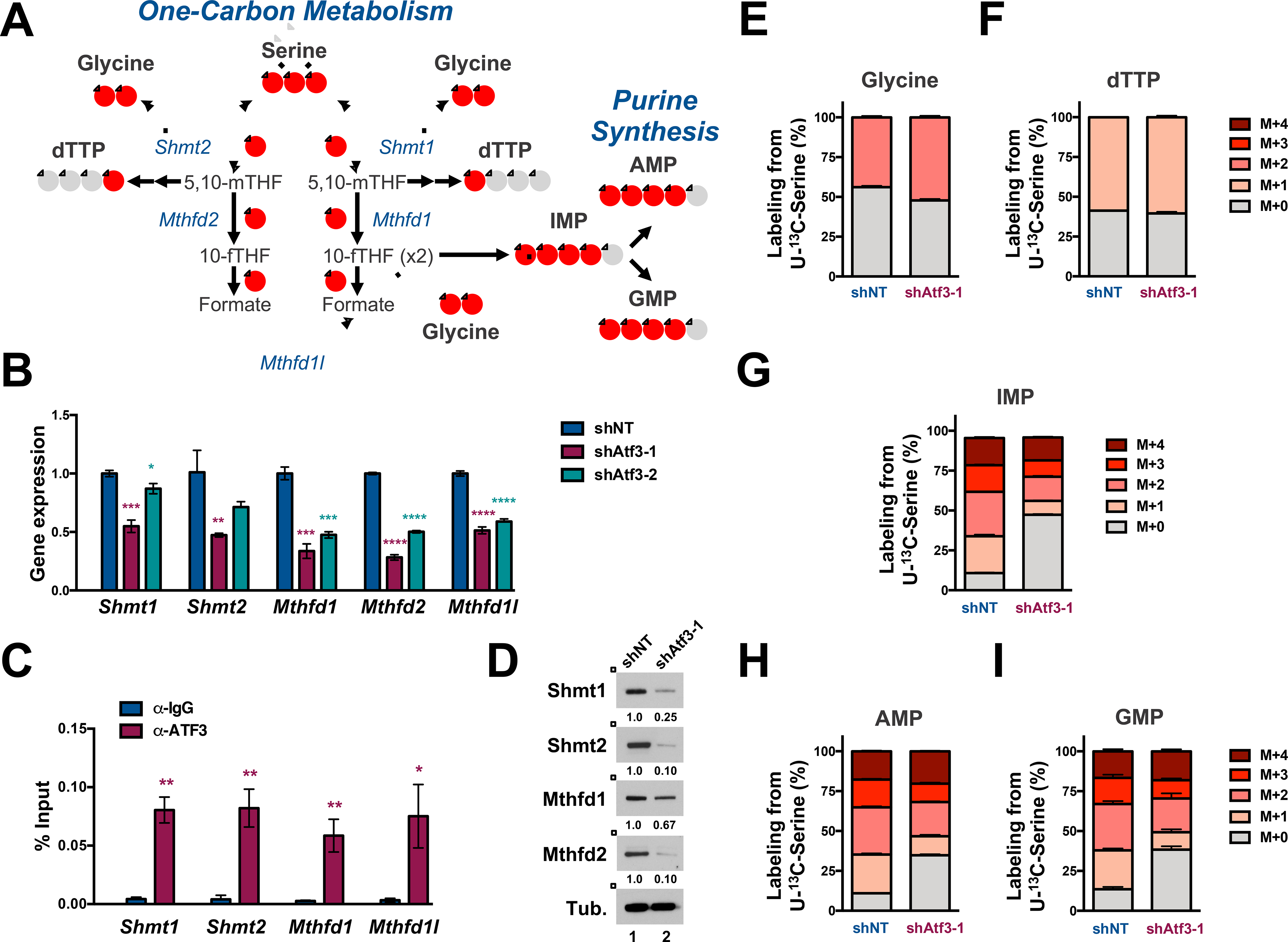
(A) Schematic of one-carbon (1C) metabolism that feed into purine synthesis. Red circles represent 13C atoms derived from U-13C-Serine. (B) qPCR analysis of indicated 1C metabolism mRNAs in shNT-, shAtf3–1- and shAtf3–2-expressing MLL-AF9 cells. (C) qPCR quantification of Atf3 binding at the indicated promoters by anti-IgG and -ATF3 ChIP analysis in MLL-AF9. The approximate promoter regions are: Shmt1 (+500 – 1500); Shmt2 (0 - +1000); Mthfd1 (−500 - +500); Mthfd1l (0 - +1000). Data are presented as the % of input. (D) Western blot analysis of GFP+ MLL-AF9 cells expressing shNT or shAtf3–1 at 3 days post-transduction with the indicated antibodies. Quantified proteins levels were normalized to tubulin. (E-I) shNT or shAtf3–1-expressing MLL-AF9 at 48 hours post-transduction were cultured for 24 hours with U-13C-serine and metabolites were subsequently extracted and analyzed by HILIC-MS. Isotopologue distribution (%) of the total steady-state levels of: (E) Glycine, (F) dTTP, (G) IMP and (H) AMP and (I) GMP. All graphed data represents the mean ± SD of three technical replicates. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S5.
To assess the impact of ATF3 on serine processing through one-carbon metabolism, we performed an isotope tracing experiment using a serine isotopologue where all three carbons are substituted for heavy carbons (U-13C-serine) in shNT- and shAtf3–1-expressing MLL-AF9 cells. GFP+ cells from each shRNA condition were grown in serine-free RPMI-1640 media supplemented with 286 μM U-13C-serine for 24 hours, after which extracted polar metabolites were subjected to HILIC-MS. At this time point, the ratio of labeled (the sum of M+1, M+2 and M+3) to unlabeled (M+0) intracellular serine was slightly increased in shAtf3–1-expressing MLL-AF9 cells compared to control cells, suggesting that Atf3 inhibition did not substantially impact the import of extracellular serine (Fig. S5G).
Despite observing a decrease in total steady-state mRNA and protein levels of Shmt1 and Shmt2 upon Atf3 inhibition we did not observe a substantial difference in the incorporation of serine-derived carbons into glycine (M+2 glycine) (Fig. 6E and S5H). We also did not observe a significant difference in the incorporation of serine-derived carbons into dTTP synthesis (Fig. 6F and S5I). However, upon examination of de novo purine synthesis, we observed that Atf3 inhibition resulted in significantly lower proportions of M+1, M+2, M+3 and M+4 isotopologues of IMP, AMP and GMP, with the exception of the M+4 GMP isotopologue (Fig. 6G–I and p-values listed in S5J). Also, shAtf3–1 expressing MLL-AF9 cells displayed significantly more unlabeled (M+0) IMP, AMP and GMP compared to shNT-expressing control cells (Fig. 6G–I, S5K–M and p-values listed in S5J). Furthermore, total levels of M+1, M+2, M+3 and M+4 isotopologues of IMP, AMP GMP, ATP and GTP (with the exception of the M+4 GMP and M+4 ATP isotopologues) were significantly decreased upon Atf3 inhibition (Fig. S5K–O). Similar to Atf3 inhibition, we found that shRNA-mediated inhibition of Mthfd2 significantly impeded MLL-AF9 cell expansion in vitro and increased CD11b expression (Fig. S5P–R).
Modulating extracellular serine concentrations minimally impacts the anti-leukemia effects of ATF3 inhibition.
We next tested how modulating extracellular serine and glycine levels impacted the anti-leukemia effects of ATF3 inhibition. Withdrawal of serine and/or glycine did not significantly alter the diminished AML cell expansion or cell death mediated by ATF3 inhibition (Fig. S6A and S6B). Serine and Glycine withdrawal did lead to significant increases in CD11b expression in control and shAtf3-expressing MLL-AF9 cells (Fig. S6C). Next, we cultured shNT-, shAtf3–1 or shAtf3–2 expressing MLL-AF9 cells in RPMI-1640-based media supplemented with standard concentrations of serine (286μM) and glycine (133μM), 5X serine, 5X glycine or 5X Serine+Glycine. Other than 5X glycine being able to marginally increase the percentage of shAtf3–2 expressing cells (i.e. %GFP+ cells), increasing the extracellular concentrations of serine and glycine minimally impacted the cell expansion or survival properties of cells expressing Atf3-targeting shRNAs (Fig. S6D and S6E). However, the combination of elevated serine and glycine, though subtle, significantly decreased CD11b expression on MLL-AF9 cells from each shRNA condition (Fig. S6F).
Exogenous deoxynucleotides mitigate the anti-leukemia effects of ATF3 inhibition
Given that ATF3 maintains pools of purine and pyrimidines and that DNA replication fork activity is highly dependent on adequate pools of nucleotides (Pai et al., 2017; Kohnken et al., 2015), we next performed a DNA fiber analysis (Nacson J et al., 2020). Both control and shAtf3–1-expressing cells were cultured in media supplemented with the nucleotide analog, 5-chloro-20-deoxyuridine (CldU, green) for 30 minutes to measure DNA fiber length (μM) and fork speed (nM/s). Cells from each condition were then removed from CldU-media and cultured in media supplemented with the nucleotide analog, 5-iodo-20-deoxyuridine (IdU, red) for 30 minutes followed by a 3 hour treatment of hydroxyurea to stall replication fork activity. Compared to controls, shAtf3–1-expressing MLL-AF9 cells displayed a significant decrease in DNA fiber length (Fig. 7A and 7B). Measurement of DNA fiber length over time indicated that Atf3 inhibition significantly slowed replication fork speed (Fig. 7C). We did not observe a change in IdU:CldU ratio following HU treatment between the two shRNA conditions signifying that Atf3 inhibition does not impact replication fork stability (Fig. S6G).
Figure 7. Nucleotide supplementation mitigates the anti-leukemia effects of ATF3 inhibition.
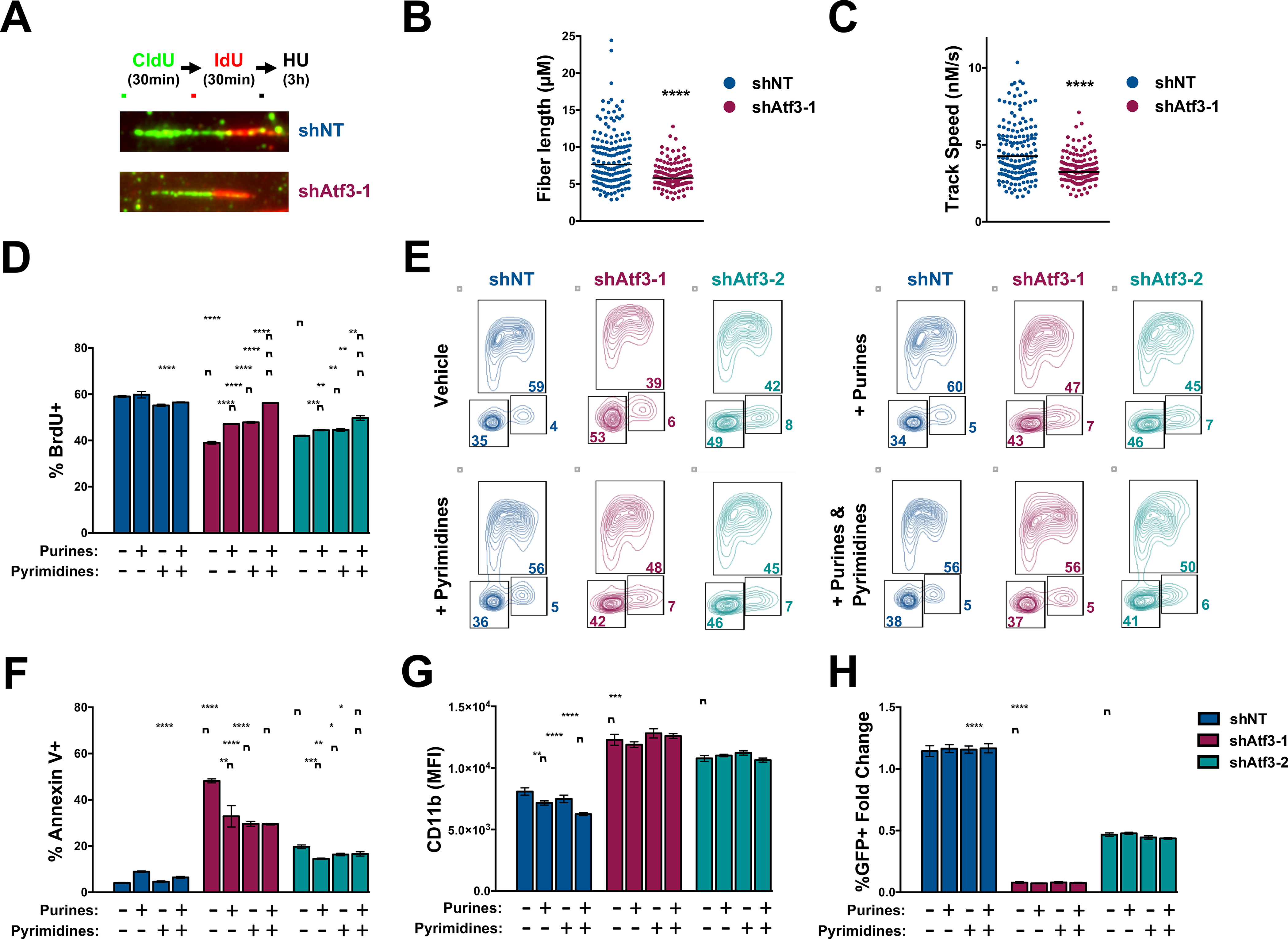
(A-C) DNA fiber analysis of shNT- or shAtf3–1-expressing MLL-AF9 cells at 96-hours post-transduction. DNA fibers were visualized by IF with fluorescently-labeled CldU- and IdU- antibodies. (A) Representative fibers for each condition and schematic of the experimental design. (B) Quantification of fiber length for the CldU portions. (C) Calculated track speeds for individual replications forks. (D-H) MLL-AF9 cells expressing control or Atf3-targeting shRNAs were cultured with vehicle, 12.5 μM purines (2’-deoxyadenosine and 2’-deoxyguanosine), 12.5 μM pyrimidines (2’-deoxycytosine and 2’-deoxyuridine 5’-monophosphate) or 12.5 μM purines and pyrimidines and then assessed by flow cytometry for: (D) %BrdU based on (E) anti-BrdU versus PI; (F) %Annexin V+; (G) CD11b MFI; and (H) changes in %GFP+ cells. Data represent the mean ± SD of three technical replicates. Asterisk key = *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. See also Figure S6.
We subsequently examined whether supplementation with exogenous deoxynucleotides (dNTs) could mitigate the anti-leukemia effects of Atf3 inhibition. MLL-AF9 cells expressing control or Atf3-targeting shRNAs were cultured without or with exogenous purines, pyrimidines or both and then assessed for cell cycle progression, cell death and CD11b expression. While MLL-AF9 cells expressing Atf3-targeting shRNAs displayed a significant decrease in BrdU incorporation compared to control cells, the addition of exogenous purines or pyrimidines partially restored BrdU incorporation and reduced G1 arrest in shAtf3–1- and shAtf3–2-expressing MLL-AF9 cells (Fig. 7D, 7E and S6H). Notably, complementation with both purines and pyrimidines almost completely restored normal cell cycling of shAtf3–1- and shAtf3–2-expressing MLL-AF9 cells (Fig. 7D, 7E and S6H). Furthermore, supplementation of purines, pyrimidines or both partly reduced the percentage of Annexin V+ cells but did not diminish the increase in CD11b expression mediated by Atf3 inhibition (Fig. 7F & 7G). Thus, dNT supplementation was able to mitigate but not completely eliminate the anti-leukemia effects of Atf3 inhibition (Fig. 7H). Supplementation of both purines and pyrimidines also mitigated the cell cycle arrest and death phenotypes mediated by ATF3 inhibition in the human AML cell lines, NOMO-1 and OCI-AML3 (Fig. S6I and S6J).
DISCUSSION
The genetic abnormalities that drive AML have been well-annotated, however, the non-mutated molecular processes that support disease pathogenesis remain largely undetermined. The recognition that AML cells, similar to many other forms of human cancer, reallocate metabolic resources to support disease has positioned cellular metabolism as a topic of investigation and potential therapeutic avenue in this disease. However, the full scope of the upstream pathways that coordinate metabolic programming as well as the cellular benefits of such rewiring have yet to be fully realized. Here, we have identified that the stress-responsive TF, ATF3 coordinates transcriptional programs that promote serine and nucleotide metabolism to support AML cell cycling, survival and the differentiation blockade.
ATF3 has been shown to play both supportive and antagonistic roles in human cancer. For example, ATF3 expression is elevated in breast and prostate cancer as well as Hodgkin’s lymphoma and supports models of prostate and colon cancer and Hodgkin’s lymphoma (Rohini et al., 2018; Thompson et al., 2009). In contrast, ATF3 is decreased in some forms of colorectal cancer and ATF3 over-expression antagonizes tumor biology in models of prostate, colon and ovarian cancer (Rohini et al., 2018; Thompson et al., 2009). We have observed that elevated ATF3 expression correlates with worse patient outcomes, particularly in those with NK-AML. Functionally, we have shown that ATF3 plays a pro-tumorigenic role in experimental models of NK-AML and AML driven by KMT2A-rearrangements.
Although ATF3 has been shown to be functional in several human cancers, details regarding its downstream mechanistic roles remain incomplete. We show that ATF3 directly regulates the expression of genes that encode key enzymatic regulators of serine synthesis, serine catabolism and de novo purine and pyrimidine synthesis and, through polar metabolite and isotope tracing experiments, that ATF3 is required to maintain these metabolic processes. Underscoring the functional importance of ATF3-mediated regulation of serine and nucleotide metabolism in AML, we have also shown that supplementation with exogenous nucleotides is able to mitigate some of the anti-leukemia effects of ATF3 inhibition. Whether ATF3 regulates these metabolic processes in other forms of human cancer will require additional studies, though, ATF3 has been purported to regulate PSAT1 transcription in a human lung cancer cell line (Riscal et al., 2016).
While our study provides the first evidence that ATF3 is a direct regulator of serine metabolism, several other TFs, such as ATF4 and NRF2 have also been shown to regulate serine metabolism in other human cancers (Adams, 2007; DeNicola et al., 2015; Ben-Sahra et al., 2016; Bjelosevic et al., 2021). ATF3 dimerizes with many other TFs to regulate distinct transcriptional programs across different tissue types (Rohini et al., 2018; Thompson et al., 2009). Notably, ATF4 has been shown to both activate ATF3 transcription as well as dimerize with ATF3 to regulate the expression of the pro-apoptotic gene, NOXA (Jiang et al., 2004; Wang et al., 2009). Furthermore, ATF3 has been shown to dimerize and subsequently suppress the trans-activation potential of NRF2 (Brown et al., 2008). We have found that ATF3 and ATF4 regulate the transcription and localize to similar regions within the promoters of SSP-encoding genes. However, localization of ATF4 but not ATF3, increases at SSP promoters in AML cells cultured in serine- and glycine-deficient media, indicating that ATF3 may be a more important regulator of SSP genes under homeostatic conditions.
Our observation that dNT supplementation was able to reverse the cell cycle defects induced by ATF3 inhibition, but not that of cell death and differentiation suggest that ATF3 regulates additional metabolic-dependent and/or -independent processes in AML. Serine and nucleotides have multiple metabolic fates beyond the generation of dNTs. For example, purines are also used in signal transduction, energy metabolism, redox homeostasis and the generation and S-adenosylmethionine (Newman and Maddocks 2017a; Maddocks et al. 2016). Serine is also utilized in the synthesis of glycine and cysteine as well as certain lipids (Mattaini et al., 2016; Gao et al., 2018). While ATF3 inhibition led to a significant reduction in steady-state AMP and GMP levels, ATP and GTP levels increased. However, ATP and GTP levels are influenced by many cellular processes including energy metabolism and signal transduction pathways. ATF3 inhibition also increased the ratio of reduced-to-oxidized glutathione with a concomitant decrease in cytosolic reactive oxygen species (Figure S6K and S6L). Furthermore, the incorporation of serine-derived carbons into the synthesis of sphingomyelin was unaffected by ATF3 inhibition (Fig. S6M). These cumulative observations suggest that ATF3 promotes the incorporation of serine-derived carbons into newly synthesized purines to support AML but no other catabolic fates of serine.
Collectively, this study has uncovered ATF3 as a previously unrecognized pro-leukemia factor in AML and key regulator of serine and nucleotide metabolism. We have also shown that inhibition of de novo serine synthesis or one-carbon metabolism, via shRNA-mediated inhibition of Phgdh or Mthfd2, respectively, significantly impedes AML cell fitness. These findings uncover that putative pharmacological targeting of the SSP and/or one-carbon pathways either alone or in combination with existing AML chemotherapies, may offer new therapeutic opportunities in AML.
LIMITATIONS OF THE STUDY.
While our studies indicate that ATF3 is a key upstream regulator of serine and nucleotide metabolism and that these processes are de-regulated in the models of AML that we employed, AML is driven by a constellation of distinct genetic mutations and/or chromosomal abnormalities. Therefore, additional studies in primary patient-derived AML samples or other genetically engineered mouse models of AML will be needed to determine which particular genetic abnormalities rely on ATF3. Furthermore, while ATF3 inhibition did not significantly impact the CFC of healthy hematopoietic stem and progenitor cells (HSPCs), a deeper, in vivo analysis will be needed to fully understand the role of ATF3, serine and nucleotide metabolism in normal HSPC biology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Stephen M. Sykes (s.m.sykes@wustl.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The genomic dataset generated in this study is available through GEO accession number GSE172453 (RNA-seq).
EXPERIMENTAL MODEL AND SUBJECT DETAIL
Cell culture
Human AML cell lines were obtained either from the American Type Culture Collection (ATCC) or the German Collection of Microorganism and Cell Cultures (DMSZ). Cells were cultured in the recommended media conditions as described previously (Zhou et al., 2017; Di Marcantonio et al., 2018). Murine AML cells were cultured in RPMI-1640 supplemented with 10% FBS and penicillin/streptomycin, 10 ng/ml mSCF (Peprotech), 6 ng/ml mIL-6 (Peprotech) and 5 ng/ml mIL-3 (Peprotech), hereafter referred to as cytokine-enriched media (CEM). Cell line authentication was carried out by IDEXX using the short tandem repeat (STAR) profiling method.
Patient-derived AML samples
Patient-derived AML samples (PD-AML) were obtained from The Ohio State Comprehensive Cancer Center and analyzed following the approved IRB protocol 16–9037 and co-cultured as described previously (Klco et al., 2013; Klco et al., 2014; Di Marcantonio et al., 2018). Briefly, thawed PD-AML cells were plated on irradiated mono-layers of HS-27 cells with Stemspan (Stem Cell Technology) supplemented with 10% FBS, 100ng/ml hSCF, 100ng/ml hFLT3 ligand, 20ng/ml hIL-3, 20ng/ml hIL-6, 20ng/ml G-CSF (cytokines from Peprotech), and 1μM StemRegenin 1 (Stem Cell Technology). Transduced cells from each condition were evaluated for GFP expression every 6 days for 24–27 days after staining with human CD45 APC-Cy7 (BD Biosciences) and Propidium Iodide (PI) by flow cytometry. Mutational spectrum of human research participants used in this study is listed in Table S1.
Bone marrow transplant leukemia model
All animal studies conducted were approved by the IACUC of the Fox Chase Cancer Center (Protocol #13–01). Leukemia mice were generated with recombinant retroviruses expressing both MLL-AF9 and a neomycin resistance cassette as described previously (Zhou et al., 2017; Di Marcantonio et al., 2018). For recombinant viral transduction, bone marrow cells recovered from leukemia mice were cultured in CEM media overnight. The next day, cells were counted and then 500,000 cells were spin-infected with recombinant lentiviruses (MOI ~ 0.4–0.6) supplemented with polybrene (8 μg/ml) in 12-well non-adherent plates. Plates were centrifuged at 2,200 rpm at 30 °C for 90 minutes. Transduced cells were then incubated for 3 hours before the viral supernatants were removed and then cells were replenished with fresh CEM. For in vivo survival assays, sorted GFP+ cells (150,000 cells/mouse) were transplanted into sub-lethally irradiated (450 rad) syngenic recipient mice 48-hours post-transduction. Recipient male mice were randomized into experimental groups based on similar age (8–12 weeks), weight and vendor and animal studies were not blinded. For western blot analysis and colony formation assays, transduced cells were subjected to FACS to isolate GFP+ cells 48-hours post-transduction. For colony formation assays, MLL-AF9, MLL-ENL and lineage-depleted (Miltenyi, 130–090-858) HSPCs, were transduced with recombinant lentiviruses co-expressing GFP and indicated shRNAs. Forty-eight hours post-transduction, FACS-purified GFP+ cells (200 MLL-AF9, 500 MLL-ENL or 1,000 HSPCs) from each shRNA condition were cultured in 1 ml of methylcellulose supplemented with cytokines (M3434, Stem Cell Technologies) for 5–7 days in triplicate.
METHOD DETAILS
Lentiviral transduction
Lentiviruses were packaged in 293TL cells by co-transfection with the pPAX and pMD vectors. Cells transduced with lentiviruses expressing GFP (MOI ~0.2–0.5) were purified by FACS at the indicated times following transduction. 500,000 cells were transduced with recombinant pLKO.1 lentiviruses co-expressing either GFP or CFP with shRNAs from the TRC shRNA library (murine shRNA Atf3–1: TRCN0000082128; murine shRNA Atf3–2: TRCN0000082132; human shRNA ATF3_1: TRCN0000329749; human shRNA ATF3_2: TRCN0000013570; murine shRNA Atf4_2: TRCN0000071727; murine shRNA Phgdh-1: TRCN0000041627; murine shRNA Phgdh-2: TRCN0000041626; murine shRNA Mthfd2–1: TRCN0000041813; murine shRNA Mthfd2–2: TRCN0000041814).
Western blot analysis
For the detection of human ATF3 and Lamin A/C, nuclear extracts were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Kit following the manufacturer’s instructions (ThermoFisher Scientific). Total cell lysates were analyzed for the detection of all other proteins. Total cell lysate or nuclear extracts were resolved by SDS-PAGE (4–12% Bis Tris gels ThermoFisher Scientific – Life technologies). Following protein transfer to a PVDF membrane, blots were blocked with TBST + 5% nonfat milk for 1 hour at room temperature and then incubated with primary antibody overnight at 4°C. Secondary antibody was incubated at RT for 1 hour and blots were developed with ECL prime (GE healthcare). The following antibodies were used: anti-ATF3 (Cell Signaling Technology, Cat#: 33593, Lot#: 1) with the dilution 1:1000, anti-ATF4 (Cell Signaling Technology, D4B8, Cat#: 11815, Lot#: 5), anti-PHGDH (Cell Signaling Technology, Cat#: 13428, Lot#: 1), anti-PSAT1 (Novus, Cat#: NBP1–32920, Lot#: 42242), anti-SHMT1 (Cell Signaling Technology, D3B3J, Cat#: 80715, Lot#: 1), anti-SHMT2 (Cell Signaling Technology, Cat#: 12762, Lot#: 1), anti-MTHFD1 (Abcam, Cat#: Ab70203, Lot#: GR3357352–4), anti-MTHFD2 (ProteinTech, Cat#: 12270–1-AP, Lot#: 00048741), anti-Laminin A/C (Cell Signaling Technology, Cat#: 4777: Lot#: 4) with the dilution 1:1000, anti-α-tubulin (Cat#: T9026, Lot#: 083M4847V) from Sigma-Aldrich with the dilution 1:5000. Secondary antibodies anti-rabbit-HRP (Cat#: 7074, Lot#: 28) and anti-mouse-HRP (Cat#: 7076, Lot#: 34) were from Cell Signaling Technologies with the dilution 1:4000. Proteins levels were quantified using Photoshop or Image J software.
Apoptosis assay
Cells were washed in PBS and stained with Annexin V and Propidium Iodide (PI) or 7AAD according to the manufacturer’s instruction (BD Biosciences). Cells were acquired and analyzed using an LSRII flow cytometer (Beckton Dickinson). All the data were analyzed using flowjo software.
Cytology
30,000 cells in a volume of 100ul were centrifuged at 300×g for 5 min onto a non-treated microscope slide. Slides were dried overnight at room temperature and then fixed with 100% methanol for 5 min. 200 uL of Wright-Giemsa (ThermoFisher Scientific) working solution (6% Wirght-Giemsa, 1% acetone in GURR buffer) was pipetted onto fixed cells and incubated at room temperature for 15 min. Slides were then rinsed, dried and mounted for microscopic analysis on a Nikon Eclipse E400.
Phagocytosis assay
The pHrodo™ Red E. coli BioParticles™ Phagocytosis Kit for Flow Cytometry (ThermoFisher Scientific) was used according to the manufacturer’s instructions. Briefly, cells were incubated with pHrodo™ BioParticles® conjugate for 15 min on either ice (negative control) or 37 degrees Celsius (experimental conditions) and then stained with antibodies followed by flow cytometric analysis.
RNA-seq
Total RNA was extracted using TRIZOL (ThermoFisher Scientific– Invitrogen). The sequencing libraries were constructed from 500 ng of total RNA using the Illumina’s TruSeq RNA Sample pre kit V2 (Illumina) following the manufacturer’s instructions. The fragment size of RNA-seq libraries was verified using the Agilent 2100 Bioanalyzer (Agilent) and the concentrations were determined using Qubit instrument (ThermoFisher Scientific – Life Technologies). The libraries were loaded onto the Illumina HiSeq 2500 at 6–10 pM on the rapid mode for 2×100 bp paired end read sequencing. The fastq files were generated on the Illumina’s BaseSpace service or locally using the Casava software package for further analysis. Quality of the reads was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) software and the QC assessed reads were aligned to human genome (ver. hg19) using Bowtie-Tophat2 algorithm (Kim et al., 2013). The resulting BAM files were quantified for differential transcript analysis using Cufflinks (Trapnell et al., 2012). The differentially expressed transcripts were selected using a statistical cut-off of p<0.001 and fold change (log2) > 0.5 and < −0.5. Fold enrichment analyses were performed using the DAVID bioinformatics tool (v6.8) and the criteria used to select significantly altered gene sets was Benjamini-Hochberg FDR of 5% and a p<0.05. The complete fold enrichment analyses are provided in File S1.
Gene expression analysis
RNA was extracted using the QIAGEN RNAeasy kit and converted to cDNA with High Capacity cDNA reverse transcription kit (Applied Biosystem). cDNA was amplified using SYBR Green and primers (listed in Supplemental Tables 1 & 2) on StepOnePlus thermocycler (Applied Biosystem).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays was performed in MLL-AF9 and THP-1 cells. Cells were crosslinked with 37% formaldehyde for 15 minutes and sonicated for 30 seconds with 30 seconds interval of rest between sonications for 30 cycles, which yielded DNA fragments of ~150 – 250bp. The chromatin was precipitated using anti-ATF3 (Abcam Cat#: 207434, Lot#: GF204423–6 and GR3204423–8), anti-ATF4 (Abcam Cat#: ab85049, Lot#: GR3229569–19) or IgG control (Cat#: 2729S, Lot#: 8) overnight at 4 °C. The eluted DNA was subjected to RT-PCR analysis with primers (listed in Supplemental Tables 1 & 2) and SYBR Green (Applied Biosystems).
DNA fiber assays
Exponentially growing cells were incubated for 30 minutes with 100 μM CldU, washed with PBS, incubated for 30 minutes with 250 μM IdU, washed with PBS and incubated for 3 hours with 2 mM hydroxyurea. Cells were then resuspended at 4000 cells/μl and lysed on slides (Superfrost Plus microscope slides, Fisher Scientific) with lysis buffer containing 200 mM Tris–HCl, 50 mM ethylenediaminetetraacetic acid, 0.5% SDS, pH 7.4 buffer. Slides were tilted 60° to spread DNA fibers, air dried and fixed in 3:1 methanol: acetic acid for 10 min and stored overnight at −20°C. DNA fibers were denatured in 2.5 M HCl for 2.5 hours, washed with PBS, then blocked with 2% bovine serum albumin (BSA) in PBS for 40 min at room temperature (RT). Slides were incubated with primary antibodies recognizing CldU (Abcam, catalog# ab6326, dilution 1:300) and IdU (BD Biosciences, catalog# 347580, dilution 1:100) for 2.5 hours at RT, washed, then incubated with AlexaFluor488 and AlexaFluor594 conjugated secondary antibodies (ThermoFisher Scientific, catalog# A11062 and A21470, dilutions 1:300) for 1 hour at RT. Images were acquired with a Nikon NIU Upright Fluorescence microscope and fiber lengths measured using ImageJ software.
Nucleotide supplementation
Mouse MLL-AF9 cells expressing Atf3-targeting shRNAs or control shRNA were sorted 48 hours after infection, seeded at the concentration of 200,000 cells/ml in 24 well plates and treated with 12.5μM of 2’-deoxyuridine 5’-monophosphate (Sigma Aldrich, D3876), 2’-deoxycytidine (Sigma Aldrich, D3897), 2’-deoxyadenosine (Sigma Aldrich, D8668) and 2’-deoxyguanosine (Sigma Aldrich, D0901) for 72hs, washed and incubated for 45 minutes with 10μM BrdU. Cells were subsequently washed, fixed and permeabilized and stained using the BrdU Flow Kits (BD Pharmingen #552598) and then analyzed by flow cytometry.
Metabolomic analysis
Metabolomics analyses were performed at The Wistar Institute Proteomics and Metabolomics Shared Resource. MLL-AF9 cells were cultured in RPMI-1640 base media + 10% dialyzed FBS and penicillin/streptomycin, 10 ng/ml mSCF, 6 ng/ml mIL-6 and 5 ng/ml mIL-3. Cells were transduced with recombinant lentiviruses co-expressing GFP and shRNAs and then FACS sorted 48 hours later followed by 24 hours of culture or non-tracing, total steady-state polar metabolite analysis. For tracing experiments, FACS-sorted cells from each shRNA-condition were sorted at 36-hours followed by 12 hours of culture. Cells were then cultured in glucose-free RPMI-1640 media supplemented with 11.1 mM uniformly labeled U-13C-glucose (Cambridge Isotope Laboratories Inc., D-Glucose-U13C6, 99%, CLM-1396) or serine-free RPMI-1640 with 286 μM uniformly labeled U-13C-serine (Cambridge Isotope Laboratories Inc., L-Serine-13C3, 99%, CLM-1574-H-PK) for 24 hours to reach isotopic steady-state. Unlabeled controls were also used for each shRNA condition. Polar metabolites were extracted using ice-cold extraction solution containing 80% MeOH, 20% water. Samples were analyzed by LC-MS/MS on a Q Exactive HF-X mass spectrometer with HESI II probe in-line with a ThermoScientific Vanquish LC System. Samples were analyzed in a pseudorandomized order. LC separation was performed under HILIC condition using a ZIC-pHILIC column, 150 × 2.1 mm, 5 μM, maintained at 45 °C (EMD Millipore). Mobile phase A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide, pH 9.2, and mobile phase B was acetonitrile. Analytical separation was performed at 0.2 ml/min flow rate using the following gradient: 0 min, 85% B; 2 min, 85% B; 17 min, 20% B; 17.1 min, 85% B; and 26 min, 85% B. Samples were analyzed by either Full MS scans with polarity switching (all samples) or Full MS/data-dependent MS/MS scans with separate acquisitions for positive and negative polarities (unlabeled samples in isotope tracing experiments; sample pool, QC, in non-tracing experiments). Relevant MS parameters include: sheath gas, 40; auxiliary gas, 10; sweep gas, 2; auxiliary gas heater temperature, 350 °C; spray voltage, 3.5/3.2 kV for positive/negative polarities; capillary temperature, 325 °C; S-lens RF, 40. Full MS scans were acquired using a scan range of 65 to 975 m/z; 120,000 resolution; automated gain control (AGC) target of 1E6; and maximum injection time (IT) of 100 ms. Data-dependent MS/MS was performed on the 10 most abundant ions; 15,000 resolution; AGC target of 5E4, maximum IT of 50 ms, isolation width of 1.0 m/z, and stepped normalized collision energy of 20, 40, 60.
Raw data were processed using Compound Discoverer 3.1 (ThermoFisher Scientific) with separate analyses for positive and negative polarities. Metabolites were identified by matching accurate mass and retention time to standards or querying MS/MS scans against the mzCloud spectral database (full match, score > 50; mzCloud.org). Identifications were transferred to labeled samples in isotope tracing experiments with consideration for all possible 13C isotopologues. Metabolite levels were quantified by integrated peak area for all detected adducts using Full MS only data. Metabolite levels from non-tracing experiments were corrected for potential errors caused by instrument drift using peak areas from technical injections of the QC pool run at the beginning, middle, and end of the sample set. Isotope tracing data were corrected for natural 13C abundance and isotope tracer purity. Metabolite levels were further normalized to total protein recovered from the polar metabolite extraction pellet.
Redox Staining
MLL-AF9 expressing either shNT, Atf3–1 or Atf3–2 were stained with the Live/ Dead Aqua staining (ThermoFisher Scientific; Catalog no. L34965) following the manufacturer’s instructions, washed and incubated with PBS + 5 μmol/L of CellROX DeepRed reagent (Life Technologies; catalog no.: C10422) at 37°C for 30 minutes. After incubation, cells were washed two times in PBS and analyzed using an LSRII flow cytometer (Beckton Dickinson).
QUANTIFICATION AND STATISTICAL ANALYSIS
For in vitro experiments utilizing mouse or human AML cell lines, the presented data is representative of a single experiment and expressed as mean ± SD of at 3–6 technical replicates and each experiment was repeated 3–5 times. For PD-AML studies, the presented data represents the mean ± SD of three wells from a single experiment. Comparisons between 2 values were performed using Student t test (unpaired 2-tailed) unless otherwise indicated and significance was determined using the following p values: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. P values from all survival curves are being analyzed using the Log-rank (Mantel-Cox) test comparing two survival curves. Statistical tests were performed using Graphpad Prism 9 software.
Supplementary Material
File S1 related toFigure 3. Results of the pathway enrichment analysis performed using DAVID v6.8. Download as a separate spreadsheet file.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse – anti Human CD45 APC-Cy7 (Clone 2D1) | BD Bioscience | Cat#:561863; RRID: AB_10897014 |
| Rabbit – anti ATF3 (Clone D2Y5W Lot 1) | Cell Signaling Technology | Cat#:33593; RRID: AB_2799039 |
| Rabbit – anti ATF4 (Lot# 5) | Cell Signaling Technology | Cat#:11815; RRID: AB_2616025 |
| Rabbit – anti PHGDH (Lot# 1) | Cell Signaling Technology | Cat#:13428; RRID: AB_2750870 |
| Rabbit – anti PSAT1 (Lot# 42242) | Novus | Cat#:NBP1-32920; RRID: AB_2172600 |
| Rabbit – anti SHMT1 (Clone D3B3J Lot# 1) | Cell Signaling Technology | Cat#: 80715; RRID: AB_2799957 |
| Rabbit – anti SHMT2 (Lot# 1) | Cell Signaling Technology | Cat#:12762; RRID: AB_2798018 |
| Mouse – anti MTHFD1 (Lot# GR3357352-4) | Abcam | Cat#:ab70203; RRID: AB_1269485 |
| Rabbit – anti MTHFD2 (Lot# 00048741) | Proteintech | Cat#:12270-1-AP; RRID: AB_2147525 |
| Mouse – anti Laminin A/C (Lot# 4) | Cell Signaling Technology | Cat#:4777; RRID: AB_10545756 |
| Mouse – anti alpha-Tubulin (Clone DM1A Lot# 083M4847V) | Sigma-Aldrich | Cat#:T9026; RRID: AB_477593 |
| Goat - anti rabbit HRP (Lot# 28) | Cell Signaling Technology | Cat#:7074; RRID: AB_2099233 |
| Horse – anti mouse HRP (Lot# 34) | Cell Signaling Technology | Cat#: 7076; RRID: AB_330924 |
| Rabbit – anti ATF3 (Lot# GF204423-6 and GR3204423-8) | Abcam | Cat#:ab207434, RRID: AB_2734728 |
| Rabbit – anti AT43 (Lot# GR3229569-19) | Abcam | Cat#:ab85049, RRID: AB_1861369 |
| Normal Rabbit IgG antibody (Lot# 8) | Cell Signaling Technology | Cat#:2729, RRID: AB_1031062 |
| Anti - BrdU (IdU) (Clone B44) | BD Biosciences | Cat#:347580, RRID: AB_10015219 |
| Rat anti – BrdU (CldU) (Clone BU1/75(ICR1)) | Abcam | Cat#:ab6326, RRID: AB_305426 |
| Rabbit – anti Mouse IgG AlexaFluor594 | Thermo Fisher Scientific | Cat#:A-11062, RRID: AB_2534109 |
| Rat – anti Rat IgG AlexaFluor488 | Thermo Fisher Scientific | Cat# A21470, RRID: AB_10561519 |
| Biological Samples | ||
| Patient derived – AML. See table S1 | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Murine SCF | Peprotech | 250-03 |
| Recombinant Murine IL-6 | Peprotech | 216-16 |
| Recombinant Murine IL-3 | Peprotech | 213-13 |
| Recombinant Human SCF | Peprotech | 300-07 |
| Recombinant Human FLT3 ligand | Peprotech | 300-19 |
| Recombinant Human IL-3 | Peprotech | 200-03 |
| Recombinant Human IL-6 | Peprotech | 200-06 |
| Recombinant Human G-CSF | Peprotech | 300-23 |
| StemRegenin 1 | Stem Cell Technology | 72342 |
| 2’-deoxiuridine 5’-monophosphate | Sigma-Aldrich | D3876 |
| 2’-deoxycytidine | Sigma-Aldrich | D3897 |
| 2’-deoxyadenosine | Sigma-Aldrich | D8668 |
| 2’-deoxyganosine | Sigma-Aldrich | D0901 |
| U-13C-Glucose | Cambridge Isotope Laboratories | CLM-1396 |
| U-13C-Serine | Cambridge Isotope Laboratories | CLM-1574-H-PK |
| Critical commercial assays | ||
| Lineage Cell Depletion Kit, Mouse | Miltenyi Biotech | 130-090-858 |
| MethoCult GF | Stem Cell Technologies | M3434 |
| NE-PER Nuclear and Cytoplasmic Extraction Kit | ThermoFisher Scientific | 78833 |
| Annexin V APC | BD Biosciences | 550474 |
| BrdU Flow Kit | BD Pharmingen | 552598 |
| Wright-Giemsa Staining (Lot# 482978) | ThermoFisher Scientific | 264-983 |
| Live/Dead Aqua staining | Thermo Fisher Scientific | L34965 |
| CellROX DeepRed | Life Technologies | C10422 |
| Gurr Buffer Tablets (Lot# 1142560) | Gibco | 10582013 |
| pHrodo™ Red E. coli BioParticles™ Phagocytosis Kit for Flow Cytometry (Lot# 2108800) | ThermoFisher Scientific | A10025 |
| TruSeq RNA Sample Preparation kit V2 | Illumina | RS-12-2001 |
| Deposited data | ||
| RNAseq data | This study | GEO172453 |
| Metabolomic data | This study | Lead Contact |
| Experimental models: cell lines | ||
| THP-1 | ATCC | TIB202 |
| HL-60 | ATCC | CCL240 |
| K-562 | ATCC | CCL243 |
| MV-4-11 | ATCC | CRL9591 |
| NOMO-1 | DSMZ | ACC542 |
| OCI-AML2 | DSMZ | ACC99 |
| OCI-AML3 | DSMZ | ACC582 |
| MOLM-14 | DSMZ | ACC777 |
| Oligonucleotides | ||
| PCR primers. See Table S1 and S2 | This study | N/A |
| Murine shRNA Atf3-1 | TRC | TRCN0000082128 |
| Murine shRNA Atf3-2 | TRC | TRCN0000082132 |
| Human shRNA ATF3-1 | TRC | TRCN0000329749 |
| Human shRNA ATF3-2 | TRC | TRCN0000013570 |
| Murine shRNA Atf4-2 | TRC | TRCN0000071727 |
| Murine shRNA Phgdh-1 | TRC | TRCN0000041627 |
| Murine shRNA Phgdh-2 | TRC | TRCN0000041626 |
| Murine shRNA Mthfd2-1 | TRC | TRCN0000041813 |
| Murine shRNA Mthfd2-2 | TRC | TRCN0000041814 |
| Software and algorithms | ||
| Prism GraphPad 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej-nih-gov.proxy.fccc.edu/ij/ |
| FloJo 10.1 | FloJo LLC | https://www.flowjo.com/ |
| Bowtie-Tophat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks |
| Compound Discoverer 3.1 | Thermo Fisher Scientific | N/A |
| Biorender | BioRENDER | https://biorender.com |
Highlights.
The ATF3 transcription factor supports certain aggressive subtypes of AML.
AML cells rely on ATF3 to maintain cell cycling and the differentiation blockade.
ATF3 is a transcriptional regulator of serine synthesis and one-carbon metabolism.
ATF3 drives serine metabolism to maintain purine and pyrimidine pools.
ACKNOWLEGMENTS
We thank the Laboratory Animal and Flow Cytometry facilities at Fox Chase Cancer Center. The Wistar Proteomics and Metabolomics Core Facility was supported by Cancer Center Support Grant CA010815 and S10 OD023586. The following individuals are supported by NIH grants: D.D.M (K99 CA241370), T.S. (R01 CA244179 and CA237286), N.J. (R01CA214799 and HL150190), H.Y.T (R50 CA221838) and S.M.S (R01 CA227830). T.S. is also supported by the Leukemia and Lymphoma Society (TRP 6565–19) and S.M.S is also supported by the American Cancer Society (RSG-18–195-01-DDC).
Footnotes
DECLARATION OF INTERESTS
S.P. is currently an employee of Merck Research Laboratories. All other authors have no competing interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams CM. (2007) Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem. 282(23):16744–16753. doi: 10.1074/jbc.M610510200 [DOI] [PubMed] [Google Scholar]
- Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sønderby CK, Winther O, Rapin N, Porse BT. (2016) BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res.January4;44(D1):D917–24. doi: 10.1093/nar/gkv1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science.351(6274):728–733. doi: 10.1126/science.aad0489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelosevic S, Gruber E, Newbold A, Shembrey C, Devlin JR, Hogg SJ, Kats L, Todorovski I, Fan Z, Abrehart TC, et al. (2021) Serine biosynthesis is a metabolic vulnerability in FLT3-ITD-driven acute myeloid leukaemia. Cancer Discov. 2021 January 12:candisc.0738.2020. doi: 10.1158/2159-8290.CD-20-0738. [DOI] [PubMed] [Google Scholar]
- Brown SL, Sekhar KR, Rachakonda G, Sasi S, Freeman ML. (2008) Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 68(2):364–368. doi: 10.1158/0008-5472.CAN-07-2170 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro I, Sampaio-Marques B, Ludovico P. (2019) Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells. 8(9):967. doi: 10.3390/cells8090967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018) The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46(D1):D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer (2015) Nat Genet. 47(12):1475–1481. doi: 10.1038/ng.3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl FF, Lewis CA, Fiske BP, Vander Heiden MG. (2019) Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab.1(9):861–867. doi: 10.1038/s42255-019-0108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marcantonio D, Martinez E, Sidoli S, Vadaketh J, Nieborowska-Skorska M, Gupta A, Meadows JM, Ferraro F, Masselli M, Challen GA, et al. (2018) PKC epsilon is a Key Regulator of Mitochondrial Redox Homeostasis in Acute Myeloid Leukemia. Clin Cancer Res. 24(3):608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X, Kang Y, Rabinowitz JD. (2016) Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway Cell Metab. 23(6):1140–1153. doi: 10.1016/j.cmet.2016.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, Basheer F, Marando L, Di Lisio L, Dias JML, et al. (2018) Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 131(15):1639–1653. doi: 10.1182/blood-2017-12-820035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, Liu J, Locasale JW. (2018) Serine Availability Influences Mitochondrial Dynamics and Function through Lipid Metabolism. Cell Rep. 22(13):3507–3520. doi: 10.1016/j.celrep.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Nemkov T, Park HJ, et al. (2019) Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin Cancer Res. 25(13):4079–4090. doi: 10.1158/1078-0432.CCR-18-3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebestreit K, Gröttrup S, Emden D, Veerkamp J, Ruckert C, Klein HU, Müller-Tidow C, Dugas M. (2012) Leukemia gene atlas--a public platform for integrative exploration of genome-wide molecular data. PLoS One. 7(6):e39148. doi: 10.1371/journal.pone.0039148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37(1):1–13. doi: 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. February;24(3):1365–77. doi: 10.1128/mcb.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, et al. (2018) Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell. November 12;34(5):724–740.e4. doi: 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N, Erdmann-Gilmore P, et al. (2013) Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. February 28;121(9):1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, Fronick C, Magrini V, Demeter RT, Fulton RS, et al. (2014)Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. March 17;25(3):379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohnken R, Kodigepalli KM, Wu L. (2015) Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015;14:176. doi: 10.1186/s12943-015-0446-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitz J, Schönfeld C, Seibert M, Stolp V, Alshamleh I, Oellerich T, Steffen B, Schwalbe H, Schnütgen F, Kurrle N, Serve H. (2019) Metabolic Plasticity of Acute Myeloid Leukemia. Cells. July31;8(8):805. doi: 10.3390/cells8080805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. (2014) Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7(4):1248–1258. doi: 10.1016/j.celrep.2014.04.045 [DOI] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. July 31;43(9):869–74. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. January24;493(7433):542–6. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Labuschagne CF, Adams PD, Vousden KH. (2016) Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell. 61(2):210–221. doi: 10.1016/j.molcel.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. (2016) J Cell Biol. 214(3):249–257. doi: 10.1083/jcb.201604085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, et al. (2008) An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. November 15;112(10):4193–201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacson J, Di Marcantonio D, Wang Y, Bernhardy AJ, Clausen E, Hua X, Cai KQ, Martinez E, Feng W, Callén E, et al. (2020) BRCA1 Mutational Complementation Induces Synthetic Viability. Mol Cell. June 4;78(5):951–959.e6. doi: 10.1016/j.molcel.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC, Maddocks ODK. (2017a) One-carbon metabolism in cancer. Br J Cancer. 116(12):1499–1504. doi: 10.1038/bjc.2017.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC, Maddocks ODK. (2017b) Serine and Functional Metabolites in Cancer. Trends Cell Biol. 27(9):645–657. doi: 10.1016/j.tcb.2017.05.001 [DOI] [PubMed] [Google Scholar]
- Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, Luengo A, Davidson SM, Ali A, Ferraro GB, et al. (2020) Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. September;10(9):1352–1373. doi: 10.1158/2159-8290.CD-19-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. (2015) p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem.290(1):457–466. doi: 10.1074/jbc.M114.616359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai CC, Kearsey SE. A (2017) Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes (Basel). 8(2):57. doi: 10.3390/genes8020057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016) Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. June 9;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. August 18;476(7360):346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. (2017) BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. November 16;551(7680):384–388. doi: 10.1038/nature24294. [DOI] [PubMed] [Google Scholar]
- Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X, Dai Z, Gao X, Wang Q, Liu Y,et al. (2018) Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. December 21;9(1):5442. doi: 10.1038/s41467-018-07868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riscal R, Schrepfer E, Arena G, Cissé MY, Bellvert F, Heuillet M, Rambow F, Bonneil E, Sabourdy F, Vincent C, et al. (2016) Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol Cell. June 16;62(6):890–902. doi: 10.1016/j.molcel.2016.04.033. Epub 2016 Jun 2. [DOI] [PubMed] [Google Scholar]
- Rohini M, Haritha Menon A, Selvamurugan N. (2018) Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int J Biol Macromol. 120(Pt A):310–317. doi: 10.1016/j.ijbiomac.2018.08.107 [DOI] [PubMed] [Google Scholar]
- Stuani L, Sabatier M, Sarry JE. (2019) Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol. 2019;17(1):57. doi: 10.1186/s12915-019-0670-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Xu D, Williams BR. (2009) ATF3 transcription factor and its emerging roles in immunity and cancer. J Mol Med (Berl). 2009;87(11):1053–1060. doi: 10.1007/s00109-009-0520x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. March1;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Löwenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009January;94(1):131–4. doi: 10.3324/haematol.13299.Epub 2008 Oct 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, et al. (2013) Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. January;41(Database issue):D171–6. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, Ye Y. (2009) ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. February17;106(7):2200–5. doi: 10.1073/pnas.0807611106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi JH, Yan C, Dong Z, Huang S, Zha Y, et al. (2016) KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. January 26;14(3):506–519. doi: 10.1016/j.celrep.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Martinez E, Di Marcantonio D, Solanki-Patel N, Aghayev T, Peri S, Ferraro F, Skorski T, Scholl C, Fröhling S, et al. (2017) JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia. May;31(5):1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 related toFigure 3. Results of the pathway enrichment analysis performed using DAVID v6.8. Download as a separate spreadsheet file.
Data Availability Statement
The genomic dataset generated in this study is available through GEO accession number GSE172453 (RNA-seq).