

Abstract
Stressful life events have been linked to declining health, and inflammation has been proposed as a physiological mechanism that might explain this association. Using 828 participants from the Dunedin Longitudinal Study, we tested whether people who experienced more stressful life events during adulthood would show elevated systemic inflammation when followed up in midlife, at age 45. We studied three inflammatory biomarkers: C-reactive protein (CRP), interleukin-6 (IL-6), and a newer biomarker, soluble urokinase plasminogen activator receptor (suPAR), which is thought to index systemic chronic inflammation. Stressful life events were not associated with CRP or IL-6. However, people who experienced more stressful life events from age 38 to 44 had elevated suPAR at age 45, and had significantly greater increases in suPAR from baseline to follow up across the same period. When examining stressful life events across the lifespan, both adverse childhood experiences (ACEs) and adult stressful life events were independently associated with suPAR at age 45. ACEs moderated the association of adult stressful life events and suPAR at age 45—children with more ACEs showed higher suPAR levels after experiencing stressful life events as adults. The results suggest systemic chronic inflammation is one physiological mechanism that could link stressful life events and health, and support the use of suPAR as a useful biomarker for such research.
Keywords: C-reactive protein, interleukin-6, stress, inflammatory biomarkers, adverse childhood experiences
1. Introduction
Higher levels of psychological stress are associated with poorer subsequent health, including greater risk of developing chronic disease, accelerated disease progression, and early mortality (Cohen, Janicki-Deverts, & Miller, 2007; Glaser, Rabin, Chesney, Cohen, & Natelson, 1999; Philips et al., 2001). This association has been documented over the course of the lifespan, in several different countries, and across a wide array of stressors, including psychological trauma, financial and relationship stress, and natural disasters (Bourassa, 2021; Browning & Heinesen, 2012; Moon, Kondo, Glymour, & Subramanian, 2011; Niiyama et al., 2014). Similarly, exposure to adverse childhood experiences (ACEs)—emotional, physical, and sexual abuse as well as neglect, violence, and household dysfunction—is associated with increased risk of physical disease in adulthood (Kalmakis & Chandler, 2015). The pathophysiological pathways responsible for these associations, however, have been more elusive. Several mechanisms have been proposed to explain how stressful life events might affect health, such as health behaviors (Hamer, Molloy, & Stamatakis, 2008), disruptions to homeostatic physiological processes (i.e., allostatic load; Danese & McEwen, 2012; Oken, Chamine, & Wakeland, 2015), and altered stress reactivity (Manuck, 1994; Carroll, Ginty, Whittaker, Lovallo, & de Rooij, 2017).
One physiological mechanism proposed to link stress and health is inflammation (Wirtz, & von Känel, 2017). Meta-analytic studies have shown that acute laboratory stressors experimentally increase systemic inflammation (Marsland, Walsh, Lockwood, & John-Henderson, 2017; Steptoe, Hamer, & Chida, 2007), and that chronic stressors—e.g., caregiving, socioeconomic disadvantage, and work-related stress—are associated with higher levels of systemic inflammation (Cohen et al., 2012; Rohleder, 2014; Tursich et al., 2014). Associations have also been observed between higher levels of inflammation and both socioeconomic disadvantage (Berger et al., 2019) and childhood adversity (Baumeister, Akhtar, Ciufolini, Pariante, & Mondelli, 2016; Chen & Lacey, 2018; Danese & Baldwin, 2017), though it is notable that these associations are generally small in effect size and can vary across individual studies (Baumeister, 2016). Evidence that higher levels of psychological stress are associated with higher levels systemic inflammation is particularly important given established associations between inflammation and poor health outcomes, including cardiovascular disease (Emerging Risk Factors Collaboration, 2010; Wirtz, & von Känel, 2017), accelerated biological aging (Rasmussen et al., 2021), cancer (Guo et al., 2013), and early mortality (Möhlenkamp et al., 2011). A recent meta-analysis found that psychosocial interventions such as cognitive behavior therapy improved immune system function across a variety of outcomes, providing further evidence that immune activation or inflammation could link stress and health and act as a malleable intervention target (Shields et al., 2020).
One challenge in studying the association between stressful life events and inflammation is the measurement of systemic inflammation. A variety of biomarkers are used to assess systemic inflammation, with limited consensus on which measures are optimal to assess future health risk (Furman et al., 2019). Commonly used inflammatory biomarkers, such as C-reactive protein (CRP) or interleukin-6 (IL-6), can reflect acute changes in immune activity, in addition to systemic chronic inflammation (Furman et al., 2019). This sensitivity routinely necessitates log transforming values or systematically excluding some research participants with particularly elevated CRP or IL-6 values before analysis, on the assumption that very high values arise from infection (Pearson et al., 2003). When examining the association between stressful life events and systemic inflammation, additional inflammatory biomarkers that index systemic chronic inflammatory processes could also have utility.
A promising and recently validated biomarker of systemic chronic inflammation is soluble urokinase plasminogen activator receptor (suPAR)—the soluble form of the membrane-bound receptor uPAR (Desmedt, Desmedt, Delanghe, Speeckaert, & Speeckaert, 2017). During proinflammatory conditions, uPAR is cleaved from the surface of immunologically active cells (e.g., monocytes, macrophages, neutrophils, endothelial cells, and some cancer cells) leading to an increase in the blood concentration of suPAR. Blood suPAR levels are correlated with levels of other inflammation markers, immune cell counts, and cytokines and as such are thought to reflect overall immune activity. Elevated suPAR statistically predicts incident and prevalent disease (Hayek et al., 2020), chronic disease progression (Rasmussen et al, 2017), accelerated biological aging (Rasmussen et al., 2021), and early death due to chronic disease (Eugen-Olsen et al., 2010). Like systemic chronic inflammation (Furman et al., 2019), suPAR is non-specifically associated with a wide range of diseases, non-communicable and infectious diseases alike, including cardiovascular disease, diabetes, cancer, rheumatic disease, pneumonia, COVID-19, and sepsis (Desmedt et al., 2017; Rovina et al., 2020). Importantly, suPAR appears to be less sensitive to acute changes in health compared to other inflammatory biomarkers, such as CRP (Lyngbæk et al., 2013; Rasmussen et al., 2021), and could therefore be a better measure of systemic chronic inflammation.
Given that suPAR appears to index chronic inflammation when accounting for other measures of inflammation (e.g., CRP; Rasmussen et al., 2019a, Rasmussen et al., 2019b), it presents the opportunity to examine the association between difficult life experiences and inflammation across the lifespan—e.g., from childhood to midlife—rather than focusing exclusively on recent stressful events. Cumulative, biological-embedding, and sensitization models (Young et al., 2019) present three plausible representations for how stressful life events at different developmental periods could influence later inflammation. A cumulative model would suggest stressful experiences from both childhood and adulthood would exert an independent, additive effect on later inflammation (Hostinar, Lachman, Mroczek, Seeman, & Miller, 2015). A biological-embedding model would instead suggest stressful life events in childhood, but not adulthood, would be associated with later inflammation. Finally, a sensitization model would suggest that people who experience childhood adversity would be at greater risk of a proinflammatory response to adult stressful life events (Miller, Chen, & Parker, 2011; Young et al., 2019). Longitudinal measurement of ACEs, stressful life events, and inflammatory biomarkers in Dunedin allow us to test which models best describe the association between stressful experiences and inflammation.
1.1. Present Study
The current study used data from 828 participants in the Dunedin Longitudinal Study (Poulton et al., 2015), a population-representative birth cohort followed from birth to age 45, to examine a number of research questions related to stressful life events and systematic inflammation. Near their 38th and 45th birthdays, we drew blood from participants to assess inflammatory biomarkers and interviewed participants regarding the stressful life events they experienced in the years since the last assessment (covering, respectively, ages 32 through 37 and 38 through 44). Our analyses focused on the period from age 38 to 45, as these were the Dunedin Study occasions at which we had measures of stressful life events and assessed the three inflammatory biomarkers. We first examined whether the number of stressful life events participants experienced was associated with chronic inflammation as assessed by suPAR levels at age 45, as well as change in suPAR from age 38 to age 45. We then compared these associations to those for CRP and IL-6. We hypothesized that experiencing more stressful life events would be associated with elevated levels of inflammation. If suPAR was a better measure of chronic inflammation than CRP and IL-6, we would expect suPAR to be more strongly associated with stressful life events than CRP or IL-6. We then tested the cumulative, biological embedding, and sensitization models that might characterize the associations between inflammation and stressful life events across the lifespan. We hypothesized that adversity from earlier in the lifespan, in the form of ACEs, would moderate the association of adult stressful life events and inflammation, such that people with more ACEs would have a stronger association between stressful life events and inflammation at age 45 (i.e., a sensitization model).
2. Method
2.1. Study Design and Population
Participants are members of the Dunedin Study, a longitudinal investigation of a population-representative birth cohort. The 1037 participants (91% of eligible births; 52% male) were all individuals born between April 1972 and March 1973 in Dunedin, New Zealand, who were eligible based on residence in the province and who participated in the first assessment at age 3 years (Poulton, Moffitt, & Silva, 2015). The cohort represented the full range of socioeconomic status (SES) in the general population of New Zealand’s South Island. As adults, the cohort matches the results from the New Zealand National Health and Nutrition Survey on key adult health indicators (Poulton et al. 2015). It matches the distribution of educational attainment among citizens of the same age from the New Zealand Census (Richmond-Rakerd et al. 2020). The cohort is predominantly white (93%), matching South Island demographics (Poulton et al., 2015). Assessments were performed at birth; at ages 3, 5, 7, 9, 11, 13, 15, 18, 21, 26, 32, 38 and, most recently at age 45 years (completed April 2019), when 938 of the 997 participants (94.1%) still alive participated. Written informed consent was obtained from participants, and the institutional review boards of the participating universities approved study protocols. Of the 938 who participated at age 45 years, 879 had blood drawn and 875 (93.3%) had plasma suPAR measured. The primary sample for this article included participants who had data on suPAR and stressful life events available at both age 38 and age 45 years (N = 828, 88.3%).
2.2. Measures
2.2.1. Inflammation.
We assessed systemic inflammation using three biomarkers, as previously described (Rasmussen et al., 2019a, Rasmussen et al., 2019b).
2.2.1.1. suPAR.
Plasma suPAR (ng/mL) was analyzed at ages 38 and 45 with the suPARnostic AUTO Flex ELISA (ViroGates A/S, Birkerød, Denmark) according to the manufacturer’s instructions. The detection limit of the assay was 0.1 ng/mL. The intraassay correlation of repeat measurements of the same sample was r = 0.98 and coefficient of variation (CV) = 2.4%, and the interassay correlation was r = 0.81 and CV = 12.8%. suPAR levels were correlated at age 38 and 45, r = .58, p < .001, and increased on average 0.6 ng/mL from age 38 to 45, with changes ranging from −5.4 to 9.3 ng/mL.
2.2.1.2. C-reactive protein.
Serum high-sensitivity CRP (hsCRP, mg/L) was measured on a Modular P analyzer (Roche Diagnostics GmbH, Mannheim, Germany) at age 38 and on a Cobas c 702 analyzer (Roche Diagnostics GmbH) at age 45, using particle-enhanced immunoturbidimetric assays. The lower detection limit of the assay was 0.3 mg/L. The intraassay and interassay CVs reported by the manufacturer were r = 0.28–1.34% and r = 2.51–5.70%, respectively. CRP increased on average 0.3 mg/L from age 38 to 45, with changes ranging from −35.6 to 88.9 mg/L. CRP values were log-transformed to account for positive skew. Transformed CRP levels were correlated at age 38 and 45, r = .48, p < .001. Previous research has also recommended excluding untransformed CRP values greater than 10 mg/L (Pearson et al., 2003) to account for acute infection. Notably, removing these values did not alter any of the substantive results reported in this study.
2.2.1.3. Interleukin-6.
At age 38, plasma IL-6 (pg/mL) was measured on a Molecular Devices (Sunnyvale, CA) SpectraMax plus 384 plate reader using R&D Systems (Minneapolis, MN) Quantikine High-sensitivity ELISA kit HS600B according to the manufacturer’s instructions. At age 45, serum IL-6 (pg/mL) was measured on a Cobas e 602 analyzer (Roche Diagnostics GmbH), using an electrochemiluminescence immunoassay. The lower detection limit of the assay was 1.5 pg/mL. The intraassay and interassay CVs reported by the manufacturer were 2.5–6.0% and 2.9–8.5%, respectively. IL-6 increased on average 0.6 pg/mL from age 38 to 45, with changes ranging from −17.3 to 27.6 pg/mL. IL-6 values were log-transformed to account for positive skew. Transformed IL-6 levels were correlated at age 38 and 45, r = .45, p < .001.
2.2.2. Stressful Life Events.
Life history calendars (Caspi et al., 1996) were used to derive a count of the number of stressful life events that participants experienced from age 32 through 37 and from 38 through 44. Participants reported the events occurring between study assessments on life history calendars at the age-38 and age-45 assessments. Stressful life events coded were (1) each relationship breakup, (2) whether someone moved frequently (10 or more moves, see Caspi et al., 2003), (3) homelessness at any time, (4) in jail at any time, (5) the death of a friend or family member, (6) a job loss, (7) experiencing a medical illness, mental illness, injury, or accident, (8) a friend or family member experiencing a medical illness, mental illness, injury, or accident, (9) legal problems, (10) a physical or sexual assault, (11) serious financial problems, (12) a natural or human-made disaster (e.g., fires, earthquakes), or (13) other. 20% of the total responses from age 38 to 44 (N = 1,442) were coded by two independent reviewers and evidenced strong interrater reliability, percent agreement = 92.2%, kappa = 0.91. Events in categories 1 to 13 were summed for each of the two study periods. These totals were then winsorized to a maximum of 30 events within each period. Participants experienced on average 5.5 stressful life events (SD = 4.6) from age 32 through 37 and 6.1 stressful life events (SD = 5.9) from age 38 through 44. The number of events during the two periods were correlated, r = .38, p < .001. (We also re-tested our hypotheses while excluding “medical illness, mental illness, injury, or accident” events, to rule out the possibility that observed associations between stressful life events and inflammation were due to higher rates of illness, rather than stressful life events in general. As shown in Supplemental Analyses 1, Supplemental Tables 1–4, substantive study results were unchanged when excluding this category of stressful life events.)
In addition, when examining the association of stressful life events and inflammation across the lifespan, we also included an additional measure of stressful life events covering ages 21 to 26 from a previous investigation using data from the Dunedin study (see Caspi et al., 2003). Age 21 to 26 stressful life events were assessed at age 26 with the aid of a life history calendar (Caspi et al., 1996). As described previously (Caspi et al., 2003), the 5-year reporting period covered events occurring after the 21st birthday and before the 26th birthday. Events included employment problems (long-term unemployment; being made redundant; losing a job because the company moved; being fired); financial problems (problems with debt, such as having items repossessed; not having enough money to pay for food or household expenses; lacking money for medical expenses; difficulty paying bills); housing problems (homelessness; multiple residential changes); health problems (a disabling physical illness lasting a month or more; a disabling injury); and relationship problems (being involved in a physically violent relationship; a break-up of a cohabiting, intimate relationship).
2.2.3. Prospective Adverse Childhood Experiences (ACEs).
As previously described (Reuben et al., 2016), archival study records from the first 15 years of participants’ lives were reviewed by four independent raters to determine whether participants experienced 10 ACEs identified in the Center for Disease Control (CDC) ACE study (CDC, 2016; Felitti et al., 1998): three types of abuse (physical, emotional, sexual), two types of neglect (physical, emotional), and five types of household dysfunction (incarceration of a family member, household substance abuse, household mental illness, loss of a parent, and household partner violence). The inter-rater agreement across all ACEs between the four raters averaged a kappa of .79 (range = .76–.82; Reuben et al., 2016). Counts greater than four were recoded to four, in line with the CDC ACE study (CDC, 2016). The distribution of ACEs in the Dunedin Study matched the distribution observed in the CDC study (Reuben et al., 2016). The mean count of ACEs in the sample was 1.1 events (SD = 1.2).
2.2.4. Clinical Characteristics: Smoking, Body Mass Index, and Anti-inflammatory Medication.
Smoking and body mass index (BMI) were included as covariates as these are associated with elevated inflammation. Current smoking was reported at age 38 and age 45 by grouping the participants into four groups: nonsmoker, smoking <10, 10–19, or ≥20 cigarettes/day, as used in prior investigations (Rasmussen et al., 2019a). BMI (kg/m2) was measured at age 38 and age 45 years. Use of anti-inflammatory medications at the time of interview was assessed at age 45 years, as previously described (Rasmussen et al., 2020). Anti-inflammatory medications included non-steroidal anti-inflammatory drugs (NSAIDs), anti-gout medication, corticosteroids (respiratory, systemic), anti-rheumatics, prophylactic aspirin, and statins. As reported in a prior investigation (Rasmussen et al., 2021), suPAR was not associated with use of anti-inflammatory medications in this study at age 45. IL-6 similarly evidenced no significant association with anti-inflammatory medications at age 45. In contrast, CRP evidenced a positive significant association with anti-inflammatory medications at age 45, r = 0.09, p = .009.
2.2.5. Childhood Covariates.
Three childhood variables (Richmond-Rakerd et al., 2021) that have previously been associated with suPAR (Rasmussen et al., 2019b) were included in the study as covariates for the initial analyses at age 45: childhood socioeconomic status (SES), IQ, and self-control. Childhood SES was measured using the 6-point Elley-Irving Socioeconomic Index for New Zealand (Elley & Irving, 1976) and represented the average of the highest SES level of either parent across the assessments of cohort families from the participants’ birth through age 15. Childhood IQ was assessed by calculating mean scores for the Wechsler Intelligence Scale for Children–Revised (WISC-R) across administration at ages 7, 9, and 11 years. Childhood self-control was assessed during participants’ first decade of life using a composite measure that included observational ratings, parent and teacher ratings, and psychiatric interviews (see Richmond-Rakerd et al., 2021).
2.3. Data Analysis
We specified three sets of multiple regression models to test our primary hypotheses. The first models examined whether the number of stressful life events participants experienced from age 38 through 44 was associated with suPAR, ln(CRP), and ln(IL-6) levels at age 45. Second, we used residualized regression to test whether the number of stressful life events from age 38 through 44 was associated with change in suPAR, CRP, and IL-6 from age 38 to age 45.
The third set of models tested the cumulative, biological embedding, and sensitization models by examining whether ACEs and the total cumulative number of adult stressful life events (ages 21 to 26 and 32 through 44) were independently associated with inflammation at age 45. We then tested whether ACEs moderated the association of stressful life events in adulthood and systemic inflammation at age 45. To create an overall assessment of stressful life events in adulthood, we standardized the measures of stressful life events from age 21 to 26, 32 to 37, and 38 to 44 and averaged the standardized variables. Similarly, to create an overall assessment of stressful life events across the lifespan, we standardized the measures of childhood ACEs and stressful life events in adulthood and averaged the standardized variables.
In all models, we first ran an unadjusted model, then a model controlling for sex, smoking, BMI, and anti-inflammatory medication. We also controlled for childhood covariates in the initial age 45 model. Models that assessed change in inflammation from age 38 to age 45 included the inflammatory marker assessed at age 38 and pre-baseline stressful life events from age 32 through 37. We report standardized regression coefficients (βs) with 95% confidence intervals. To account for missing data, we used full information maximum likelihood estimation (Graham, 2009) in MPLUS version 8.3 (Muthén & Muthén, 2012). A second data analyst checked the analyses for reproducibility by deriving code from the manuscript and applying it to an original copy of the data set.
3. Results
Participants’ characteristics and demographic variables are reported in Table 1. Participants included in the current study (N = 828) did not differ significantly from other participants alive at age 45 on childhood ACEs, socioeconomic status, IQ, or low self-control (see Supplemental Figure 1). The three measures of systemic inflammation were significantly associated at age 45 (suPAR and CRP, r = .27; suPAR and IL-6, r = .18; CRP and IL-6, r = .50, all ps < .001).
Table 1.
Main Study Variables
| N = 828 | Mean ± SD |
|---|---|
| Sex | 49.4% women |
| Childhood variables | |
| No. of ACEs | 1.0 ± 1.2 |
| Socioeconomic status | 3.8 ± 1.1 |
| IQ | 101.2 ± 13.8 |
| Self-control | −0.05 ± 0.9 |
| Age 26 assessment | |
| No. of stressful life events age 21 through 26 | 1.6 ± 1.7 |
| Age 38 assessment | |
| CRP (mg/L) | 2.4 ± 3.9 |
| IL-6 (pg/mL) | 1.5 ± 1.5 |
| suPAR (ng/mL) | 2.4 ± 0.9 |
| No. of stressful life events age 32 through 37 | 5.5 ± 4.6 |
| Age 45 assessment | |
| Smoking status1 | |
| Non-smokers, n (%) | 667 (80.7%) |
| Smokers, n (%) | 160 (19.3%) |
| Body mass index (kg/m2) | 28.5 ± 5.8 |
| Anti-inflammatory medication use, n (%) | 241 (29.1%) |
| CRP (mg/L) | 2.7 ± 5.6 |
| IL-6 (pg/mL) | 2.1 ± 2.6 |
| suPAR (ng/mL) | 3.0 ± 1.0 |
| No. of stressful life events age 38 through 44 | 6.1 ± 5.9 |
Note: CRP and IL-6 values reported were untransformed. ACEs = adverse childhood experiences; CRP = C-reactive protein; IL-6 = interleukin-6; SES = socioeconomic status; suPAR = soluble urokinase plasminogen activator receptor.
One participant was missing this variable at age 45
3.1. Are Stressful Life Events Associated with Systemic Inflammation at Age 45?
Participants experiencing more stressful life events from age 38 through 44 had higher age-45 suPAR levels (Table 2). This association remained significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use. Figure 1 shows higher numbers of stressful life events from age 38 through 44 accompanied by higher age-45 suPAR levels.
Table 2.
The Association of Stressful Life Events from Age 38 through 44 and Inflammatory Biomarker Levels at Age 45 years
| suPAR | CRPa | IL-6a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p | β | 95% CI | p | β | 95% CI | p | |
| Bivariate association | 0.20 | [0.13, 0.27] | <.001 | 0.05 | [−0.02, 0.12] | .133 | 0.03 | [−0.03, 0.11] | .273 |
| Adjusted for sex, smoking, BMI, and anti-inflammatory medication | 0.14 | [0.07, 0.20] | <.001 | 0.03 | [−0.03, 0.09] | .326 | 0.00 | [−0.06, 0.06] | .966 |
| Adjusted for childhood covariates | 0.13 | [0.07, 0.19] | <.001 | 0.03 | [−0.03, 0.09] | .333 | 0.00 | [−0.06, 0.06] | .953 |
Note: Childhood covariates included childhood socioeconomic status, IQ, and self-control. 95% CI = 95% confidence interval; BMI = body mass index; CRP = C-reactive protein; IL-6 = interleukin-6; suPAR = soluble urokinase plasminogen activator receptor.
Log-transformed (natural logarithm)
Figure 1.
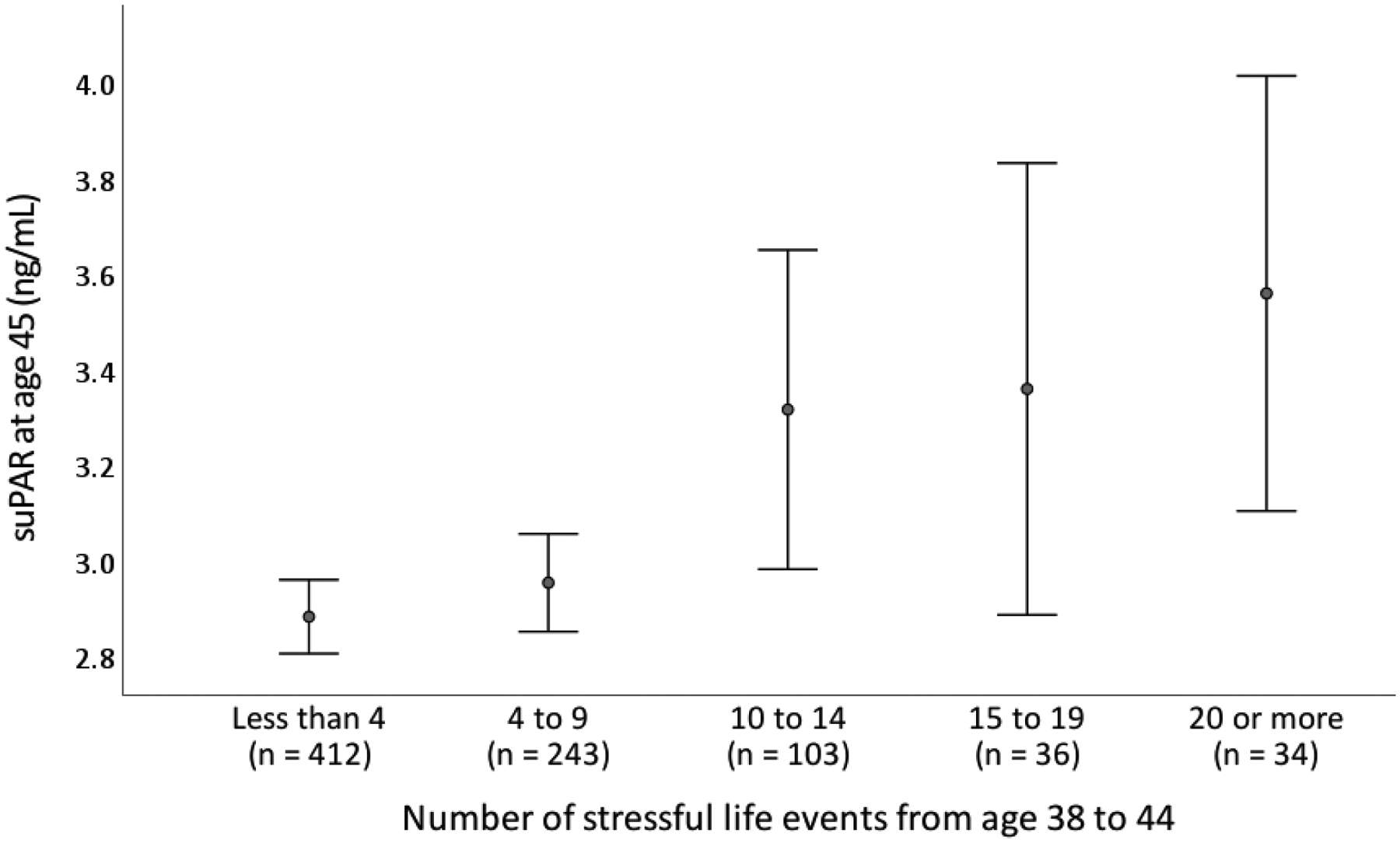
Number of stressful life events from age 38 through 44 and suPAR levels at age 45. Circles represent means and error bars represent 95% confidence intervals (± 2 SDs). suPAR = soluble urokinase plasminogen activator receptor measured in ng/mL.
In contrast to suPAR, participants’ stressful life events were not significantly associated with age-45 CRP or IL-6 levels in terms of bivariate associations, or when controlling for sex, smoking, BMI, and anti-inflammatory medication use (Table 2). None of the associations between stressful life events and systemic inflammation were substantively changed when further adjusting for childhood covariates (childhood SES, IQ, and self-control; Table 2).
3.2. Are Stressful Life Events Associated with Change in Systemic Inflammation from Age 38 to 45?
Next, we examined whether stressful life events were associated with change in suPAR from age 38 through 45 using residualized regression. When controlling for age-38 suPAR, participants who experienced more stressful life events from age 38 through 44 evidenced significantly greater suPAR at age 45 (Table 3). This association remained significant when adjusting for sex, smoking, BMI, anti-inflammatory medication use, and the number of pre-baseline stressful life events experienced from age 32 through 37 (Table 3). Stressful life events from age 32 through 37 were also significantly associated with age-38 suPAR (β = 0.13, 95% CI [0.06, 0.20], p < .001), suggesting that stressful life events were associated with suPAR levels at this earlier occasion as well. Supplemental Analyses 2 presents results of the primary analyses when excluding two outliers.
Table 3.
The Association of Stressful Life Events from Age 38 through 44 and Change in suPAR
| Outcome: suPAR at age 45 | Model 1 | Model 2 | ||||
|---|---|---|---|---|---|---|
| N = 828 | β | 95% CI | p | β | 95% CI | p |
| Stressful life events age 38 through 44 | 0.10 | [0.04, 0.15] | .001 | 0.08 | [0.02, 0.14] | .008 |
| suPAR at age 38 | 0.57 | [0.52, 0.61] | < .001 | 0.50 | [0.45, 0.55] | < .001 |
| Sex | −0.08 | [−0.13, −0.02] | .007 | |||
| Smoking at 45 | 0.16 | [0.10, 0.22] | < .001 | |||
| BMI at 45 | 0.11 | [0.05, 0.16] | < .001 | |||
| Anti-inflammatory medication use at 45 | 0.04 | [−0.02, 0.09] | .203 | |||
| Stressful life events age 32 through 37 | −0.01 | [−0.07, 0.05] | .707 | |||
Note: BMI = body mass index; suPAR = soluble urokinase plasminogen activator receptor.
In contrast, stressful life events from age 38 through 44 were not associated with change in either CRP (β = 0.02, 95% CI [−0.04, 0.08], p = .470) or IL-6 (β = −0.02, 95% CI [−0.08, 0.04], p = .511). This was also the case when controlling for sex, smoking, BMI, anti-inflammatory medication use, and stressful life events from age 32 through 37 (CRP: β = 0.03, 95% CI [−0.03, 0.09], p = .345; IL-6: β = −0.01, 95% CI [−0.07, 0.06], p = .809).
3.3. Stressful Life Events Across the Lifespan
We next examined whether stressful life events across the lifespan were associated with systemic inflammation levels at age 45.
3.3.1. ACEs, Stressful Life Events, and Inflammation: Biological Embedding and Cumulative Models.
To examine the cumulative and biological embedding models of stress and health across the lifespan, we tested whether ACEs (birth to age 15) and stressful life events in adulthood (ages 21 to 26 and 32 through 44) were independently associated with age-45 suPAR levels. In bivariate models, ACEs (β = 0.21, 95% CI [0.15, 0.28], p < .001) and stressful life events in adulthood (β = 0.23, 95% CI [0.16, 0.29], p < .001) were both associated with age-45 suPAR. In a multivariate model, ACEs and stressful life events in adulthood were also independently associated with age-45 suPAR (Table 4). These associations remained when including sex, smoking, BMI, and anti-inflammatory medication use (Table 4). When ACEs and adult stressful life events were combined into a single measure, people with more stressful life events across the lifespan evidenced higher levels of age-45 suPAR (β = 0.28, 95% CI [0.21, 0.34], p < .001), and this association remained when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = 0.17, 95% CI [0.10, 0.23], p < .001). This pattern of results suggests a cumulative effect of stressful life events, rather than biological embedding model.
Table 4.
Stressful Life Events Across the Lifespan (Childhood ACEs from birth to age 15 and Stressful Life Events from age 21 to 26 and age 32 through 44) and suPAR at age 45
| Outcome: suPAR at age 45 | ||||||
|---|---|---|---|---|---|---|
| N = 828 | β | 95% CI | p | β | 95% CI | p |
| Prospective ACEs | 0.18 | [0.11, 0.24] | < .001 | 0.10 | [0.04, 0.17] | .002 |
| Adult stressful life events | 0.19 | [0.13, 0.26] | < .001 | 0.12 | [0.06, 0.19] | < .001 |
| Sex | −0.15 | [−0.21, −0.09] | < .001 | |||
| Smoking at 45 | 0.28 | [0.22, 0.34] | < .001 | |||
| BMI at 45 | 0.14 | [0.07, 0.20] | < .001 | |||
| Anti-inflammatory medication use at 45 | 0.05 | [−0.01, 0.11] | .111 | |||
Note: ACEs = adverse childhood experiences; BMI = body mass index; suPAR = soluble urokinase plasminogen activator receptor.
We next tested these models with CRP at age 45 as the outcome. In bivariate models, stressful life events in adulthood (β = 0.08, 95% CI [0.01, 0.15], p = .023) were associated with age-45 CRP, whereas ACEs were not (β = 0.06, 95% CI [−0.01, 0.13], p = .069). In a multivariate model including both ACEs and adult stressful life events, stressful life events in adulthood remained associated with age-45 CRP (β = 0.07, 95% CI [0.00, 0.14], p = .047). However, stressful life events in adulthood were no longer significantly associated with CRP at age 45 when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = 0.03, 95% CI [−0.03, 0.09], p = .313). Similarly, people with more stressful life events across the lifespan evidenced relatively greater CRP at age 45 (β = 0.09, 95% CI [0.03, 0.16], p = .007), however this association was no longer significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = 0.02, 95% CI [−0.05, 0.08], p = .612).
Finally, we tested models with IL-6 at age 45 as the outcome. In bivariate models, neither ACEs (β = 0.04, 95% CI [−0.03, 0.11], p = .285), nor stressful life events in adulthood (β = 0.02, 95% CI [−0.05, 0.09], p = .543), were significantly associated with age-45 IL-6. These associations remained non-significant in a multivariate model including both ACEs and stressful life events in adulthood and when including sex, smoking, BMI, and anti-inflammatory medication use. People with more stressful life events across the lifespan did not evidence significantly greater IL-6 at age 45 (β = 0.04, 95% CI [−0.03, 0.11], p = .239), and this association remained non-significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = −0.02, 95% CI [−0.08, 0.05], p = .625).
3.3.2. Do ACEs Sensitize People to Stressful Life Events in Adulthood?
Next, to test the sensitization model we examined whether ACEs moderated the association between adult stressful life events and suPAR levels. We observed a significant interaction, such that the more ACEs a participant had experienced in childhood the stronger the association between stressful life events in adulthood and age-45 suPAR (Table 5). The interaction remained significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use (Table 5). When splitting the sample into those with no ACEs (n = 351), one ACE (n = 261), or multiple ACEs (2+; n = 216), the strength of the association between suPAR at 45 and adult stressful life events was stronger among people who had experienced more ACEs—people without ACEs (β = 0.14, p = .009), people with one ACE (β = 0.15, p = .016), people with multiple ACEs (β = 0.28, p < .001). These associations are presented in Figure 2 and suggest that ACEs could sensitize people to pro-inflammatory effects of stressful life events in adulthood.
Table 5.
Adult Stressful Life Events (from age 21 to 26 and age 32 through 44) and suPAR at age 45 Moderated by ACEs
| Outcome: suPAR at age 45 | ||||||
|---|---|---|---|---|---|---|
| N = 828 | β | 95% CI | p | β | 95% CI | p |
| Prospective ACEs | 0.16 | [0.09, 0.23] | < .001 | 0.09 | [0.02, 0.15] | .008 |
| Adult stressful life events | 0.17 | [0.11, 0.24] | < .001 | 0.11 | [0.04, 0.17] | .001 |
| Prospective ACEs × Adult Stressful Life Events | 0.13 | [0.06, 0.19] | < .001 | 0.10 | [0.04, 0.17] | .001 |
| Sex | −0.16 | [−0.22, −0.09] | < .001 | |||
| Smoking at 45 | 0.27 | [0.21, 0.34] | < .001 | |||
| BMI at 45 | 0.13 | [0.06, 0.19] | < .001 | |||
| Anti-inflammatory medication use at 45 | 0.05 | [−0.01, 0.12] | .085 | |||
Note: Stressful life events in adulthood included stressful life events from age 21 to 26, 32 to 37, and 38 to 44. ACEs = adverse childhood experiences; BMI = body mass index; suPAR = soluble urokinase plasminogen activator receptor.
Figure 2.
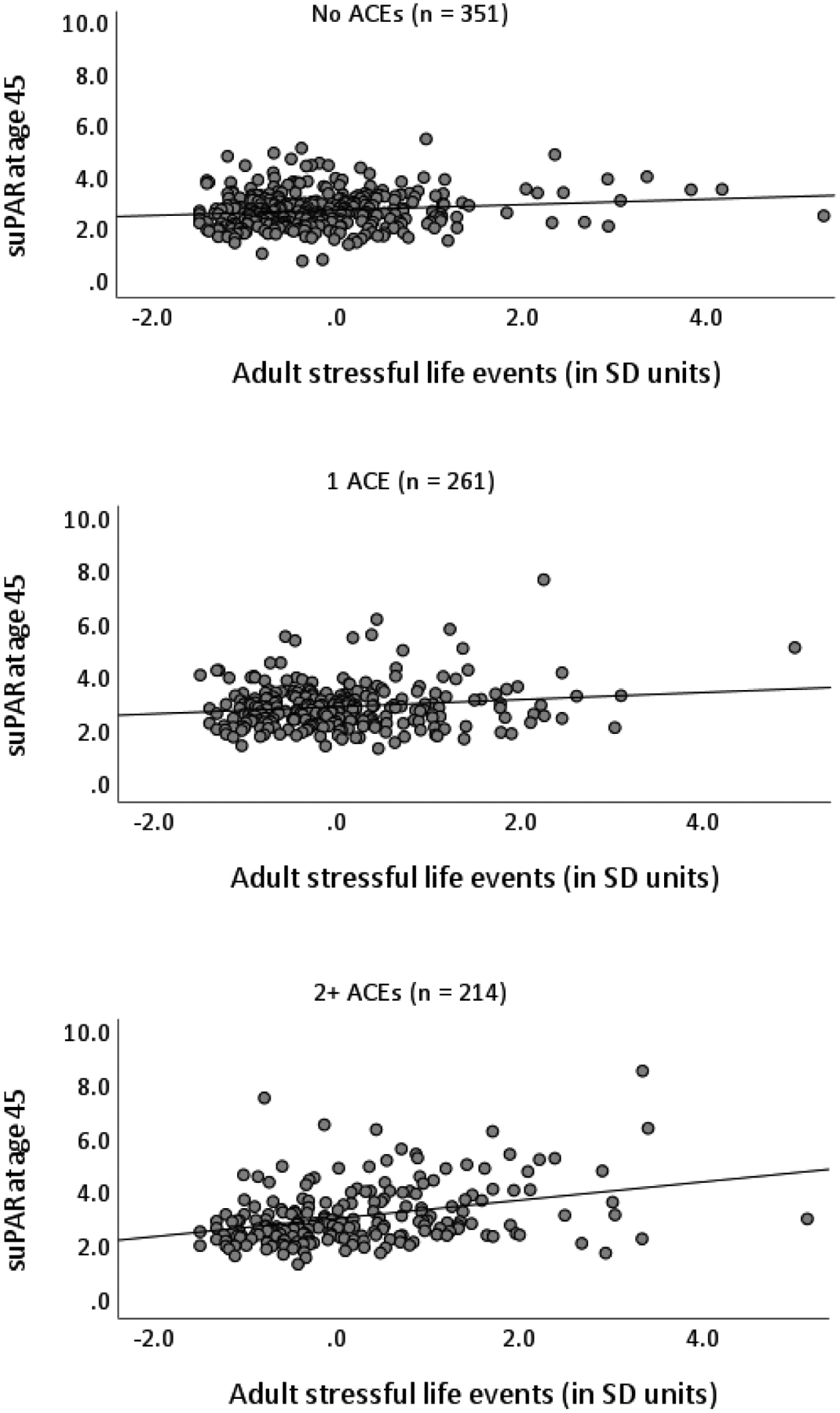
Scatterplots for the number of stressful life events participants experienced from age 32 to 44 and suPAR (soluble urokinase plasminogen activator receptor, measured in ng/mL) at age 45 separated by number of prospective adverse childhood experiences (ACEs) experienced in childhood (0, 1, and 2+). Two outliers in suPAR described in the main text are not included. SD = standard deviation.
We next tested these models with CRP at age 45 as the outcome. We observed a significant interaction, such that the more ACEs a participant had experienced in childhood the stronger the association between stressful life events in adulthood and age-45 CRP (β = 0.08, 95% CI [0.01, 0.15], p = .032). However, this interaction was no longer significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = 0.03, 95% CI [−0.03, 0.10], p = .271).
Finally, we tested models with IL-6 at age 45 as the outcome. We observed a significant interaction, such that the more ACEs a participant had experienced in childhood the stronger the association between stressful life events from age 32 through 44 and age-45 IL-6 (β = 0.09, 95% CI [0.02, 0.16], p = .011). The interaction remained significant when controlling for sex, smoking, BMI, and anti-inflammatory medication use (β = 0.07, 95% CI [0.01, 0.13], p = .027). As with suPAR, the association between stressful life events in adulthood and IL-6 at age 45 was stronger among people who experienced more ACEs in childhood.
3.4. Additional Analyses: Stressful Life Events across the Lifespan and Childhood Protective Factors
At the request of the reviewers, we also investigated whether the childhood covariates (childhood SES, IQ, and self-control) might moderate the association of stressful life events across the lifespan (combining ACEs and stressful life events in adulthood) and inflammation at age 45. Childhood IQ did not moderate the association between lifetime stressful life events and age-45 suPAR (β = −0.04, 95% CI [−0.11, 0.02], p = .219), whereas both childhood self-control (β = 0.09, 95% CI [0.03, 0.16], p = .006) and childhood SES did (β = −0.14, 95% CI [−0.22, −0.07], p < .001). People with less self-control as children had a stronger association between lifetime stressful life events and age-45 suPAR. Similarly, people who grew up in homes with lower SES had a stronger association between lifetime stressful life events and age-45 suPAR. These associations remained when controlling for sex, smoking, BMI, and anti-inflammatory medication use (low self-control: β = 0.08, 95% CI [0.02, 0.15], p = .019; childhood SES: β = −0.12, 95% CI [−0.19, −0.05], p = .001). None of the childhood covariates moderated the association of lifetime stressful life events with CRP or IL6 at age 45.
4. Discussion
The current study examined the association between stressful life events and systemic inflammation using a sample of 828 adults from the Dunedin Study. The findings indicated that people who experienced a greater number of stressful life events evidenced higher midlife systemic inflammation, in the form of elevated suPAR. These associations included both suPAR levels at age 45, as well as increases in suPAR from age 38 to 45. In contrast, no associations were observed between stressful life events and CRP or IL-6. The results suggest that systemic chronic inflammation as assessed by suPAR warrants more attention in the study of stressful life events and ill health. Measures of chronic inflammation such as suPAR could represent cumulative inflammation over the course of years and provide utility in life course and cohort research studies that assess individuals over years and decades. In such contexts, suPAR’s stability over time could make it useful as an inflammatory biomarker, particularly when assessing responses to chronic stressors.
When testing the timing of stressful life events across the lifespan and later inflammation, we found the most support for the cumulative and sensitization models (Young et al., 2019). Experiencing stressful life events in childhood, in the form of ACEs, was associated with increased suPAR in midlife. Notably, this association was above and beyond the association with stressful life events in adulthood. Stressful events in childhood and adulthood appear to exert a cumulative, additive effect on suPAR in midlife, though the fact that ACEs continued to evidence an association with inflammation even when accounting for adult stressful life events could also suggest the unique importance of childhood adversity—more in line with a biological embedding model. ACEs also appeared to sensitize people to later stressful life events, such that people who experienced more ACEs evidenced higher levels of suPAR in response to stressful life events compared to those who did not. These results suggest that childhood adversity is associated with heightened inflammatory responses decades later, matching previous research from this sample (Danese, Pariante, Caspi, Taylor, & Poulton, 2007) and highlighting the relevance of assessing stressful life events across the lifespan.
These results have theoretical, methodological, and clinical implications. Theoretically, the results suggest that systemic inflammation might be one biological pathway through which stressful life events become embedded within people’s physiology to affect downstream health. A number of behavioral and physiological pathways have been proposed to explain the association between stressful life events and health. For example, people who experience stressful life events, such as trauma and divorce, are more likely to smoke, less likely to engage in physical activity (Bourassa, Ruiz, & Sbarra, 2019; Feldner Babson, & Zvolensky, 2007; Zen, Whooley, Zhao, & Cohen, 2012), and more likely to evidence poor cardiovascular functioning following such events (Bourassa, Hasselmo, & Sbarra, 2016; Buckley, Holohan, Greif, Bedard, & Suvak, 2004; Pole, 2007). Systemic inflammation may complement or work in concert with such pathways to result in poorer health. For example, higher levels of inflammation can contribute to poorer cardiovascular health (Emerging Risk Factors Collaboration, 2010), and may explain why people who experience trauma and develop PTSD are at greater risk for cardiovascular disease and early mortality (Boscarino, 2008; Edmondson & von Känel, 2017).
In terms of methodological implications, the current results suggest that it may be useful to assess systemic inflammation using chronic inflammatory biomarkers, such as suPAR. The traditional inflammatory biomarkers included in this study—CRP and IL-6—did not evidence consistent associations with stressful life events, which stands in contrast to previous findings showing associations between traumatic and stressful life events with CRP and IL-6 (Baumeister et al., 2016; Chen & Lacey, 2018; Danese & Baldwin, 2017). However, research in this field has not always yielded consistent results, with some studies reporting non-significant associations for CRP or IL-6 with stress and traumatic experiences (Baumeister et al., 2016; Chiang et al., 2019; Hartwell et al., 2013; Low, Matthews, & Hall, 2013). One possible explanation is that these traditional biomarkers of inflammation may mix chronic and acute effects due to their sensitivity to short-term influences and involvement in the acute-phase response. High variability could result in mixed findings across studies if the underlying associations are small in size, as some meta-analyses have found (Baumeister et al., 2016). Currently, there are no standard biomarkers for measuring systemic chronic inflammation, which is inadequately measured by combining canonical biomarkers of acute inflammation (Furman et al., 2019). suPAR is generated under proinflammatory conditions through cleavage and shedding of uPAR (Thunø, Macho, Eugen-Olsen, 2009) and appears to be more stable, less sensitive to acute changes and circadian rhythm, and less rapidly upand down-regulated (Andersen, Eugen-Olsen, Kofoed, Iversen, Haugaard, 2008; Lyngbæk et al., 2013). This stability could make suPAR better suited to index underlying chronic inflammation.
Although additional replication is needed, these results suggest suPAR may be a useful biomarker with which to investigate the association between stressful events and health. This is particularly relevant given the established associations linking suPAR to poorer health (Botha et al., 2015; Eugen-Olsen et al., 2010; Persson et al., 2013; Rasmussen et al., 2021). Importantly, we have previously shown that suPAR is elevated in individuals who have been exposed to early-life stress even in the absence of elevated CRP or IL-6 in this cohort and the UK E-Risk cohort (Rasmussen et al., 2019a). suPAR remained associated with accelerated aging, physical and cognitive decline (Rasmussen et al., 2021), and mortality, even after adjustment for CRP or IL-6 (Rasmussen et al., 2016; Botha et al., 2015). However, we also observed the strongest associations between early-life stress and inflammation when utilizing all three biomarkers to maximize information yield on inflammation (Rasmussen et al., 2019a). Future studies could provide additional information as to whether combining suPAR with other inflammatory markers or if using suPAR independently provides the most research and clinical utility in the study of stressful life events.
Clinically, these results highlight that systemic inflammation is a potential intervention target to improve health for people under stress. Behavioral interventions that reduce people’s psychological distress following trauma or other stressors might be able to reduce inflammation and improve people’s health as a result (Shields et al., 2020). Changes in systemic inflammation might also serve as a useful surrogate endpoint in intervention studies seeking to reduce poor health among people who experience stressful life events (Moffitt & Klaus-Grawe Think Tank, 2013). Establishing the potential for interventions to improve health following stressful life events would require the inclusion of biomarkers, such as suPAR, in future behavioral intervention studies for people who experience stressful life events to provide evidence of reversibility (see Malarkey, Jarjoura, & Klatt, 2012; Thornton, Andersen, Schuler, & Carson, 2009; Villalba et al., 2019).
The results of this study should be understood within the context of its limitations. First, the current study used data from a longitudinal cohort study assessed from birth to age 45, but all three inflammatory biomarkers were only assessed at ages 38 and 45. Future replication of these results at the age-52 Dunedin follow-up would provide additional confidence in our findings. Similarly, the sample is predominantly composed of New Zealanders of European ancestry. Replications in more diverse populations are needed. Second, the current study assessed stressful life events using a coding system that provided a count of events, but did not assess the degree to which identified events were stressful to individuals. It is possible that the timing, salience, or type of stressful event may have played a role in the observed association. Similarly, although the study includes stressful life events across every decade from birth to age 45, at least in part, there are period of time that do not have stressful life events assessed. Future studies would benefit from assessing stressful life events at each study occasion. In addition, ACEs and adult stressful life events were assessed using different methods, which may have affected the results. Third, the detected effect sizes for suPAR were modest, though this might be expected in a general population of generally healthy persons at midlife. Fourth, the distributional properties of suPAR are appealing for research purposes, but the optimal threshold for its use as a clinical biomarker has not yet been determined, limiting clinical utility. Finally, the current study was an observational study and cannot rule out non-causal alternative explanations.
4.1. Conclusions
The findings from the current study suggest suPAR could be a particularly useful inflammatory biomarker with which to investigate the association between chronic stressors and systemic inflammation. The association between stressful life events and inflammation was particularly strong among people who had experienced ACEs in childhood, suggesting that the timing of stressful life events across the lifespan has relevance to inflammation in midlife.
Supplementary Material
Highlights.
In this longitudinal study, stressful life events were associated with increased suPAR from age 38 to 45
CRP and IL-6 did not evidence significant associations with stressful life events
Stressful events were more strongly linked to suPAR among people with childhood adversity.
Increased inflammation may be one mechanism linking stressful life events to poorer health
Acknowledgments
The Dunedin Multidisciplinary Health and Development Research Unit is supported by the New Zealand Health Research Council and New Zealand Ministry of Business, Innovation and Employment (MBIE). The Dunedin Study received support from the US-National Institute on Aging [grants R01AG032282, P30AG028716, P30AG034424] and the UK Medical Research Council [grant MR/P005918/1]. KJB received support from the National Institute on Aging [grant T32-AG000029]. LJHR received support from the Lundbeck Foundation [grant R288-2018-380]. We thank the Dunedin Multidisciplinary Health and Development Study members, Unit research staff, and Study founder Phil Silva, PhD, University of Otago. The study’s preregistration materials can be accessed online at https://sites.google.com/site/moffittcaspiprojects/home/projectlist/bourassa_rasmussen_2019.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: JEO is a named inventor on patents on suPAR as a prognostic biomarker. The patents are owned by Copenhagen University Hospital Amager and Hvidovre, Denmark, and is licensed to ViroGates A/S. J.E.-O. is a co-founder, shareholder, and CSO of ViroGates A/S.
References
- Andersen O, Eugen-Olsen J, Kofoed K, Iversen J, & Haugaard SB (2008). Soluble urokinase plasminogen activator receptor is a marker of dysmetabolism in HIV-infected patients receiving highly active antiretroviral therapy. Journal of Medical Virology, 80(2), 209–216. 10.1002/jmv.21114 [DOI] [PubMed] [Google Scholar]
- Baumeister D, Akhtar R, Ciufolini S, Pariante CM, & Mondelli V (2016). Childhood trauma and adulthood inflammation: A meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Molecular Psychiatry, 21(5), 642–649. 10.1038/mp.2015.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger E, Castagné R, Chadeau-Hyam M, Bochud M, d’Errico A, Gandini M, … & Panico S, (2019). Multi-cohort study identifies social determinants of systemic inflammation over the life course. Nature Communications, 10(1), 1–10. 10.1038/s41467-019-08732-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botha S, Fourie CM, Schutte R, Eugen-Olsen J, Pretorius R, & Schutte AE (2015). Soluble urokinase plasminogen activator receptor as a prognostic marker of all-cause and cardiovascular mortality in a black population. International Journal of Cardiology, 184, 631–636. 10.1016/j.ijcard.2015.03.041 [DOI] [PubMed] [Google Scholar]
- Boscarino JA (2008). A prospective study of PTSD and early-age heart disease mortality among Vietnam veterans: implications for surveillance and prevention. Psychosomatic Medicine, 70(6), 668–676. 10.1097/PSY.0b013e31817bccaf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa KJ (2021). Financial Stressors During the Great Recession and Subsequent Risk of Early Mortality. Psychosomatic Medicine. Advanced Online Publication. 10.1097/psy.0000000000000943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa KJ, Hasselmo K, & Sbarra DA (2016). Heart rate variability moderates the association between separation-related psychological distress and blood pressure reactivity over time. Psychological Science, 27(8), 1123–1135. 10.1177/0956797616651972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa KJ, Ruiz JM, & Sbarra DA (2019). Smoking and physical activity explain the increased mortality risk following marital separation and divorce: evidence from the English Longitudinal Study of Ageing. Annals of Behavioral Medicine, 53(3), 255–266. 10.1093/abm/kay038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning M, & Heinesen E (2012). Effect of job loss due to plant closure on mortality and hospitalization. Journal of Health Economics, 31(4), 599–616. 10.1016/j.jhealeco.2012.03.001 [DOI] [PubMed] [Google Scholar]
- Buckley TC, Holohan D, Greif JL, Bedard M, & Suvak M (2004). Twenty-four-hour ambulatory assessment of heart rate and blood pressure in chronic PTSD and non-PTSD veterans. Journal of Traumatic Stress, 17(2), 163–171. 10.1023/B:JOTS.0000022623.01190.f0 [DOI] [PubMed] [Google Scholar]
- Carroll D, Ginty AT, Whittaker AC, Lovallo WR, & de Rooij SR (2017). The behavioural, cognitive, and neural corollaries of blunted cardiovascular and cortisol reactions to acute psychological stress. Neuroscience & Biobehavioral Reviews, 77, 74–86. 10.1016/j.neubiorev.2017.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Thornton A, Freedman D, Amell JW, Harrington H, … & Silva PA (1996). The life history calendar: a research and clinical assessment method for collecting retrospective event-history data. International Journal of Methods in Psychiatric Research, 6(2), 101–114. [DOI] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, … & Poulton R (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science, 301(5631), 386–389. 10.1126/science.1083968 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. (2016). About the CDC-Kaiser ACE study. Centers for Disease Control and Prevention, 8. Accessed via: https://www.cdc.gov/violenceprevention/childabuseandneglect/acestudy/about.html [Google Scholar]
- Chen M, & Lacey RE (2018). Adverse childhood experiences and adult inflammation: Findings from the 1958 British birth cohort. Brain, Behavior, and Immunity, 69, 582–590. 10.1016/j.bbi.2018.02.007 [DOI] [PubMed] [Google Scholar]
- Chiang JJ, Park H, Almeida DM, Bower JE, Cole SW, Irwin MR, McCreath H, Seeman TE, & Fuligni AJ (2019). Psychosocial stress and C-reactive protein from mid-adolescence to young adulthood. Health Psychology, 38(3), 259–267. 10.1037/hea0000701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, & Turner RB (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proceedings of the National Academy of Sciences, 109(16), 5995–5999. 10.1073/pnas.1118355109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, & Miller GE (2007). Psychological stress and disease. JAMA, 298(14), 1685–1687. 10.1001/jama.298.14.1685 [DOI] [PubMed] [Google Scholar]
- Danese A, & Baldwin JR (2017). Hidden wounds? Inflammatory links between childhood trauma and psychopathology. Annual Review of Psychology, 68, 517–544. 10.1146/annurev-psych-010416-044208 [DOI] [PubMed] [Google Scholar]
- Danese A, & McEwen BS (2012). Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiology & behavior, 106(1), 29–39. 10.1016/j.physbeh.2011.08.019 [DOI] [PubMed] [Google Scholar]
- Danese A, Pariante CM, Caspi A, Taylor A, & Poulton R (2007). Childhood maltreatment predicts adult inflammation in a life-course study. Proceedings of the National Academy of Sciences, 104(4), 1319–1324. 10.1073/pnas.0610362104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt S, Desmedt V, Delanghe JR, Speeckaert R, & Speeckaert MM (2017). The intriguing role of soluble urokinase receptor in inflammatory diseases. Critical Reviews in Clinical Laboratory Sciences, 54(2), 117–133. 10.1080/10408363.2016.1269310 [DOI] [PubMed] [Google Scholar]
- Edmondson D, & von Känel R (2017). Post-traumatic stress disorder and cardiovascular disease. The Lancet Psychiatry, 4(4), 320–329. 10.1016/S2215-0366(16)30377-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elley WB, & Irving JC (1976). Revised socioeconomic index for New-Zealand. New Zealand Journal of Educational Studies, 11(1), 25–36. 10.1111/obr.12073 [DOI] [Google Scholar]
- Emerging Risk Factors Collaboration. (2010). C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. The Lancet, 375(9709), 132–140. 10.1016/S0140-6736(09)61717-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugen-Olsen J, Andersen O, Linneberg A, Ladelund S, Hansen TW, Langkilde A, … & Lyngbaek S (2010). Circulating soluble urokinase plasminogen activator receptor predicts cancer, cardiovascular disease, diabetes and mortality in the general population. Journal of Internal Medicine, 268(3), 296–308. 10.1111/j.1365-2796.2010.02252.x [DOI] [PubMed] [Google Scholar]
- Feldner MT, Babson KA, & Zvolensky MJ (2007). Smoking, traumatic event exposure, and post-traumatic stress: A critical review of the empirical literature. Clinical Psychology Review, 27(1), 14–45. 10.1016/j.cpr.2006.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, & Marks JS (1998). Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults: The Adverse Childhood Experiences (ACE) Study. American Journal of Preventive Medicine, 14(4), 245–258. 10.1016/S0749-3797(98)00017-8 [DOI] [PubMed] [Google Scholar]
- Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, … & Miller AH (2019). Chronic inflammation in the etiology of disease across the life span. Nature medicine, 25(12), 1822–1832. 10.1038/s41591-019-0675-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Rabin B, Chesney M, Cohen S, & Natelson B (1999). Stress-induced immunomodulation: implications for infectious diseases?. JAMA, 281(24), 2268–2270. 10.1207/10.1001/jama.281.24.2268 [DOI] [PubMed] [Google Scholar]
- Guo YZ, Pan L, Du CJ, Ren DQ, & Xie XM (2013). Association between C-reactive protein and risk of cancer: a meta-analysis of prospective cohort studies. Asian Pacific Journal of Cancer Prevention, 14(1), 243–248. 10.7314/APJCP.2013.14.1.243 [DOI] [PubMed] [Google Scholar]
- Hamer M, Molloy GJ, & Stamatakis E (2008). Psychological distress as a risk factor for cardiovascular events: pathophysiological and behavioral mechanisms. Journal of the American College of Cardiology, 52(25), 2156–2162. 10.1016/j.jacc.2008.08.057 [DOI] [PubMed] [Google Scholar]
- Hartwell KJ, Moran-Santa Maria MM, Twal WO, Shaftman S, DeSantis SM, McRae-Clark AL, & Brady KT (2013). Association of elevated cytokines with childhood adversity in a sample of healthy adults. Journal of Psychiatric Research, 47(5), 604–610. 10.1016/j.jpsychires.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayek SS, Leaf DE, Samman Tahhan A, Raad M, Sharma S, Waikar SS, Sever S, Camacho A, Wang X, Dande RR, Ibrahim NE, Baron RM, Altintas MM, Wei C, Sheikh-Hamad D, Pan JS, Holliday MW Jr, Januzzi JL, Weisbord SD, Quyyumi AA, … Reiser J (2020). Soluble Urokinase Receptor and Acute Kidney Injury. The New England Journal of Medicine, 382(5), 416–426. 10.1056/NEJMoa1911481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostinar CE, Lachman ME, Mroczek DK, Seeman TE, & Miller GE (2015). Additive contributions of childhood adversity and recent stressors to inflammation at midlife: Findings from the MIDUS study. Developmental Psychology, 51(11), 1630–1644. 10.1037/dev0000049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmakis KA, & Chandler GE (2015). Health consequences of adverse childhood experiences: a systematic review. Journal of the American Association of Nurse Practitioners, 27(8), 457–465. 10.1002/2327-6924.12215 [DOI] [PubMed] [Google Scholar]
- Low CA, Matthews KA, & Hall M (2013). Elevated C-reactive protein in adolescents: Roles of stress and coping. Psychosomatic Medicine, 75(5), 449–452. 10.1097/PSY.0b013e31828d3f1d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyngbæk S, Andersson C, Marott JL, Møller DV, Christiansen M, Iversen KK, … & Jeppesen JL (2013). Soluble urokinase plasminogen activator receptor for risk prediction in patients admitted with acute chest pain. Clinical Chemistry, 59(11), 1621–1629. 10.1373/clinchem.2013.203778 [DOI] [PubMed] [Google Scholar]
- Lyngbæk S, Marott JL, Sehestedt T, Hansen TW, Olsen MH, Andersen O, … & Jeppesen J (2013). Cardiovascular risk prediction in the general population with use of suPAR, CRP, and Framingham Risk Score. International Journal of Cardiology, 167(6), 2904–2911. 10.1016/j.ijcard.2012.07.018 [DOI] [PubMed] [Google Scholar]
- Malarkey WB, Jarjoura D, & Klatt M (2013). Workplace based mindfulness practice and inflammation: a randomized trial. Brain, Behavior, and Immunity, 27, 145–154. 10.1016/j.bbi.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuck SB (1994). Cardiovascular reactivity in cardiovascular disease: “Once more unto the breach”. International Journal of Behavioral Medicine, 1(1), 4–31. 10.1207/s15327558ijbm0101_2 [DOI] [PubMed] [Google Scholar]
- Marsland AL, Walsh C, Lockwood K, & John-Henderson NA (2017). The effects of acute psychological stress on circulating and stimulated inflammatory markers: a systematic review and meta-analysis. Brain, Behavior, and Immunity, 64, 208–219. 10.1016/j.bbi.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, & Parker KJ (2011). Psychological stress in childhood and susceptibility to the chronic diseases of aging: Moving toward a model of behavioral and biological mechanisms. Psychological Bulletin, 137(6), 959–997. 10.1037/a0024768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt TE andthe Klaus-Grawe 2012 Think Tank (2013). Childhood exposure to violence and lifelong health: Clinical intervention science and stress biology research join forces. Development and Psychopathology, 25, 116–1634. 10.1017/S0954579413000801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhlenkamp S, Lehmann N, Moebus S, Schmermund A, Dragano N, Stang A, … & Heinz Nixdorf Recall Study Investigators. (2011). Quantification of coronary atherosclerosis and inflammation to predict coronary events and all-cause mortality. Journal of the American College of Cardiology, 57(13), 1455–1464. 10.1016/j.jacc.2010.10.043 [DOI] [PubMed] [Google Scholar]
- Moon JR, Kondo N, Glymour MM, & Subramanian SV (2011). Widowhood and mortality: A meta-analysis. PloS One, 6(8). 10.1371/journal.pone.0023465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén LK, and Muthén BO. Mplus User’s Guide. Seventh Edition. Los Angeles, CA: Muthén & Muthén; 1998–2012 [Google Scholar]
- Niiyama M, Tanaka F, Nakajima S, Itoh T, Matsumoto T, Kawakami M, … & Sakata K (2014). Population-Based Incidence of Sudden Cardiac and Unexpected Death Before and After the 2011 Earthquake and Tsunami in Iwate, Northeast Japan. Journal of the American Heart Association, 3(3), e000798. 10.1161/JAHA.114.000798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken BS, Chamine I, & Wakeland W (2015). A systems approach to stress, stressors and resilience in humans. Behavioural Brain Research, 282, 144–154. 10.1016/j.bbr.2014.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, Criqui M, … & Rifai N (2003). Markers of inflammation and cardiovascular disease application to clinical and public health practice: A statement for healthcare professionals from the centers for disease control and prevention and the American Heart Association. Circulation, 107(3), 499–511. doi: 10.1161/01.CIR.0000052939.59093.45 [DOI] [PubMed] [Google Scholar]
- Persson M, Östling G, Smith G, Hamrefors V, Melander O, Hedblad B, & Engström G (2014). Soluble urokinase plasminogen activator receptor: a risk factor for carotid plaque, stroke, and coronary artery disease. Stroke, 45(1), 18–23. 10.1161/STROKEAHA.113.003305 [DOI] [PubMed] [Google Scholar]
- Phillips DP, Liu GC, Kwok K, Jarvinen JR, Zhang W, & Abramson IS (2001). The Hound of the Baskervilles effect: natural experiment on the influence of psychological stress on timing of death. BMJ, 323(7327), 1443–1446. 10.1136/bmj.323.7327.1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pole N (2007). The psychophysiology of posttraumatic stress disorder: A meta-analysis. Psychological Bulletin, 133(5), 725–746. 10.1037/0033-2909.133.5.725 [DOI] [PubMed] [Google Scholar]
- Poulton R, Moffitt TE, & Silva PA (2015). The Dunedin Multidisciplinary Health and Development Study: overview of the first 40 years, with an eye to the future. Social Psychiatry and Psychiatric Epidemiology, 50(5), 679–693. 10.1007/s00127-015-1048-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJH, Caspi A, Ambler AA, Danese A, Elliott M, Eugen-Olsen J, … & Moffitt TE (2021). Association between elevated suPAR, a new biomarker of chronic inflammation, and accelerated aging. Journal of Gerontology: Medical Sciences. 76(2):318–327. 10.1093/gerona/glaa178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJH, Ladelund S, Haupt TH, Ellekilde G, Poulsen JH, Iversen K, … & Andersen O (2016). Soluble urokinase plasminogen activator receptor (suPAR) in acute care: a strong marker of disease presence and severity, readmission and mortality. A retrospective cohort study. Emergency Medicine Journal, 33(11), 769–775. 10.1136/emermed-2015-205444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJH, Moffitt TE, Arseneault L, Danese A, Eugen-Olsen J, Fisher HL, … & Williams B (2019a). Association of adverse experiences and exposure to violence in childhood and adolescence with inflammatory burden in young people. JAMA Pediatrics, 174(1), 38–47. 10.1097/10.1001/jamapediatrics.2019.3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJH, Moffitt TE, Eugen-Olsen J, Belsky DW, Danese A, Harrington H, … & Caspi A (2019b). Cumulative childhood risk is associated with a new measure of chronic inflammation in adulthood. Journal of Child Psychology and Psychiatry, 60(2), 199–208. 10.1111/jcpp.12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJH, Schultz M, Gaardsting A, Ladelund S, Garred P, Iversen K, … & Lebech AM (2017). Inflammatory biomarkers and cancer: CRP and suPAR as markers of incident cancer in patients with serious nonspecific symptoms and signs of cancer. International Journal of Cancer, 141(1), 191–199. 10.1002/ijc.30732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A, Moffitt TE, Caspi A, Belsky DW, Harrington H, Schroeder F, … & Danese A (2016). Lest we forget: comparing retrospective and prospective assessments of adverse childhood experiences in the prediction of adult health. Journal of Child Psychology and Psychiatry, 57(10), 1103–1112. 10.1111/jcp p.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond-Rakerd LS, Caspi A, Ambler A, d’Arbeloff T, de Bruine M, Elliott M, … & Moffitt TE (2021). Childhood self-control forecasts the pace of midlife aging and preparedness for old age. Proceedings of the National Academy of Sciences, 118(3), e2010211118. 10.1073/pnas.2010211118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond-Rakerd LS, D’Souza S, Andersen SH, Hogan S, Houts RM, Poulton R, … & Moffitt TE (2020). Clustering of health, crime and social-welfare inequality in 4 million citizens from two nations. Nature Human Behaviour, 4(3), 255–264. 10.1038/s41562-019-0810-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohleder N (2014). Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosomatic Medicine, 76(3), 181–189. 10.1097/PSY.0000000000000049 [DOI] [PubMed] [Google Scholar]
- Rovina N, Akinosoglou K, Eugen-Olsen J, Hayek S, Reiser J, & Giamarellos-Bourboulis EJ (2020) Soluble urokinase plasminogen activator receptor (suPAR) as an early predictor of severe respiratory failure in patients with COVID-19 pneumonia. Critical Care, 24(1):187. doi: 10.1186/s13054-020-02897-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields GS, Spahr CM, & Slavich GM (2020) Psychosocial Interventions and Immune System Function: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Psychiatry, 77(10), 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steptoe A, Hamer M, & Chida Y (2007). The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain, Behavior, and Immunity, 21(7), 901–912. 10.1016/j.bbi.2007.03.011 [DOI] [PubMed] [Google Scholar]
- Thornton LM, Andersen BL, Schuler TA, & Carson III WE (2009). A psychological intervention reduces inflammatory markers by alleviating depressive symptoms: secondary analysis of a randomized controlled trial. Psychosomatic Medicine, 71(7). 10.1097/PSY.0b013e3181b0545c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunø M, Macho B, & Eugen-Olsen J (2009). suPAR: the molecular crystal ball. Disease markers, 27(3), 157–172. 10.3233/DMA-2009-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tursich M, Neufeld RW, Frewen PA, Harricharan S, Kibler JL, Rhind SG, & Lanius RA (2014). Association of trauma exposure with proinflammatory activity: A transdiagnostic meta-analysis. Translational Psychiatry, 4(7), e413. 10.1038/tp.2014.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba DK, Lindsay EK, Marsland AL, Greco CM, Young S, Brown KW, … & Creswell JD (2019). Mindfulness training and systemic low-grade inflammation in stressed community adults: Evidence from two randomized controlled trials. PloS one, 14(7), e0219120. 10.1371/journal.pone.0219120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz PH, & von Känel R (2017). Psychological stress, inflammation, and coronary heart disease. Current Cardiology Reports, 19(11), 111. 10.1007/s11886-017-0919-x [DOI] [PubMed] [Google Scholar]
- Young ES, Farrell AK, Carlson EA, Englund MM, Miller GE, Gunnar MR, … & Simpson JA (2019). The dual impact of early and concurrent life stress on adults’ diurnal cortisol patterns: A prospective study. Psychological Science, 30(5), 739–747. 10.1177/0956797619833664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen AL, Whooley MA, Zhao S, & Cohen BE (2012). Post-traumatic stress disorder is associated with poor health behaviors: Findings from the heart and soul study. Health Psychology, 31(2), 194–201. 10.1037/a002598 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.