

Abstract
We report a database of tautomeric structures that contains 2819 tautomeric tuples extracted from 171 publications. Each tautomeric entry has been annotated with experimental conditions reported in the respective publication, plus bibliographic details, structural identifiers (e.g., NCI/CADD identifiers FICTS, FICuS, uuuuu, and Standard InChI), and chemical information (e.g., SMILES, molecular weight). The majority of tautomeric tuples found were pairs; the remaining 10% were triples, quadruples, or quintuples, amounting to a total number of structures of 5977. The types of tautomerism were mainly prototropic tautomerism (79%), followed by ring–chain (13%) and valence tautomerism (8%). The experimental conditions reported in the publications included about 50 pure solvents and 9 solvent mixtures with 26 unique spectroscopic or nonspectroscopic methods. 1H and 13C NMR were the most frequently used methods. A total of 77 different tautomeric transform rules (SMIRKS) are covered by at least one example tuple in the database. This database is freely available as a spreadsheet at https://cactus.nci.nih.gov/download/tautomer/.
Graphical Abstract
INTRODUCTION
Tautomerism is a phenomenon in which a set of molecules can interconvert by movement of a hydrogen or group of atoms and/or molecular rearrangement. The movement of hydrogen atoms along with the migration of pi-bonds is called prototropic tautomerism. The intermolecular arrangements leading to isomerization due to ring opening or cyclization are known as ring–chain tautomerism. (We have recently compiled 11 sets of rules for ring–chain tautomerism in SMIRKS notation.1) Another type of isomerization, which involves rapid reorganization of single and double bonds without migration of any atom or group, is termed valence tautomerism.
Tautomers usually have different physicochemical properties such as logP, hydrophobicity, pKa, solubility, electrostatic potential, similarity index, etc. with concomitant computation of such properties and molecular descriptors yielding different results from one tautomer to another.2 They may behave differently in docking tools, pose challenges in macromolecular X-ray structures, particularly for small-molecule ligands in protein–ligand complexes, and wreak havoc in compound registration systems and vendor catalogs.3 Therefore, consideration of tautomers has been of high interest to the drug design community for decades.4–7 This leads to the question: How does one select the tautomer(s) of a molecule that allow one to most accurately predict its properties? One of the issues in this context has been the lack of a publicly accessible database providing a significant number of quantitative ratios or qualitative data of tautomeric forms in different solvents.
While a significant body of work exists of individual experimental studies (e.g., spectroscopy on tautomeric molecules), quantum chemical analyses (e.g., energy and structure computations), and―to a limited extent―chemoinformatics studies (e.g., rule-based tautomer prediction), very few systematic collections of experimental results in this field have been undertaken so far. A set of 785 transformations belonging to 11 types of tautomeric reactions with equilibrium constants measured in different solvents and at different temperatures was recently used in an effort to build QSPR models of equilibrium constants of tautomeric molecules.8 To the best of our knowledge, there is currently no database publicly available that provides details of several thousands of molecules and their experimentally investigated tautomers under a wide variety of experimental conditions along with a detailed chemoinformatics analysis. We acknowledge, however, the Tautomer Codex database (Tautobase) and its references provided directly to us by Wahl and Sander before its recent publication.9 It provides a very useful complement of water-based tautomerism measurements in addition to the diversity of solvents represented in our database. Here, we report on a tautomer database we have created from the literature in an attempt to compile experimental and quantitative tautomeric preferences together with chemical and bibliographic information as well as an analysis along a set of more than 70 tautomeric transforms. We hope this resource, which we made freely available for download at https://cactus.nci.nih.gov/download/tautomer/ starting October 2017, will allow the scientific community to more easily explore the phenomenon of tautomerism by finding several thousand such molecules in one location. (A preprint version of this paper with a more-extended discussion is available at 10.26434/chemrxiv.10790369.v1.)
METHODS AND DATA
Data Set of Database.
The current tautomer database consists of 2819 entries, each comprising an n-tuple of tautomers (n = 2–5) studied in a particular set of experimental conditions (pH, solvent, solvent mixture, temperature, experimental technique used). All these tuples together comprise a total of 5977 records. The data were extracted from 171 publications, which included a number of reviews (see full list in the Spreadsheet S1 of the Supporting Information). The initial extraction from these literature sources was done by a contract mechanism (Parthys Reverse Informatics, http://www.reverseinformatics.com), whereas a significant workup and curation of the initial data was performed by the authors.
For each entry for all n-tuples in the tautomer database, the corresponding NCI/CADD Chemical Structure Identifiers10 were calculated using the chemoinformatics toolkit CACTVS11 (in which they have been implemented as standard molecular properties). The nature of these identifiers, which are based on the standard CACTVS molecular hashcodes, is based on turning off or on sensitivity to the following five chemical features: fragments, isotopes, charges, tautomers, and stereochemistry. In this database, we used the FICTS, FICuS, and uuuuu identifiers out of a possible 32 possible set of variants (see the original publication10 for explanation of the nomenclature). The FICTS identifier, in which all five features are turned on, represents the original input structure as is. It is sensitive to fragments (such as counterions), isotopes, charges, and stereochemistry in the input structure as well as to the specific tautomer drawn. The FICuS identifier is tautomer invariant (but sensitive to all four other features), meaning that different tautomers have the same FICuS hashcode. For the uuuuu identifier, all five features are turned off, implying that molecules differing by fragment, isotope, charge, tautomer, or stereochemistry have the same uuuuu (which can thus be regarded as a sort of parent structure identifier). The FICuS identifier is conceptually similar to the InChIKey, though the latter handles tautomerism less comprehensively than FICuS due to an only limited range of tautomerism transforms implemented in its current version (v. 1.05). Additionally, it is not currently possible for the user to add entirely new types of tautomerism to the InChI[Key] calculation. This and other shortcomings of the current InChI in the handling of tautomerism has led to an IUPAC-sanctioned project of redesigning the handling of tautomerism for an InChI V2,12 for which this tautomer database forms an experimental backdrop of sorts and whose authors are involved in the IUPAC project.
In order to describe the tautomeric transformation(s) between the members of each of the tautomeric n-tuples in the database, we used a total of 77 rules. This set is closely related to, and essentially a major subset of, the 86 rules described in the context of redesigning of the handling of tautomerism for InChI V2 in the accompanying paper.13 All these rules were encoded in SMIRKS line notation.14 They were all processed in CACTVS, which comes with a default set of 20 prototropic rules covering a wide range of common and some rarer types of tautomerism. Twelve of the 20 rules have a representative in this database. To these, we added a subset of 8 SMIRKS from our recently published 11 types of ring⇌chain rules (encoded in a total of 38 SMIRKS),1 plus 57 out of 61 heretofore unpublished rules, which are detailed in the accompanying publication.13
We use the following nomenclature (again aligned with the accompanying paper13) for the three types of rules discussed in this paper: (1) Prototropic tautomerism rules are called PT_nn_mm, where nn and mm are the number of the rule and a possible variant, respectively. The names of most rules end with a 00 indicator, indicating that there is only one variant. (2) Ring–chain tautomerism rules are named RC_nn_mm, where nn and mm have the same meaning as described above. (3) Valence tautomerism rules are termed VT_nn_mm according to the same scheme.
To determine the single transform or sequence of transforms connecting the tuple members with each other, we applied the following procedure: In the first step, we enumerated all possible tautomers from each tautomeric tuple. In the second step, we generated a tautomer network among those enumerated tautomers. In such a network, we typically have several pathways that connect one tautomer to the other by different tautomeric transforms. As the final step, we searched for the shortest pathway, defined by the smallest number of transformation steps within the tautomeric pair. If two different paths had the same number of steps, we used a notation of the type of {PT_03_00/PT_06_00} > PT_09_00. This means the pathway can either use PT_03_00 or PT_06_00 in the first step, followed by PT_09_00 in the second step.
Database Description.
The database is provided to the user as a spreadsheet in Excel format. Each entry consists of three major segments: conditions, tautomer, and publication. Each segment has several fields as listed in Table 1. For each additional (second, third, …) tautomer of a compound, fields in the second (and third, etc.) instance of the tautomer-specific columns are populated with data, otherwise left empty. The following provides a brief explanation of some key columns in the spreadsheet. Others should be self-explanatory. A legends worksheet is also available in the spreadsheet providing explanations for all columns.
Table 1.
Summary of Data Fields Used in Tautomer Databasea
| Conditions | Tautomer_1 | Publication_1 |
|---|---|---|
| ref | Entry_ID1 | Filename_1 |
| Size | Type_1 | Publication_DOI_1 |
| Solvent | Transf_1_2 | Publication_ID_1 |
| Solvent_Proportion | ID_Hash_1 | Authors_1 |
| Solvent_Mixture | FICTS_1 | Affiliation_1 |
| Temperature | HASHISY_1 | Title_1 |
| pH | FICuS_1 | Section_1 |
| Experimental_Method | uuuuu_1 | Page_Number(s)_1 |
| Solvent_Mixture | Std_InChIKey_1 | Notes_1 |
| Std_InChI_1 | Cmpd_Number_1 | |
| SMILES_1 | ||
| Mol_Formula_1 | ||
| Mol_Weight_1 | ||
| IUPAC_Name_1 | ||
| Quantitative_ratio_1 | ||
| Qualitative_Prevalence_1 | ||
| Prevalence_Category_1 |
Entries with the value “nul” in any column indicate that it was not possible to extract sufficiently specific information from the publication.
Size: Number of tautomers reported in the publication as being in equilibrium. In a few publications, only the main tautomer of the compound was described; in such cases, we entered a second entry based on a possible (calculated) tautomer.
Solvent: Solvent in which the tautomers were observed. This can be a mixture of solvents. If their concentration is indicated, then it is also mentioned in the solvent column.
Solvent_Proportion: Fraction of solvents or their mixtures; typically measured on a mass, molar, or volume ratio scale (though in some cases, the scale used was not clear from the publication).
Solvent_Mixture: Indicates whether a single solvent or mixture was used. This column has a “yes” if the “Solvent” column indicates a solvent mixture, otherwise “no”.
Temperature: Temperature (K) at which the tautomers were observed or the experiment was carried out. In the case of mass spectroscopy experiments, the temperature of the injector was used as the experimental temperature.
pH: pH of the medium at which the tautomers were observed or experiment was conducted.
Experimental_Method: This describes the spectroscopic or physical methods that were used in the experimental determination of the tautomers. It may be a single method or a combination of several methods by which the tautomers were established in the experiments. If the experimental details were not available in the review, then those were extracted from the original references.
Entry_ID1: Unique ID composed from the publication reference (journal name, year, volume, page numbers) along with the tautomer ID in that publication (if given) and the nature of the tautomerism (e.g., “Keto⇌enol”).
Type_1: The chemical nature of the tautomer, e.g., keto, hydroxy, imine, enamine, etc. An entry with “nul” in this column indicates that it was difficult to assign any specific name from the molecule’s common name or based on similar structures in the database.
Transf_1_2: The rule(s) (prototropic [PT], ring⇌chain [RC], or valence tautomerism [VT]) which transform(s) tautomer_1 into tautomer_2 (single or multiple steps). A forward slash “/” is used to indicate alternative rules for any step. Curly braces “{}” are used to group together alternative rules if these appear in multi-step transforms. The greater than sign “>“ is used to separate steps in multi-step transformations. An entry with “no_transform” in this column indicates that these pairs are not covered by our rules because these examples are releated to zwitterionic and complex protonated structures; hence, we did not develop any rules for them.
ID_Hash_1: A hashed unique ID generated for each tautomer by the original contractor (Reverse Informatics). Some entries added later by us do not have an ID_Hash.
FICTS_1: Tautomer-sensitive NCI/CADD structural identifier of tautomer_1.
HASHISY_1: Tautomer-sensitive CACTVS structural identifier of tautomer_1.
FICuS_1: Tautomer-insensitive NCI/CADD identifier, which therefore is the same for all tautomers of the same molecule.
uuuuu_1: NCI/CADD identifier for the parent compound of tautomer_1.
Quantitative_ratio_1: Quantitative ratio of tautomer_1 compared to other tautomers. This can be a single number, a range, or an upper or lower bound between 0 and 1. Decimal numbers are reported up to the third decimal digit.
Qualitative_Prevalence_1: Qualitative prevalence category of tautomer_1 reported in the publication. These keywords describe the prevalence of one tautomer over other tautomer(s) and are mostly extracted from the papers, assigned based on the quantitative data or the spectra or other informations in the text of paper. “nul” is used if no such keywords were available in the papers or reviews.
Prevalence_Category_1: In order to make both quantitatively and qualitatively reported prevalences of tautomers comparable, at least in a categorical way, we numerically categorized tautomer_1 into five classes: 0, 1, 2, 3, and 4 based on its quantitative ratio and/or qualitative prevalence as described below.
Numeric classification of qualitative prevalence’s keywords:
0: Not observed
1: Less favored, less stable, minor, observed
2: Equally, favored, major, in equilibrium, preferred, similar spectra
3: More favored, more stable, predominant, strongly favored
4: Exclusively observed, only observed, only tautomer, identical tautomer
Numeric classification of quantitative amount of tautomers
0: Quantitive_ratio = 0.0–0.0099
1: Quantitive_ratio = 0.01–0.30
2: Quantitive_ratio = 0.31–0.69
3: Quantitive_ratio = 0.70–0.99
4: Quantitive_ratio = 1
If there were three or more tautomers reported, there would be corresponding columns in the spreadsheet with “3” or “_3″, e.g., Transf_1_3, etc.
DATABASE ANALYSIS
Provenance and Relationship of Tuples.
We did not identify any direct tuples’ duplicates in terms of both chemical structure and experimental conditions. Purely chemical duplicates were found for 479 tautomeric tuples in the database, but they differ in conditions such as temperature, solvent, pH, or spectroscopy method.
Size Distribution of Tuples.
The database contains tautomeric tuples ranging in size from 2 to 5 (2530, 250, 28, 11 cases, respectively) (Figure 1).
Figure 1.
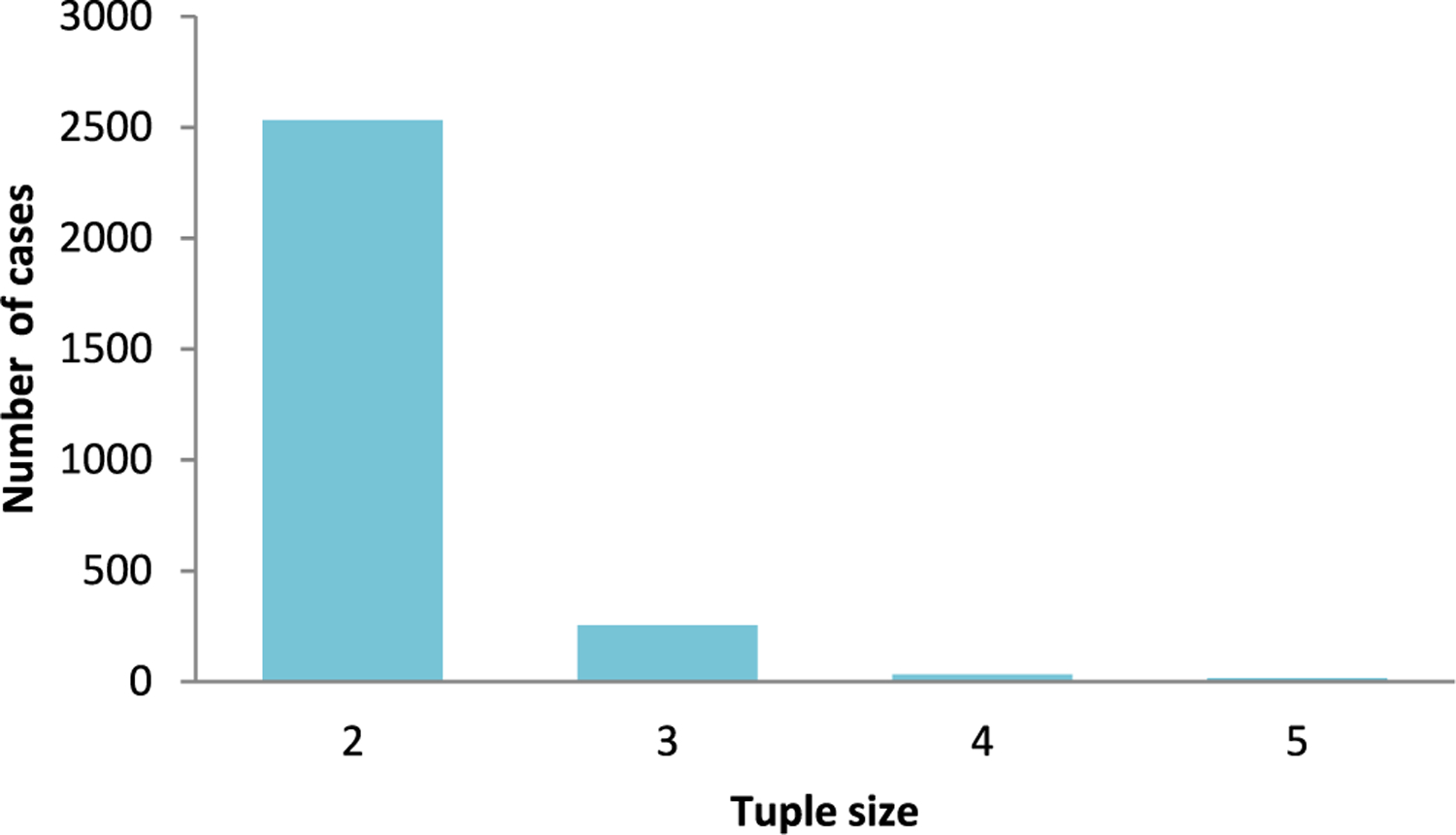
Distribution of sizes of tautomer tuples in the database.
Solvent.
The database contains tautomeric equilibrium studies performed in solvents (87% of the cases), in the solid state (6%), neat liquid (<1%), gas phase/matrix (5%), and vapor phase (<1%). The majority of experiments were conducted in some kind of solvent or solvent mixture. About 50 different types of solvents were reported in the papers (Figure 2a). The database has 12 solvent mixtures, in which 12 different types of solvents were used (Figure 2b).
Figure 2.
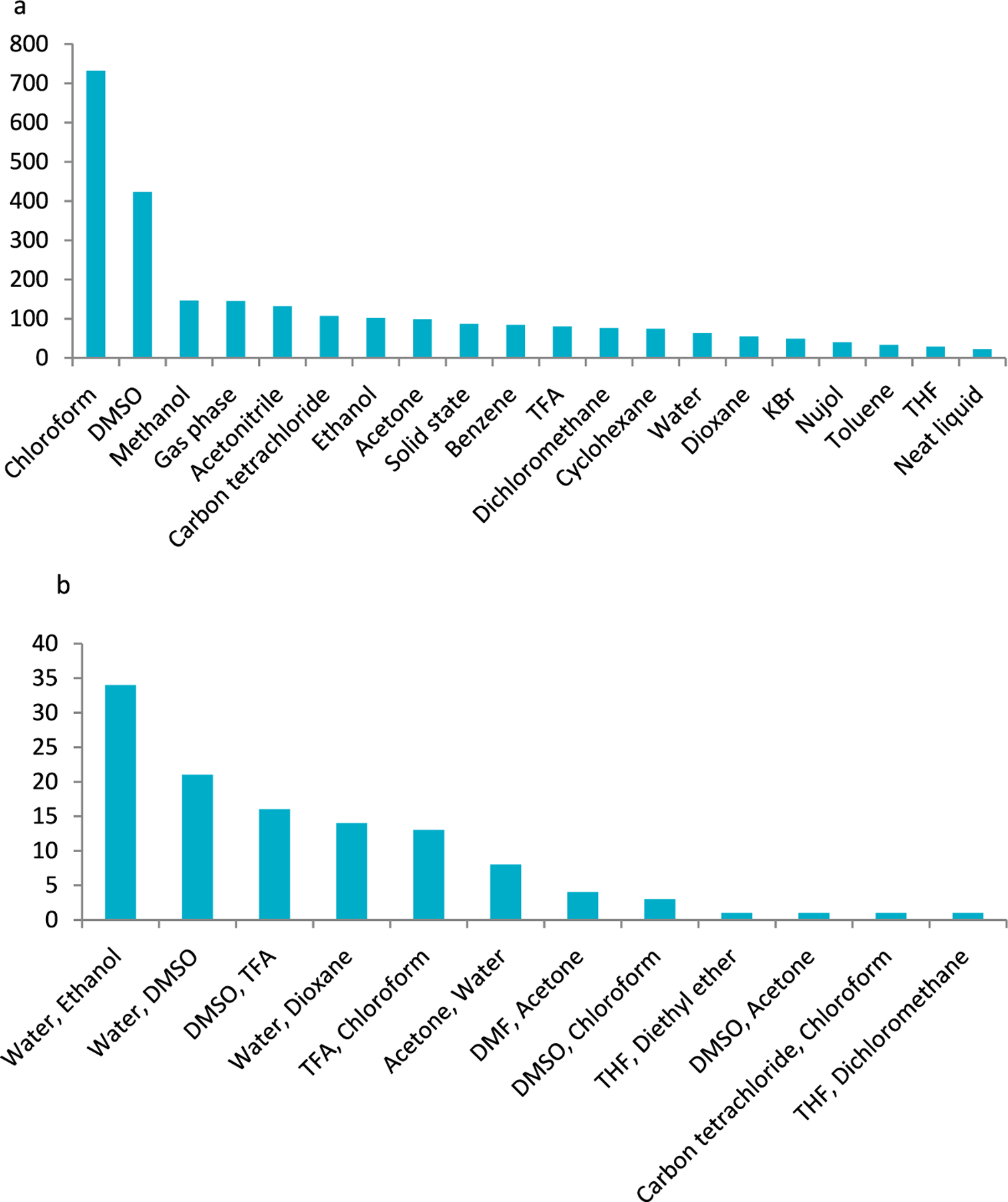
Frequency distribution of most commonly used (a) solvents and (b) solvent mixtures.
Temperature Distribution.
Experimental temperature information was available for 1389 entries in the form of either exact value, range, room (RT), or ambient temperature. About 82% of the studies represented in the database were carried out at a temperature range of 250–350 K (Figure 3). The majority of those (50%) were carried out in the range of 251–300 K. There were only 53 entries below 201 K and 23 entries at a higher temperature (e.g., 523 K).
Figure 3.
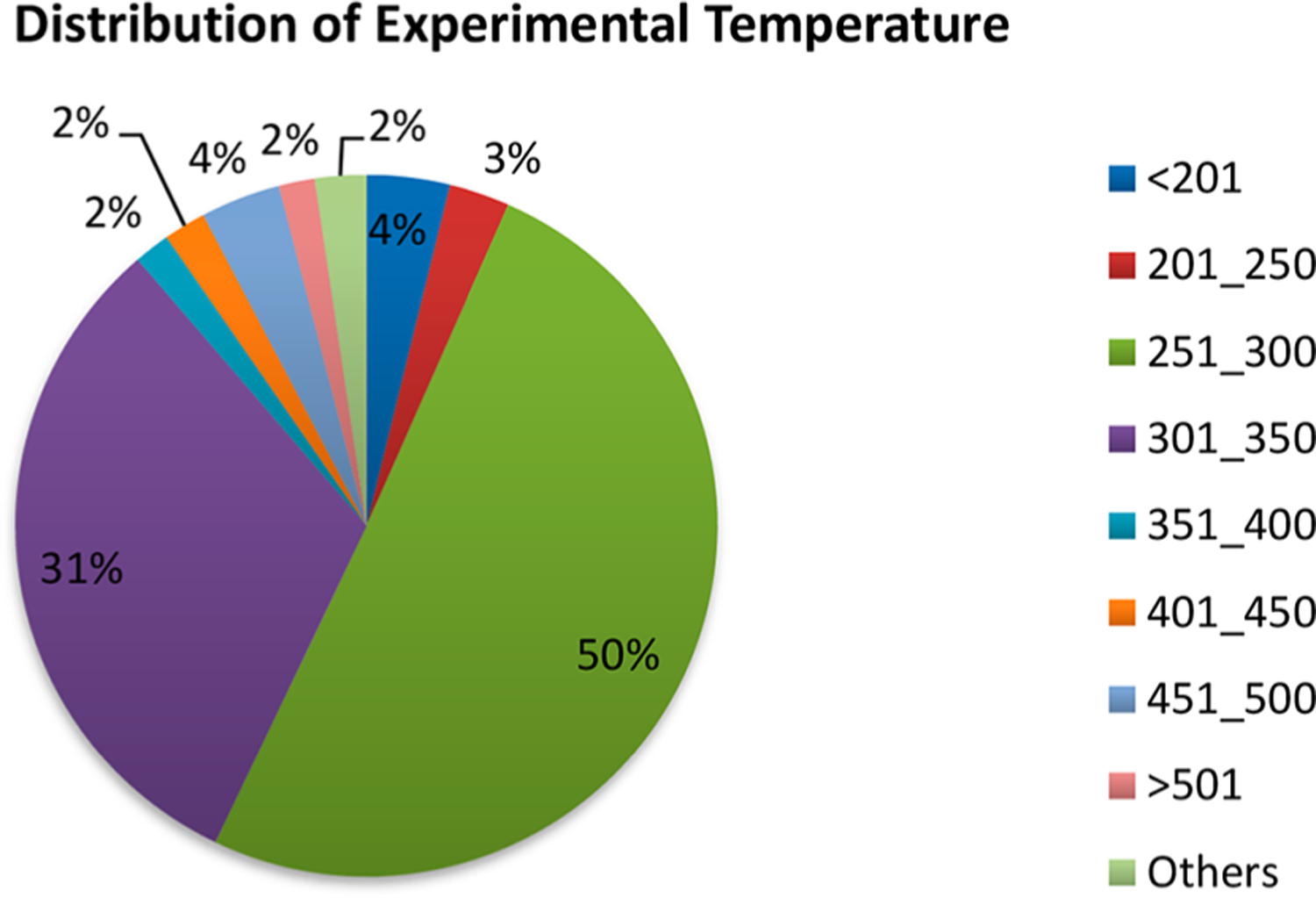
Temperature range distribution of experimental studies. (The range of 251–300 K includes studies that simply reported “room temperature”.)
pH Distribution.
The database has experimental pH details for 100 entries, 63% of which were reported to have used an acidic medium (Figure 4a). For 91 entries of 2-tautomer sets and 9 entries of 3-tautomer sets reported in pH based studies, medium polar to polar solvents (98%) or their mixtures (2%) were used (Figure 4b). These studies used the following spectroscopy methods: 1H NMR, flash photolysis, Raman, UV, and UV/vis. Of these, UV/vis spectroscopy was used in 79% of the cases with methanol, acetonitrile, and DMSO-water.
Figure 4.
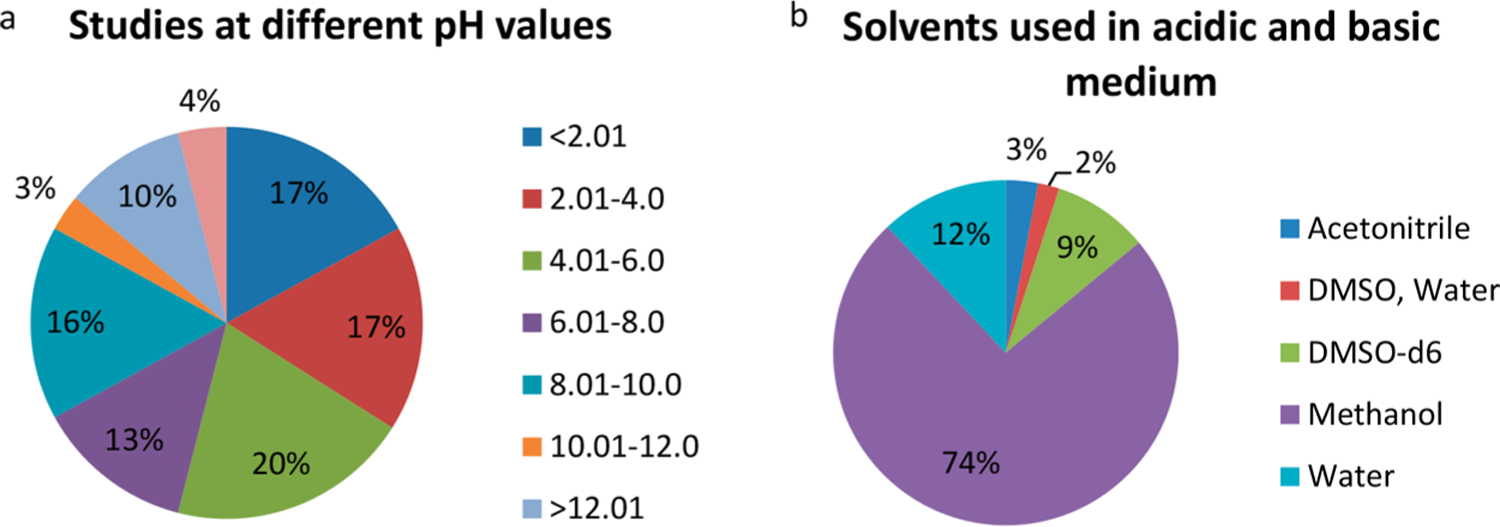
(a) Distribution of studies at different pH ranges. (b) Distribution of different solvents used in pH based studies.
Experimental Methods.
In most of the studies (85%), a single spectroscopy or physical method was used, while in the remainder of the studies two to three methods were used, often by way of an additional method used as support of the primary method. In the multiple method studies, spectroscopic methods from 1H, 13C, 14N, 15N, 17O, and/or 31P NMR spectroscopy were the most common (∼75% of the cases). Out of the total 29 unique methods, 1H NMR (1014), 13C NMR (340), UV (253), IR (172), and UV/vis (139) were the top five methods (Figure 5a). In the multiple method studies (Figure 5b), 1H NMR and 13C NMR were frequently used together (131). In addition, 1H NMR was commonly used together with other methods such as 31P, 15N and 17N NMR, and IR. Some of the spectroscopic methods used different types of solvents; for example, 1H NMR, UV, IR, 31P NMR, and 13C NMR methods were performed in 41, 22, 19, 20, and 14 different solvents and solvent mixtures, respectively. Chloroform and DMSO were the most important solvents in 1H NMR (279 and 227 cases, respectively) and 13C NMR (182 and 84 cases, respectively) (Spreadsheet S2, Supporting Information). In IR, chloroform (59) and nujol (38) were used extensively. In UV/vis, methanol (89) and acetonitrile (8) were used extensively. In UV, ethanol (76) and water (31) were used extensively.
Figure 5.
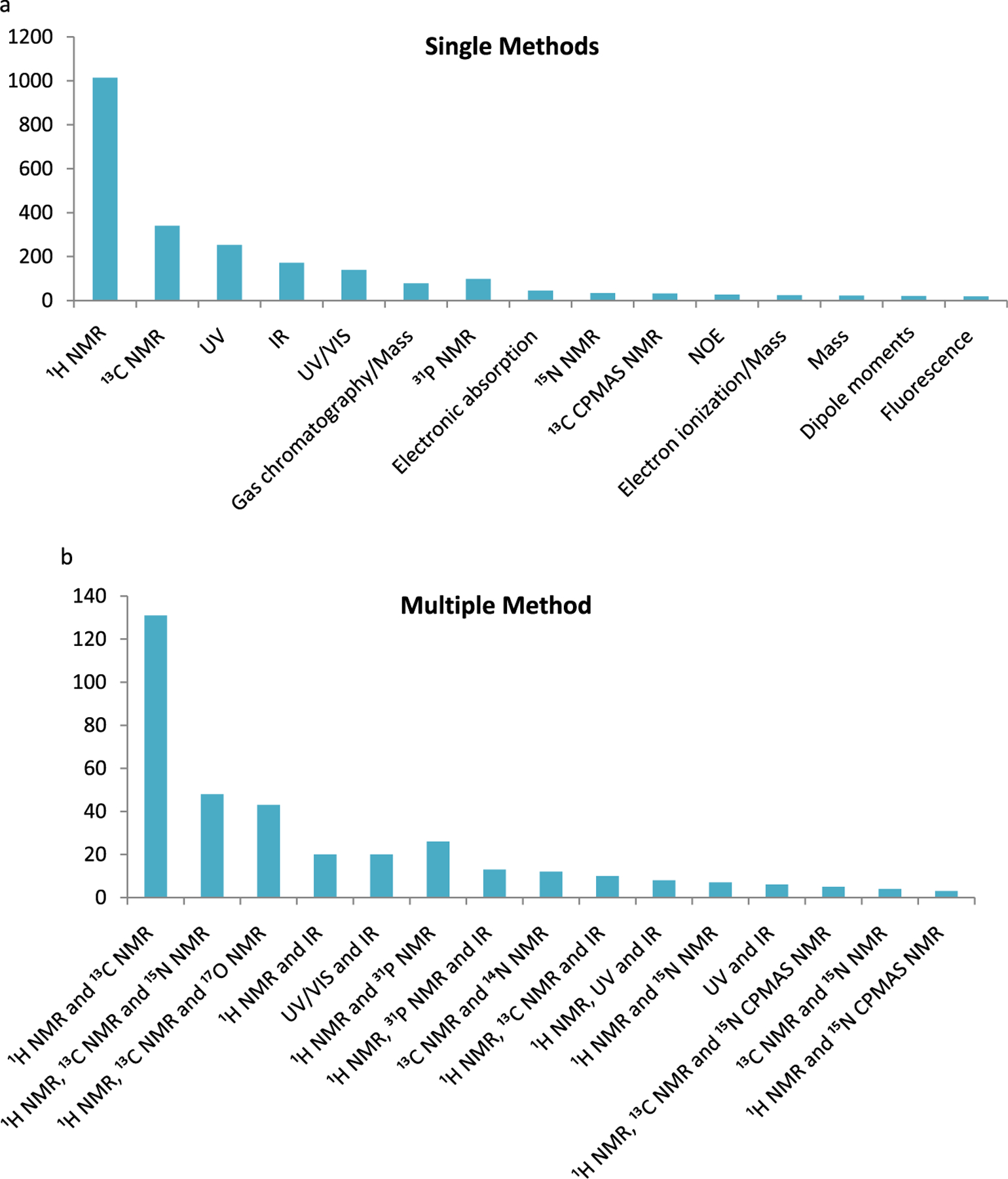
Frequency distribution of (a) single experimental methods and (b) multiple experimental methods (only top 15 methods are shown).
Analysis by Tautomeric Transform Rules.
As already mentioned, we used as the starting point for the tautomeric rule compilation (a) 20 standard prototropic rules (default CACTVS rules PT_02_00 – PT_21_00) and (b) 11 ring⇌ chain (RC_01_00 – RC_11_00) rules (in 38 SMIRKS strings) that have been published by our group recently.1 In addition, we have compiled13 61 new tautomeric rules derived from various literature sources. These new rules consist of 34 prototropic rules (PT_22_00 – PT_49_00) including two variants with mm > 00 and variants of PT_11_mm for long-range hydrogen migration, where mm ranges from 01 to 04, 16 ring⇌chain rules (RC_03_03, RC_03_04, RC_04_04, and RC_12_00 – RC_24_00), and_ 11 valence rules (VT_01_00, VT_01_01 – VT_10_00). (See footnotes of Table 2 for rule naming and numbering nomenclature.)
Table 2.
Frequency Distribution of Prototropic, Ring⇌Chain, and Valence Tautomerism Rules for Single-Rule Transformations and Those with Combined or Alternative Rulesa,b,c
| Standard Rules |
||||
|---|---|---|---|---|
| Type | Rule number | Rule name | Single rule | Combined or alternative rule |
| Prototropic Rules | PT_02_00 | 1,5 (thio)keto/(thio)enol | 0 | 230 |
| PT_03_00 | simple (aliphatic) imine | 0 | 323 | |
| PT_04_00 | special imine | 0 | 127 | |
| PT_05_00 | 1,3 aromatic heteroatom H-shift | 0 | 184 | |
| PT_06_00 | 1,3 heteroatom H-shift | 708 | 891 | |
| PT_07_00 | 1,5 (aromatic) heteroatom H-shift (1) | 391 | 463 | |
| PT_08_00 | 1,5 (aromatic) heteroatom H-shift (2) | 0 | 88 | |
| PT_09_00 | 1,7 (aromatic) heteroatom H-shift | 89 | 256 | |
| PT_10_00 | 1,9 (aromatic) heteroatom H-shift | 0 | 72 | |
| PT_11_00b | 1,11 (aromatic) heteroatom H-shift | 0 | 33 | |
| PT_12_00 | 1,3 furanones | 0 | 84 | |
| PT_16_00 | nitroso/oxime | 0 | 14 | |
| Ring–Chain Rules | RC_03_00 | 5_exo_trig | 0 | 50 |
| RC_03_01 | 5_exo_trig | 0 | 50 | |
| RC_03_02 | 5_exo_trig | 19 | 0 | |
| RC_04_01 | 6_exo_trig | 0 | 49 | |
| RC_04_02 | 6_exo_trig | 0 | 49 | |
| RC_09_00 | 5_endo_trig | 67 | 0 | |
| RC_10_00 | 6_endo_trig | 10 | 15 | |
| RC_10_01 | 6_endo_trig | 29 | 15 | |
| New Rules |
||||
| Type | Rule number | Rule name | Single rule | Combined or alternative rule |
| Prototropic Rules | PT_22_00 | imine/imine | 3 | 0 |
| PT_23_00 | 1,5 furanones | 12 | 0 | |
| PT_24_00 | 1,4 N-oxide/N-hydroxide | 8 | 0 | |
| PT_25_00 | 1,6 N-oxide/N-hydroxide (1) | 4 | 0 | |
| PT_26_00 | 1,6 N-oxide/N-hydroxide (2) | 5 | 0 | |
| PT_27_00 | acene | 13 | 0 | |
| PT_27_01 | thiophene analogue of acene | 15 | ||
| PT_28_00 | nitro/aci-nitro via aromatic ring (1): 1,7 H-shift | 2 | 0 | |
| PT_29_00 | nitro/aci-nitro via aromatic ring (1): 1,5 H-shift | 3 | 0 | |
| PT_29_01 | o-tolualdehyde | 2 | 0 | |
| PT_30_00 | nitramide/N-nitronic acid | 1 | 0 | |
| PT_31_00 | sulfone-based aliphatic compounds | 1 | 0 | |
| PT_32_00 | nitrile/ketenimine: 1,3 H-shift | 8 | 0 | |
| PT_33_00 | nitrile/ketenimine: 1,5 H-shift | 8 | 0 | |
| PT_34_00 | triad phosphorus–carbon | 5 | 0 | |
| PT_35_00 | sulfenyl/sulfinyl: 1,2 H-shift | 2 | 0 | |
| PT_36_00 | oxime/nitrone: 1,2 H-shift | 5 | 0 | |
| PT_37_00 | sulfenyl/S-oxide: 1,4 H-shift | 1 | 0 | |
| PT_38_00 | sila-hemiaminal/silanoic amide | 2 | 0 | |
| PT_39_00 | nitrone/azoxy or Behrend rearrangement | 19 | 0 | |
| PT_40_00 | tetrad phosphorus–carbon | 1 | 0 | |
| PT_41_00 | pyridine 1-oxide/1-hydroxypyridine | 2 | 0 | |
| PT_42_00 | Δ3-/Δ4-pyrro(thio/seleno)lin-2-one | 27 | 0 | |
| PT_43_00 | isobenzofuran/phthalan | 4 | 0 | |
| PT_44_00 | 2-subsituted-pyrrole | 6 | 0 | |
| PT_45_00 | isopropylidenecycloalkane/isopropylcycloalkene | 17 | 0 | |
| PT_46_00 | 4-picoline | 1 | 0 | |
| PT_47_00 | isoindole/isoindolenine | 24 | 0 | |
| PT_48_00 | benzofuranone | 4 | 0 | |
| PT_49_00 | N-hydroxyindole | 6 | 0 | |
| Ring–Chain Rules | RC_03_03 | boronic acid/oxaborole | 19 | 0 |
| RC_03_04 | 5_exo_trig | 15 | 0 | |
| RC_04_04 | 6_exo_trig | 25 | 0 | |
| RC_12_00 | 5_endo_tet or iminophosphorane/benzoxazaphospholine | 39 | 0 | |
| RC_13_00 | 6_endo_dig | 1 | 0 | |
| RC_14_00 | thiadiazoline rearrangement | 9 | 0 | |
| RC_15_00 | 5_exo_trig: 1,4 H-shift | 3 | 0 | |
| RC_16_00 | boryl/borate | 2 | 0 | |
| RC_17_00 | boryl/borate: ion-complex | 2 | 0 | |
| RC_18_00 | 5_exo_tet or hydroxyphosphorane | 4 | 0 | |
| RC_19_00 | nitroolifin/1,2-oxazine N-oxide | 6 | 0 | |
| RC_20_00 | 5_endo_trig: 1,4 H-shift or aminoethyl nitrone/imidazolidin-1-ol | 6 | 0 | |
| RC_21_00 | cyclobutane/enamine | 3 | 0 | |
| RC_22_00 | 5_endo_trig: 1,5 H-shift | 12 | 0 | |
| RC_23_00 | 6_endo_trig: 1,4 H-shift | 1 | 0 | |
| RC_24_00 | λ5-/λ3-phosphane | 2 | 0 | |
| Valence Rules | VT_01_00 | monothio-o-benzoquinone/benzoxathiete | 2 | 0 |
| VT_01_01 | α-dithione/1,2-dithiete | 12 | 0 | |
| VT_02_00 | tetrazole/azide | 84 | 0 | |
| VT_03_00 | isothiocyanate/triazinethione | 8 | 0 | |
| VT_04_00 | tetrazine/azodiazo | 21 | 0 | |
| VT_05_00 | 1,2,3-triazole/diazoamidine | 8 | 0 | |
| VT_06_00 | norcaradiene/cycloheptatriene or benzene-oxide/oxepin | 18 | 0 | |
| VT_07_00 | phospha-münchnones | 11 | 0 | |
| VT_08_00 | 1,2,3,4-tetrazinium/azodiazonium | 15 | 0 | |
| VT_09_00 | diazaphosphazole/phosphinoimine | 25 | 0 | |
| VT_10_00 | phosphine/phosphonium salt | 25 | 0 | |
Different classes of tautomerism are defined by prefixing each rule with PT, RC, or VT for prototropic tautomerism, ring⇌chain tautomerism, and valence tautomerism, respectively. The second placeholder in the rule name between the underscores indicates the rule number in that category (i.e., “02” in PT_02_00), and the last number in the name indicates a variant of that rule (i.e.“01” in VT_01_01, “03” in RC_03_03). A rule ending with “_00” occurs only in one variant for that rule. This naming scheme allows us to add more variants in that rule if it is required in the future.
We also have four variants of PT_11_mm for long-range hydrogen migration, where mm ranges from 01 to 04.
SMIRKS of these tautomeric rules are given in Spreadsheet S3 of the Supporting Information.
Table 2 shows the frequency of the applicability of all these rules to the entries in our database showing both the cases where the transformation between the experimental tautomers only required the application of a single rule as well as of cases that needed additional, or allowed alternative, rules in the single- or multi-step transformation between observed tautomers.
The most commonly encountered prototropic, ring⇌chain, and valence rules are shown in Figure 6. The majority of transformations from our database occur in a single step (60%), while the others involve the use of additional rules to complete the transformation. About 35% of the transformations are achieved by the application of PT_06_00 and PT_07_00 in a single step. Some rules (PT_02_00 to PT_05_00, PT_08_00, PT_10_00 to PT_16_00) appeared only in multi-step transformations or as alternative rules to others. Here, 353 cases needed an additional one step (for a total of two steps), and 27 other cases required two or more steps (for a total of three or more steps) to complete the observed tautomeric transformations.
Figure 6.
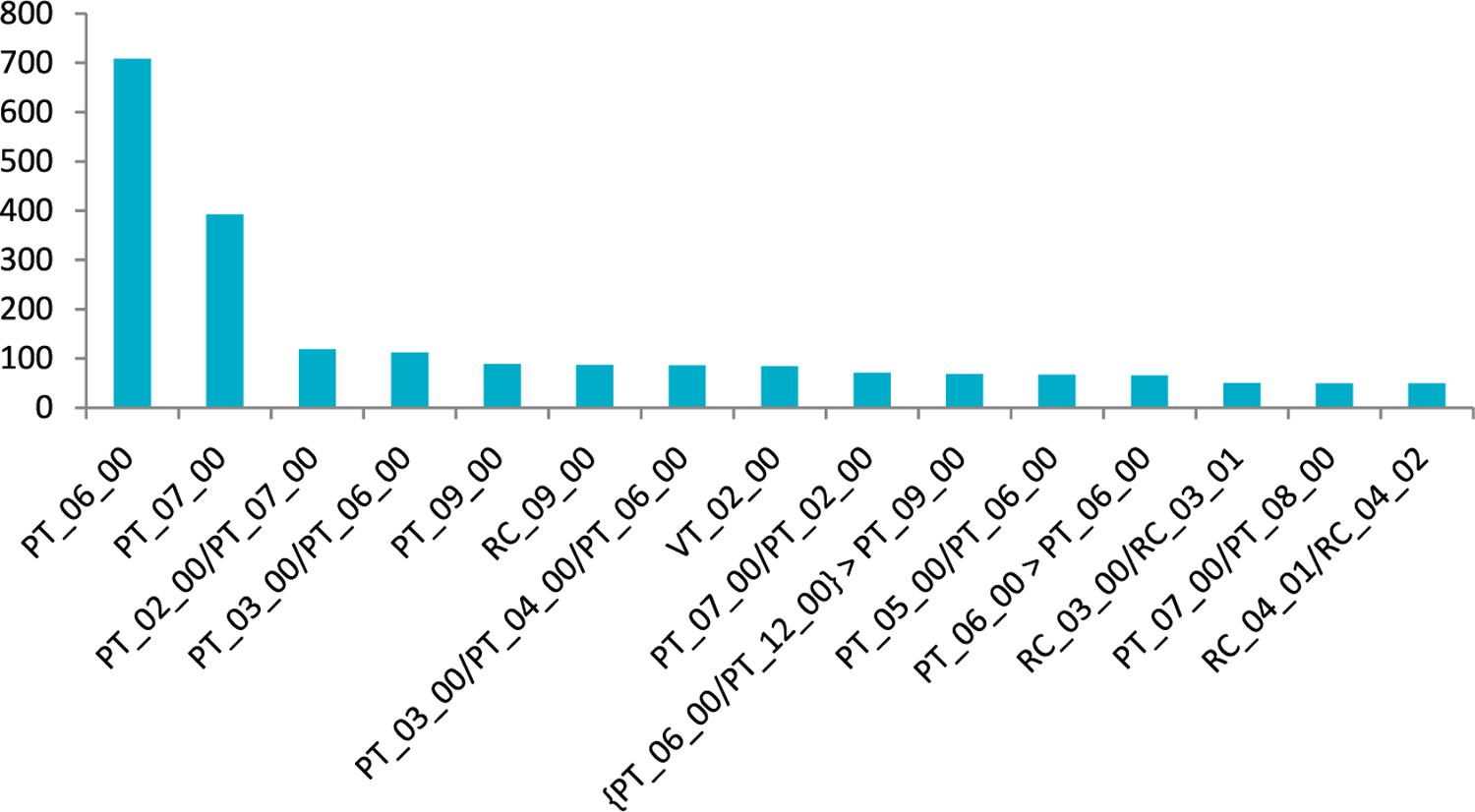
Frequency distribution of commonly observed rules.
Most frequently, a hydrogen atom migrates in a tautomeric transformation from its initial position in the molecule to an odd numbered (relative) position (such as 3, 5, 7, 9, or 11), designated as “1,3 H-shift,” “1,5 H-shift”, etc. Migration to an even position (such as 2, 4, or 6) is rare. In most cases, hydrogen migrated via 1,3 H-shift (1,120), followed by 1,5 H-shift (707) and 1,7 (91) H-shift, respectively, in the single-step transformations. One notes that this distance traveled by the hydrogen is well correlated with the observed frequency of H-shifts. The 1,3 H-shift can alternatively be achieved via long distance migration using 1,5 H-shift (30) or 1,7 H-shift (118), respectively. Likewise, 1,5 H-shift based transformations can be in competition with 1,7 H-shift and 1,9 H-shift in single-step equilibria. For two-step transformations, we observed the order by frequency of occurrence shown in Table 3. We note that, as in single-step transformations, shorter distance hydrogen migrations are more prevalent than longer ones.
Table 3.
Types of Hydrogen Shifts Observed in the One-Step and Two-Step Hydrogen Migrationsa
| One-step hydrogen migrationsa | Count | Two-step hydrogen migrationsb | Count |
|---|---|---|---|
| 1,2 | 7 | 1,3 > 1,3 | 135 |
| 1,3 | 1120 | 1,3 > 1,7 | 91 |
| 1,4 | 12 | 1,5 > 1,5 | 42 |
| 1,5 | 707 | 1,5 > 1,11 | 27 |
| 1,6 | 15 | 1,3 > 1,5 | 5 |
| 1,7 | 91 | 1,5 > 1,7 | 3 |
| 1,3/1,5 | 30 | 1,5 > 1,3 | 2 |
| 1,3/1,7 | 118 | 1,7 > 1,7 | 1 |
| 1,5/1,9 | 15 | Others | 48 |
| 1,5/1,7 | 2 | – | – |
| Others | 609 | – | – |
“/” indicates alternative H-shifts possible for the same trans- formation.
“>” denotes that a first H-shift is followed by a second one to achieve the transformation.
The database contains significantly fewer cases (388) of ring⇌chain tautomerism than of prototropic tautomerism. They generally belong to cyclization to 4-, 5-, and 6-membered ring systems, which can occur either via an endocyclic or exocyclic process where the double bond becomes part of the ring or the side chain, respectively. In 180 cases of endocyclic ring⇌chain transformations, ring closure happened at digonal (sp), trigonal (sp2), or tetrahedral (sp3) centers. The three rules RC_12_00, RC_18_00, and RC_24_00 do not follow the concept of ring closing and ring opening according to Baldwin’s rules. In contrast to other rules, RC_24_00 involves tautomerization between trivalent (chain) and pentavalent (ring) tautomers. There are some rules that involve a 1,2 H-shift (i.e., RC_24_00), 1,4 H-shift (i.e., RC_15_00, RC_20_00 and RC_23_00), and 1,5 H-shift (RC_22_00) during ring closure.
In 193 cases of exocyclic ring⇌chain transformations, the ring closing and opening took place at trigonal or tetrahedral centers. There were some instances of ring⇌chain tautomerism in the thiadiazoline (RC_14_00), boryl/borate (RC_16_00 and RC_17_00), and λ5/λ3-phosphane (RC_24_00) systems that did not involve any unsaturated electrophilic center (or endocyclic or exocyclic bonds) during interconversions but rather involved saturated sulfur, boron, and phosphorus centers, respectively.
The ring–chain rules did not occur in combination with any prototropic or ring⇌chain rule; i.e., in all cases, transformation proceeded in a single step. Generally, ring⇌chain tautomerism showed a high prevalence for the chain form over of the ring form. There were 20 cases of ring⇌chain tautomerism where three tautomers are in equilibrium with each other in solution, the two ring tautomers existing as cis and trans isomers, respectively.
There are 228 cases of valence tautomerism in the database. They all involved ring opening or closing in 4-, 5-, or 6- membered ring systems without migration of any hydrogen atom. The ring-opened tautomers of four rules (VT_02_00, VT_04_00, VT_05_00, and VT_08_00) have a charge-separated moiety in their structures, and this charge disappears in the ring-closed tautomers. In contrast hereto, a charge-separated moiety is present in the ring-closed tautomer of both VT_07_00 and VT_10_00. The tautomeric equilibrium via VT_06_00 involves ring-contraction (6-membered) and ring-expansion (7-membered) in the tautomers. VT_09_00 is the only one rule that involves a valency change during tautomerization: between trivalent phosphinoimine and pentavalent diazaphosphazole tautomers. Among the 11 valence tautomerism rules, our database contains significant counts only for the tetrazole⇌azide tautomerism (VT_02_00), with the tetrazole tautomer being more favored in a polar aprotic solvent and the azide tautomer in the nonpolar solvent.
Type of Tautomerism.
Many of the transforms listed in Table 2 align quite closely with chemotypes the way the organic chemist would usually perceive them. However, others among these transforms, as they are expressed as general SMIRKS patterns,13 cover a broader range of compound types. For example, transform PT_06_00 (1,3 heteroatom H-shift) recognizes C, O, N, S, P, Se, and Te in its SMIRKS pattern, thus covering quite diverse types of compounds and tautomerism based on those. Conversely, the interconversion between the hydrazine and the azo species of a compound can be affected at the transform level by a 1,3 H-shift, 1,5 H-shift, and 1,7 H-shift, which are encoded in different transforms. Table 4 shows the distribution of the records along more than 50 chemical types of tautomerism (see molecular examples in Table S1, Supporting Information, which also contains a more extensive discussion of the tautomer types). Table 5 shows commonly identified sets of three tautomers with their occurrences. Table 6 shows the distribution of some of the common tautomers across the five different prevalence categories described above (0–4).
Table 4.
Types of Tautomeric Equilibria in Tautomeric Pairs with Their Occurrencesd
| Type of tautomerism | Count |
|---|---|
| Azo⇌Hydrazone | 333 |
| Ring⇌Chaina | 318 |
| Enol⇌Keto | 138 |
| Oxo-enamine⇌Oxo-imine | 113 |
| Diketo⇌Keto-enol | 108 |
| Enol-imine⇌Oxo-enamine | 104 |
| Amine⇌Imine | 83 |
| Keto-enethiol⇌Thioketo-enol | 82 |
| Azide⇌Tetrazoleb | 82 |
| nul⇌nulc | 78 |
| Ring⇌Chainb (Valence) | 77 |
| Enamine⇌Imine | 72 |
| Oxo-enamine⇌Phenol-imine | 65 |
| Pyridol⇌Pyridone | 58 |
| NH⇌NH | 57 |
| Phenol-quinone⇌Phenol-quinone | 51 |
| Enol-imine⇌Oxo-imine | 40 |
| Benzoxazaphospholine⇌Iminophosphoranea | 39 |
| CH⇌NH | 35 |
| Keto-enol⇌Keto-enol | 33 |
| NH-imidazole⇌NH-imidazole | 31 |
| Lactam⇌Lactim | 31 |
| Amine-imine⇌Amine-imine | 27 |
| Cyclohexadienone⇌Phenol | 27 |
| 3H-2-one⇌5H-2-one | 27 |
| Enethiol⇌Thioketo | 26 |
| N-hydroxide⇌N-oxide | 25 |
| Diazaphosphazole⇌Phosphinoimineb | 25 |
| Phosphine⇌Phosphonium saltb | 25 |
| IsoindoIe⇌Isoindolenine | 24 |
| 1,4-Dihydro⇌1,6-Dihydro | 19 |
| Nitrone⇌Nitrone | 19 |
| Isopropylcycloalkene⇌Isopropylidenecycloalkane | 17 |
| Thioamide⇌Thioimidol | 16 |
| Keteneimine⇌Nitrile | 16 |
| Tropolone⇌Tropolone | 12 |
| 2H⇌6H | 12 |
| Amide⇌Imidol | 12 |
| Amino⇌Imino | 12 |
| Arene-imine⇌Azepineb | 12 |
| Anaquinoid⇌Paraquinonimine | 10 |
| NH⇌OH | 10 |
| 1,2-Dihydro⇌1,4-Dihydro | 9 |
| Carbamoylimino⇌Guanidinea | 9 |
| Thiol⇌Thione | 9 |
| Nitroso-enamine⇌Oxim-imine | 7 |
| 1,2-Dihydro⇌2,5-Dihydro | 6 |
| Cycloheptatriene⇌Norcaradieneb | 6 |
| Pyrrole⇌Pyrrolidine | 6 |
| 1,4-Dihydro⇌4,6-Dihydro | 5 |
| Nitrone⇌Oxime | 5 |
| Triazole⇌Triazole | 4 |
| 2H⇌4H | 4 |
| Enol-enamine⇌Oxo-enamine | 4 |
| Amino-thieno⇌Imine-thieno | 4 |
| Isobenzofuran⇌Phthalan | 4 |
| CH⇌OH | 3 |
| 1,4-Dihydro⇌4,5-Dihydro | 3 |
| N(1)H⇌N(3)H | 3 |
| Amine⇌Zwitterion | 3 |
| Selenol⇌Selone | 3 |
| Imine⇌Imine | 3 |
| Nitro⇌aci-Nitro | 3 |
| 5,6-Dihydro⇌5,6-Dihydro | 2 |
| 5,6-Dihydro-2H⇌5,6-Dihydro-4H | 2 |
| C1-H⇌C3-H | 2 |
| Thiol⇌Zwitterion | 2 |
| Sulfenyl⇌Sulfinyl | 2 |
| Sila-hemiaminal⇌Silanoic-amide | 2 |
| λ3-Phosphane⇌λ5-Phosphanea | 2 |
| 1H⇌2H | 1 |
| 2H⇌2H | 1 |
| 1,6-Dihydro⇌3,6-Dihydro | 1 |
| 4H⇌6H | 1 |
| Nitroso-imine⇌Oxim-imine | 1 |
| C3-H⇌N(5)H | 1 |
| Oxo-thione⇌nulc | 1 |
| Pyridol⇌Zwitterion | 1 |
| 1H⇌3H | 1 |
| N-nitronic acid⇌Nitramide | 1 |
| Enol⇌Ylide | 1 |
| S-oxide⇌Sulfenyl | 1 |
Ring―chain tautomerism type (total count for ring―chain tautomerism of two tautomers including Benzoxazaphospholine⇌ Iminophosphorane, Carbamoylimino⇌Guanidine, and λ5-Phosphane⇌λ3-Phosphane pairs is 368).
Valence tautomerism type.
“nul” indicates cases of tautomeric equilibria for which no name for one or the other or both tautomers was given in the references, and we were not able to assign any specific name.
Examples for each of these types are given in Table S1 of the Supporting Information.
Table 5.
Types of Tautomeric Equilibria in Three-Tautomer Sets with Their Occurrences
| Type of tautomerism | Count |
|---|---|
| Enethiol⇌Enethiol⇌Thioketo | 51 |
| Enol-imine⇌Oxo-enamine⇌Oxo-imine | 42 |
| Phenol-quinone⇌Phenol-quinone⇌Phenol-quinone | 25 |
| CH⇌NH⇌OH | 21 |
| Chain⇌Ring⇌Ring | 20 |
| 5-Hydroxytriazine⇌Orthoquinonoid⇌Paraquinonoid | 11 |
| Thioamide⇌Thioimidol⇌Thioimidol | 11 |
| nul⇌nul⇌nula | 9 |
| Enol⇌Enol⇌Keto | 6 |
| Enol⇌Keto⇌Keto | 6 |
| Enamine⇌Imine⇌nul | 6 |
| Enamine⇌Enamine⇌Imine | 6 |
| 1,2-Dihydro⇌1,4-Dihydro⇌1,5-Dihydro | 5 |
| 1,7-Dihydro-7-oxo⇌4,7-Dihydro-7-oxo⇌7-Hydroxy | 5 |
| Nitroso-enamine⇌Nitroso-imine⇌Oxim-imine | 4 |
| Enol⇌Keto⇌Zwitterion | 4 |
| Triazole⇌Triazole⇌Triazole | 4 |
| Azo⇌Hydrazone⇌Zwitterion | 3 |
| Diketo⇌Keto-enol⇌Keto-enol | 3 |
| Others | 8 |
See Table 4.
Table 6.
Distribution of Some Common Tautomeric Pairs in Different Prevalence Categories
| Prevalence_Category |
Prevalence_Category |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tautomer_1 | 0 | 1 | 2 | 3 | 4 | Tautomer_2 | 0 | 1 | 2 | 3 | 4 |
| Azo | 39 | 73 | 99 | 79 | 43 | Hydrazone | 54 | 127 | 80 | 38 | 34 |
| Enol | 63 | 35 | 20 | 14 | 5 | Keto | 20 | 34 | 10 | 43 | 30 |
| Oxo-enamine | 9 | 2 | 37 | 44 | 21 | Oxo-imine | 25 | 70 | 7 | 2 | 9 |
| Diketo | 27 | 41 | 9 | 25 | 6 | Keto-enol | 6 | 37 | 22 | 39 | 4 |
| Enol-imine | 6 | 53 | 0 | 29 | 16 | Oxo-enamine | 16 | 29 | 2 | 51 | 6 |
| Keto-enethiol | 0 | 59 | 19 | 3 | 0 | Thioketo-enol | 0 | 4 | 19 | 58 | 0 |
| Amine | 10 | 32 | 5 | 28 | 8 | Imine | 18 | 33 | 5 | 24 | 3 |
| Enamine | 19 | 13 | 26 | 9 | 5 | Imine | 5 | 14 | 26 | 8 | 19 |
| Oxo-enamine | 16 | 15 | 34 | 0 | 0 | Phenol-imine | 0 | 0 | 34 | 15 | 16 |
| Pyridol | 3 | 38 | 8 | 8 | 0 | Pyridone | 2 | 15 | 12 | 27 | 2 |
| Enethiol | 0 | 6 | 3 | 1 | 16 | Thioketo | 18 | 1 | 4 | 3 | 0 |
| Enol-imine | 6 | 53 | 0 | 29 | 16 | Oxo-enamine | 16 | 29 | 2 | 51 | 6 |
| Lactam | 10 | 1 | 4 | 15 | 1 | Lactim | 4 | 11 | 5 | 9 | 1 |
| 5H-2-one | 2 | 4 | 2 | 13 | 6 | 3H-2-one | 6 | 13 | 2 | 4 | 2 |
| Cyclohexadienone | 9 | 1 | 5 | 4 | 8 | Phenol | 9 | 3 | 5 | 0 | 10 |
| Isoindole | 1 | 8 | 5 | 10 | 0 | Isoindolenine | 0 | 10 | 5 | 8 | 1 |
| N-hydroxide | 1 | 3 | 5 | 9 | 1 | N-oxide | 1 | 10 | 5 | 2 | 1 |
| Keteneimine | 5 | 11 | 0 | 0 | 0 | Nitrile | 0 | 6 | 0 | 9 | 1 |
| Ringa | 28 | 125 | 114 | 64 | 7 | Chaina | 11 | 65 | 171 | 65 | 26 |
| Benzoxazaphospholinea | 0 | 11 | 17 | 10 | 1 | Iminophosphoranea | 1 | 10 | 17 | 11 | 0 |
| Diazaphosphazoleb | 2 | 4 | 1 | 17 | 1 | Phosphinoimineb | 1 | 17 | 1 | 4 | 2 |
| Phosphineb | 3 | 9 | 5 | 8 | 0 | phosphonium saltb | 4 | 4 | 5 | 12 | 0 |
| Ringb | 6 | 22 | 8 | 26 | 15 | Chainb | 18 | 28 | 8 | 17 | 6 |
| Tetrazoleb | 5 | 29 | 8 | 35 | 5 | Azideb | 4 | 70 | 1 | 3 | 4 |
Ring-chain tautomerism type.
Valence tautomerism type.
SUMMARY AND CONCLUSIONS
A significant variety of structures, chemotypes, analytical procedures, and experimental conditions including solvents has been compiled to form the Tautomer Database. We hope that this database of experimental data and its included analysis by chemoinformatics methods (by way of annotation with tautomeric transform rules) may provide a set of data useful for future work in the field of tautomerism. This would include tools such as software and chemical identifiers that could be used to avoid tautomeric duplication in chemical databases and compound registration systems. We also hope it may help in developing approaches to predict the most “medicinally” relevant and “reasonable” tautomer forms. This data set could be a useful training set for machine learning models based on quantum mechanics15,16 to rapidly identify the lowest energy tautomer.
Supplementary Material
ACKNOWLEDGMENTS
We have to send copious thanks to Wolf-Dietrich Ihlenfeldt for his initial work with CACTVS and its treatment of tautomerism, as well as for his support in our generating and testing the new rules. We gratefully acknowledge Thomas Sander and Oya Wahl for providing us with a copy of their Tautomer Codex database, which helped in the generation of a handful of additional rules. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute. All authors received funding from the NCI, NIH, Intramural Research Program. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.9b01156.
Spreadsheet S1: List of the publications used in tautomer database generation (XLSX)
Spreadsheet S2: Distribution of solvents or their mixtures, or general experimental environments, by spectroscopic methods (XLSX)
Spreadsheet S3: SMIRKS of tautomeric rule (XLSX) Representative examples of chemical types of tautomerism (Table S1) (PDF)
Spreadsheet S4: Tautomer database_itself (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.9b01156
The authors declare no competing financial interest.
REFERENCES
- (1).Guasch L; Sitzmann M; Nicklaus MC Enumeration of Ring–Chain Tautomers Based on SMIRKS Rules. J. Chem. Inf. Model 2014, 54 (9), 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Martin YC Let’s Not Forget Tautomers. J. Comput.-Aided Mol. Des 2009, 23 (10), 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Guasch L; Yapamudiyansel W; Peach ML; Kelley JA; Barchi JJ; Nicklaus MC Experimental and Chemoinformatics Study of Tautomerism in a Database of Commercially Available Screening Samples. J. Chem. Inf. Model 2016, 56 (11), 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Masand VH; Mahajan DT; Gramatica P; Barlow J Tautomerism and Multiple Modelling Enhance the Efficacy of QSAR: Antimalarial Activity of Phosphoramidate and Phosphorothioamidate Analogues of Amiprophos Methyl. Med. Chem. Res 2014, 23 (11), 4825–4835. [Google Scholar]
- (5).Milletti F; Vulpetti A Tautomer Preference in PDB Complexes and Its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model 2010, 50 (6), 1062–1074. [DOI] [PubMed] [Google Scholar]
- (6).Kalliokoski T; Salo HS; Lahtela-Kakkonen M; Poso A The Effect of Ligand-Based Tautomer and Protomer Prediction on Structure-Based Virtual Screening. J. Chem. Inf. Model 2009, 49 (12), 2742–2748. [DOI] [PubMed] [Google Scholar]
- (7).Oellien F; Cramer J; Beyer C; Ihlenfeldt W-D; Selzer PM The Impact of Tautomer Forms on Pharmacophore-Based Virtual Screening †. J. Chem. Inf. Model 2006, 46 (6), 2342–2354. [DOI] [PubMed] [Google Scholar]
- (8).Gimadiev TR; Madzhidov TI; Nugmanov RI; Baskin II; Antipin IS; Varnek A Assessment of Tautomer Distribution Using the Condensed Reaction Graph Approach. J. Comput.-Aided Mol. Des 2018, 32 (3), 401–414. [DOI] [PubMed] [Google Scholar]
- (9).Wahl O; Sander T Tautobase: An Open Tautomer Database. J. Chem. Inf. Model 2020, DOI: 10.1021/acs.jcim.0c00035. [DOI] [PubMed]
- (10).Sitzmann M; Filippov IV; Nicklaus MC Internet Resources Integrating Many Small-Molecule Databases 1. SAR QSAR Environ. Res 2008, 19 (1–2), 1–9. [DOI] [PubMed] [Google Scholar]
- (11).Xemistry Chemoinformatics https://www.xemistry.com/ (accessed29–01–2020).
- (12).IUPAC projects https://iupac.org/projects/project-details/?project_nr=2012-023-2-800 (accessed29–01–2020).
- (13).Dhaked DK; Ihlenfeldt W-D; Patel H; Delanneé V; Nicklaus MC Toward a Comprehensive Treatment of Tautomerism in Chemoinformatics Including InChI V2. J. Chem. Inf. Model 2020, DOI: 10.1021/acs.jcim.9b01080. [DOI] [PMC free article] [PubMed]
- (14).Daylight Theory Manual https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed29–01–2020).
- (15).Smith JS; Isayev O; Roitberg AE ANI-1: An Extensible Neural Network Potential with DFT Accuracy at Force Field Computational Cost. Chem. Sci 2017, 8 (4), 3192–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Smith JS; Isayev O; Roitberg AE ANI-1, A Data Set of 20 Million Calculated off-Equilibrium Conformations for Organic Molecules. Sci. Data 2017, DOI: 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.