

Abstract
The SARS-CoV-2 that caused Covid-19 has spread since the end of 2019. Its major effects resulted in over 4 million deaths around the whole world by August 2021. Therefore, understanding virulence mechanisms is important to prevent future outbreaks and for COVID-19 drug development. The envelope protein is an important structural protein affecting virus assembly and budding. The E protein pentamer is a viroporin, serving as ions transferring channel in cells. In this work, we applied MD simulations, topological and electrostatic analyses to study the effects of palmitoylation on the E protein pentamer. The results indicate the cation transferring direction is more like lumen to the cytosol. The structure of palmitoylated E protein pentamer is more stable while the loss of palmitoylation caused the pore radius to reduce and even collapse. The electrostatic forces on the two sides of palmitoylated E protein pentamer are more beneficial to attract cations in the lumen and to release cations into the cytosol. The results indicate the importance of palmitoylation, which can help the drug design for the treatment of COVID-19.
Keywords: SARS-CoV-2, envelope protein, viroporin, electrostatic calculation, Delphi
Graphical Abstract
1. Introduction
Coronaviruses (CoVs) are notorious as the pathogens of numerous diseases in a wide range of vertebrates, including human beings. In 2002, the SARS-CoV-related severe acute respiratory syndrome (SARS) took 4 months to overwhelm 29 countries and killed 774 reported patients. It almost paralyzed the Asian economy1. After 10 years, the outbreak of a new coronavirus, named the Middle East Respiratory Syndrome coronavirus (MERS-CoV) spread in 27 countries, causing a 35% death rate1b,2. In December 2019, a novel coronavirus named Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) was discovered and now has already spread across the whole world. The disease caused by it is known as coronavirus disease 2019 (COVID-19)3. So far, the disease has infected 106 million people and caused over 4 million deaths in the whole world4. In addition to severe pneumonia, the SARS-CoV-2 can also cause multi-organ damages, including cardiovascular disease5, reproductive risk6, mental illness7, and smell disfunction8. Thus, it is urgent to understand the virulence mechanisms to prevent future outbreaks and to develop the remedies for COVID-19.
The SARS-CoV-2 is a positive RNA strand virus that origins from the family Coronaviridae3a, 9. The genome of coronavirus encodes four major structural proteins, which are the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and envelope (E) protein10. The S protein mainly regulates the binding with the ACE2 (Angiotensin-Converting Enzyme) receptor of cells11. The N protein and the M protein serve as RNA genome binding protein12 and the most abundant structural protein of viral envelope, respectively13. The E protein is the smallest among four major structural proteins. Other than the S, N, and M proteins, the E protein is abundantly expressed in the infected cells but only a small percentage is assembled into viral envelope14. The majority of the protein is located at the ER (endoplasmic reticulum), Golgi complex, and ERGIC (ER-Golgi intermediate compartment)15. The E protein is related to intracellular trafficking and participates in the viral assembly and budding14, 16. The recombined CoVs which lack E protein express crippled viral maturation and incompetent progeny17. The study about the E protein is important to understand virulence mechanisms.
The SARS-CoV E protein contains a short hydrophilic terminal, a large hydrophobic transmembrane domain (TMD), and a long hydrophilic carboxyl end (Figure S2)18. Synthetic peptides of TMD of E protein can form dimers, trimers, and pentamers19, among which the pentametric structure is widely accepted and studied as viroporin13a, 14-15. Similarly, the TMD of SARS-CoV-2 E protein was also found to form a five-helix buddle surrounding a narrow pore19. The narrow pore is suggested as viroporin to regulate multiple stages of viral life circles. Viroporins can transport different ions but usually prefer cations (H+, K+, Na+, and Ca2+)20. As reported, the cell infected by coronavirus exhibited a remarkable increase of pH in the Golgi complex. The increased pH protects spike protein and promotes the release of the virus from the cell16. This finding supports that the E protein pentamer of coronavirus could be an H+ channel, transferring H+ from lumen to cytosol. The higher pH in the lumen may help the release of the virus. To modulate protein functions, Post-Translational Modifications (PTMs) are necessary. The PTMs of the E protein of coronavirus include glycosylation and palmitoylation (Palm)13a, 21. N-glycosylation, as reported, appears in a minor form of SARS-CoV E protein, which has both C- and N- terminals exposed on the luminal side22. Nevertheless, the E protein with both C- and N- terminal on the cytoplasmic side is not modified by glycosylation. By contrast, all three cysteine residues (C40, C43, C44) in the SARS-CoV E protein are modified by palmitoylation23. The mutants of these cysteines in E protein on mouse hepatitis virus A59 (MHV-A59) weakens the ability to form virus-like particles24. Additionally, MHV was prone to degradation when carrying triple mutants24a. Additionally, The palmitoylated protein with the higher hydrophobicity benefits membrane anchoring and association25. The studies on human coronavirus also proved the poor localization of non-palmitoylated E protein in membranes, The studies on human coronavirus also proved the poor localization of non-palmitoylated E protein in membranes, while the palmitoylated E proteins are not affected 24a. These findings proved the importance of palmitoylation on the E protein.
To better understand how palmitoylation affects the function of coronavirus E protein, SARS-CoV-2 E protein pentameric structure was used as the reference (E protein without palmitoylation). The palmitoylation was added on the cysteine residues 40, 43, and 44 to each monomer as the palmitoylated E protein pentamer (E protein with palmitoylation). Afterward, Molecular Dynamic (MD) simulations were run to observe the effects of palmitoylation. Besides, the topological and electrostatic studies were applied to analyze the function of palmitoylation. In the end, we simulated several layers of H+ ions on two sides to detect the differences in the force on cations. These studies were summarized in the conclusion section to discuss the effects of palmitoylation on the function of passing ions on the E protein pentamer.
2. Method
2.1. Modeling and simulation
2.1.1. Modeling
The sequence of E protein monomer of SARS-CoV-2 from 2019 was used as the query sequence (RefSeq: YP_009724392.1) (Figure S1). The 3-dimensional pentameric structure of the E protein of the SARS-CoV-2 structure was based on the pentameric structure of the SARS-CoV E protein of the PDB 5X2926 and was achieved by Robetta server27. Figure S1 shows the sequence and structural alignment of SARS-CoV-2 and SARS-CoV. The sequence similarity is 96 %. The modeled pentameric structure was used as the initial configuration (E protein without palmitoylation) for the MD simulation. The palmitoylation was attached to the cysteine residues 40, 43, and 44 to each monomer (Figure 1) as the E protein pentamer with palmitoylation. The palmitate groups were added using CHARMM-GUI28 by CHARMM36 force field29.
Figure 1:

The diagram of E protein pentamer in the membrane. The C-terminal of E protein pentamer is on the top, cytosolic side, and the N-terminal of E protein pentamer is on the bottom, luminal side. The membrane lipid is represented in grey lines. The E proteins are represented in the carton (blue). The palmitate groups (palmitoylation) are colored purple.
2.1.2. Simulation
The CHARMM-GUI28 webserver was used to create the simulating system (Figure 1). The two structures were then embedded in the membrane dioleoyl phosphatidylcholine (DOPC) around the center along the Z-axis. The DOPC is used widely in experiments and close to the membrane where E protein was found30. The membrane is a square with a side length of 150 Å long along the x and y-axis. The system was solvated with water of type TIP3P31 of thickness 15 Å on either side of the membrane. The NaCl was used to ionize the system with a concentration of 150 mM. Parameterization of the atoms in the system was attained with the CHARMM36 force field29. Periodic boundary condition was applied to the simulating box and the Particle Mesh Ewald (PME)32 was used for the long-range electrostatic interactions. The final system was then subjected to the MD simulation with NAMD 2.1233. The whole simulations include two steps. The first is equilibration and the second is production run. NPT was used for the equilibration. The temperature was set to 300 K and the pressure was set to 1 atm using a Langevin thermostat with a damping coefficient of 1/ps and a Nosé–Hoover Langevin piston barostat with a decay period of 25 fs. The temperature was reassigned every 500 steps. During the equilibration, the constraint was applied to the entire E protein pentamer with palmitoylation and the headgroup of the DOPC lipid. In the production run, NPT ensemble was continued for 100 ns. The constraints on E-proteins and headgroups of DOPC membrane were released during the production run. For nonbonded interactions (electrostatic and van der Waal), the cutoff was set to 12 Å. The switching distance was set to 10 Å. In the production run, the NPT ensemble was continued for the production run (100 ns). Constraints on E-proteins and headgroups of DOPC membrane were released during the production run. E protein pentamers with and without palmitoylation simulations were repeated two more times. The analyses were based on the production runs.
2.2. Topological study
As shown in Figure S1 and S2, the transmembrane domain (TMD) of E protein pentamer is marked from 17V to 37L (21 residues) in five monomers for RMSF (Root-Mean-Square Fluctuations) analysis based on the stable state (60 ns −100 ns). The diagram (Figure 1) showed that the cytosolic side (C-terminal) is on the top while the luminal side (N-terminal) is on the bottom13a.
2.2.1. RMSF and RMSD
The RMSF of the α-carbons of the residues (21 residues) on the TMD is achieved based on the last 40 ns simulations by Visual Molecular Dynamics (VMD)34 (Equation 1).
| (1) |
Where represents the residue ID, the T represents the total simulation time (Here is the number of frames), represents the residues in the time of position. The is the reference position of residue , calculated by the time-average position.
The RMSD (Root-Mean-Square Deviation) is to measure the average distance between two protein structures, calculated by equation 234
| (2) |
Where , is the weighting factor, and N is the total number of atoms. The is the position of atom i at time t after least square fitting the structure to the reference structure. The is the reference position of residue i defined by the reference structure (Here we used the initial structure as the reference).
2.2.2. General comparison of the projections of the E protein pentamer’s top and bottom views
The E protein pentamers with and without palmitoylation were compared to show the conformational differences in both top and bottom views. The structures were from the last frame (at 100 ns) of the simulation.
2.2.3. Minimal pore radius and pore volume testing
The pore radius is tested by HOLE236 for the E protein pentamer with and without palmitoylation in 60 - 100 ns simulations. The minimal pore radius is the minimum radius of the ion channel formed by the E protein pentamer transmembrane domain. To avoid the spherical probe ball running in the wrong direction, the moving direction is restrained to follow the Z-axis36. The pore radius was tested three times for a single frame and the maximum value was taken as the minimal pore radius. It is because of the existent of the gradient when HOLE increases the probe size. To better understand the effect of palmitoylation on channel size, the TMD was split into 3 parts, including the C-terminal part (residue 30-39), middle part (residue 20-30), and N-terminal part (residue 11-20) to test the minimal radius separately. The volume is calculated by the integration through the pore.
2.2.4. Electrostatic potential study
The structures of palmitoylated and non-palmitoylated E protein pentamer were from the last frame (at 100 ns) of the simulations. The electrostatic potential calculations of E protein pentamer were calculated by Delphi37. The charge and the radius of atoms were calculated by the force field CHARMM36 and assigned by pdb2pqr38. The dielectric constants were set as 2 for proteins and memebrane39, while 80 for water. The salt concentration was set as 150 mM, and the probe radius was set as 1.4 Å. The protein filling percentage was set as 70% and the resolution was set as 1.5 grids/Å. The electrostatic potentials on the surfaces were visualized by Chimera40 for both cytosolic (top view) and luminal sides (bottom view). The color range was set from −1.0 kT/e (red) to 1.0 kT/e (blue). The potential surfaces were visualized by Chimera40 and the electric field lines were visualized by VMD34 to demonstrate the interactions.
2.2.5. Principal component analysis
The mass centers of the transmembrane domain in 60 ns – 100 ns (1000 frames) were selected for Principal component analysis (PCA). The X, Y, Z of the mass center were treated as variables to performance PCA. The analyses were applied separately for non-palmitoylated E protein and palmitoylated E protein.
2.2.6. Electrostatic force study
To quantitively demonstrate the electrostatic force on the cations along with the distance with membrane, H+ ions were selected. On both cytosolic and luminal sides, ten spherical layers of H+ ions were simulated (Figure S3). The nearest layer is 1 Å to the membrane while the farthest is 10 Å. For each layer, there are 288 H+ ions, the center is on the Z-axis, which passed the mass center of the transmembrane domain. In each layer, the radiuses of H+ circles are from 3 Å to 10 Å. In a certain circle, for every 10°, a hydrogen ion is placed. The electrostatic force on H+ was split along the Z-axis (Z component), to demonstrate the component force of the direction. The Z component force direction was set from the bottom (luminal side) to the top (cytosolic side). The electrostatic force was calculated by DelphiForce41 and the parameters were the same as that of potential surface calculations.
3. Result and discussion
3.1. Topological study
From the RMSD study of the transmembrane domain (Figure S4), both systems have achieved stability after 10 ns simulation. The RMSD of the repeated simulations for both E protein pentamer with and without palmitoylation are also reached stable after 40 ns. The repeated simulations also show that the E protein pentamer reached stable after 40 ns. The structures in Figure 2 were the E protein pentamers extracted from the last frame (at 100 ns) of the MD simulations of both systems. The structures were aligned by Chimera40 and the two sides of the ion channel were compared from the top and bottom views. From the general comparison, the palmitoylated E protein pentamer stabilizes the pentametric structure in the larger pore size on the cytosolic sides while the loss of palmitoylation reduces the pore radius in the cytosolic side and reshapes the structure into an irregular conformation. By contrast, there is no significant difference in the bottom view. In Figure S6, we also compared the structures in the two repeated simulations on the top view (structures are from the frame at 100ns). The comparison shows the same result. To further investigate the effects of palmitoylation on the whole ion channel during simulations, the HOLE236 program was applied to test the minimal pore radius of the C-terminal part, middle apart, and the N-terminal part of the ion channel.
Figure 2:

The general comparison of the E protein pentamer on the top and bottom view. The non-palmitoylated E protein pentamer is red colored while the palmitoylated E protein pentamer is colored blue. The palmitate groups are colored purple.
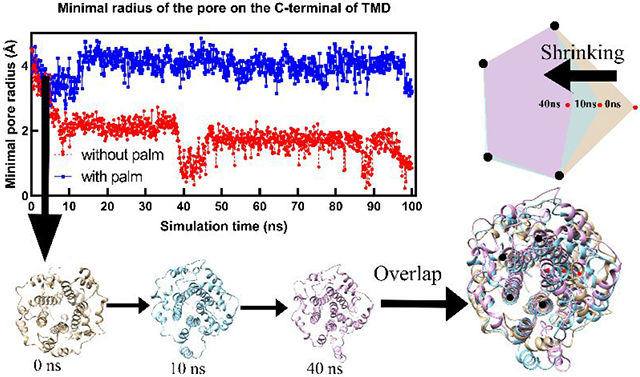
As shown in Figure 3 (A), in equilibrium state (60 ns – 100 ns), the C-terminal part of the palmitoylated E protein pentamer exhibits a minimal pore radius of 3.96 Å (standard deviation: 0.31 Å ). by contrast, that in non-palmitoylated E protein pentamer only has a 1.56 Å minimal pore radius (standard deviation: 0.35 Å). The loss of palmitoylation directly reduces the pore radius. Without palmitoylation, the collapses of the ion channel happened twice at 10 ns and 40 ns, respectively. The structures (Figure 3B) were extracted from the simulation at 0 ns, 10 ns, and 40 ns for structural comparison (Figure 3C). The diagram (Figure 3D) clearly shows one of the helix monomers' movements (red dots) during the simulation, causing the pore radius to shrink. The volume calculated by the C-terminal part of TMD also shows that the E protein pentamer with palmitoylation possesses a higher capacity for ion transfer while the loss of palmitoylation makes the capacity of E protein pentamer at a lower level (Figure S8). Besides, palmitoylation at C-terminal also affects the middle part of TMD (Figure S8). The minimal pore radius in the middle part of TMD of palmitoylated E protein pentamer is larger than that without palmitoylation. While in the N-terminal part (Figure S9), the palmitoylation does not affect. This finding is consistent with the general comparison, which shows the higher area in the top view for palmitoylated E protein pentamer but no significant difference in the bottom view.
Figure 3:

The minimal pore radius of the E-protein pentamer in the C-terminal during the simulation (A) and the structure comparison of E protein pentamer without palmitoylation in 0 ns, 10 ns, and 40 ns (B, C, and D).
To investigate the reason for the collapses, the salt bridges and RMSF were calculated for the E protein pentamers. Interestingly, the salt bridge in both E protein pentamer with and without palmitoylation is the same one (ARG61 chain D-ASP72 chain E), The occupancy of that in E protein pentamer with palmitoylation is close to 1 while that in E protein without palmitoylation is around 0.4 (Figure 4). The distance of ARG61 (chain D)-ASP72(chain E) is over 3 Å, revealing the salt bridge belongs to the weak electrostatic binding. Due to the salt bridge threshold of 4 Å, the residue pairs of ARG61-ASP72 in other adjacent chains are not displayed. We further extend the threshold to calculate the distance of ARG61(chain A)-ASP72(chain B) (Figure S12). The result shows the ARG61(chain A)-ASP72(chain B) in the palmitoylated E protein pentamer possess lower distance of 6 Å while that in non-palmitoylated E protein is over 10 Å. It means the structure of E protein pentamer without palmitoylation is unstable compared with the E protein pentamer with palmitoylation. The RMSF study also shows the α-Carbons in the TMD residues of the non-palmitoylated E protein pentameric structure are higher than that in palmitoylated E protein pentamer (Figure S11). Both results reveal the instability of the non-palmitoylated E protein pentamer. The loss of the palmitoylation caused an unstable pentametric structure, leading to the narrow pore radius and collapses of the ion channel. In previous studies, the palmitoylation of the E protein pentamer has also been shown with high importance in murine coronavirus assembling24b. The related palmitoylated residues have also been studied in the previous works24a. However, they did not apply the mechanism studies. Unlike previous works, this work focused on the mechanism of how palmitoylation affects the E protein pentamer in computational studies. The function of palmitoylation in stabilizing proteins has also been found in Rous Sarcoma Virus (RSV) transmembrane glycoprotein42, CCR5 Receptor43, and TEAD proteins44. The mechanism found in this work may be also related to the other palmitoylated transmembrane proteins.
Figure 4:

The number of salt bridges of E-protein pentamer in adjacent monomers with and without Palm during 60 ns – 100 ns simulations (A) and the sole salt bridge (ARG61-ASP72) in both E protein pentamer with and without palmitoylation(B).
3.2. Electrostatic study
As shown in Figure 5, the top side (cytosolic side) of the electrostatic surface of the E protein pentamer is positively charged while the bottom side (luminal side) of that is negatively charged. This configuration provides the electrostatic forces acting on cations in two sides are from lumen to cytosol, which is suitable for attracting cation on the luminal side and releasing cation on the cytosolic side. Combined with cation preference20e for viroporin, if the cation is H+ ions, the release of H+ from the lumen to the cytoplasm by E protein pentamer would cause higher pH in the lumen. The result is consistent with the previous finding the loss of E protein pentamer causes the high pH in the Golgi complex16. From the top view, the center of the E protein is positively charged (Figure 5). The palmitoylation on the cytosolic side increases the positively charged area. The enlarged area strengthens the repulsive force on the cation on the top side. By contrast, even the luminal side (Figure 5 bottom view) of E protein pentamer is not palmitoylated, the surface of that does not show significant differences. The electrostatic surface and electric field lines of the membrane with the E protein pentamer were visualized in Figure 6. From the top view, the positively charged area is extended by palmitoylation. It is consistent with the results of the E protein pentamer in Figure 5. For the bottom view of the membranes, the E protein pentamers with and without palmitoylation show no significant differences in the electrostatic potential at the surfaces. However, the electric field lines in the E protein pentamer with palmitoylation show intensive interactions with the surrounding membrane. Compared with findings in Figure 6, the palmitoylation on the top can enhance the interactions on the bottom.
Figure 5:

The electrostatic surface of E-protein pentamer with and without palmitoylation in the top and bottom view. The color range was set from −1.0 kT/e (red) to 1.0 kT/e (blue).
Figure 6:

The electrostatic surfaces and electric field lines of membrane/E-protein with and without palmitoylation. The membrane structure (with E protein pentamer) is represented in the first column, the electrostatic surfaces are represented in the second column, and the filed lines are presented in the third column. The color range was set from −1.0 kT/e (red) to 1.0 kT/e (blue).
3.3. The principal component analysis
In Figure 6 (first column), the clear movement of the palmitoylated E protein pentamer is observed. So, the principal component analysis was applied on both palmitoylated E protein and non-palmitoylated E protein pentamer separately. The results (Figure S13) show that in 4 stages (stage1: 60 ns – 70 ns; stage 2: 70 ns – 80 ns; stage 3: 80 ns – 90 ns; stage 4: 90 ns – 100 ns), the center of the non-palmitoylated E protein pentamer moved based on one center while the palmitoylated E protein pentamer shows a clear motion trajectory in 4 stages. This finding infers that the palmitoylated E protein pentamer possesses higher motility in the membrane.
3.4. The electrostatic force on H+
To better analyze the electrostatic force on the cations when approaching the E protein pentamer, several layers of H+ ions (along the Z-axis) were simulated to place on both sides of the membrane (Figure S3). The farthest layer of H+ is 10 Å away from the membrane. The electrostatic forces were split into the X, Y, Z components. The Z component is the main force to attract or repel ions. On the cytosolic side (Figure 7A), the Z component of the electrostatic force shows a constant decrease on palmitoylated E protein pentamer when the H+ leaving the membrane and the direction is from lumen to cytoplasm. By contrast, the loss of the palmitoylation caused the non-constant Z components when H+ ions are leaving the membrane. Besides, the standard deviations in non-palmitoylated E protein pentamer are much higher than that in palmitoylated E protein pentamer. It means, for each layer, the potentials in different positions are different, which results in non-constant forces acting on cations when the ions approach the membrane. It does not help repel cation into the cytoplasm. However, there is no significant difference between non-palmitoylated and palmitoylated E protein pentamer on the luminal side (N-terminal) (Figure 7B). Though the standard deviations of palmitoylated E protein pentamer are higher than that in non-palmitoylated E protein pentamer, they are only one-third of that in the C-terminal for non-palmitoylated E protein pentamer. In general, the electrostatic force of palmitoylated E protein is more supportive for transferring cation from lumen to cytoplasm.
Figure 7:

The Z component of the electrostatic force on hydrogen ions in both C-terminal (A) and N-terminal (B) sides of the E-protein pentamer. The distance is the distance with the membrane. The error bars represent the standard deviation.
4. Conclusion
The E protein is an important SARS-CoV-2 structural protein. The pentameric structure of E protein serves as viroporin. Palmitoylation is a common and important post-translational modification on the E protein. MD simulations are performed on the E protein pentamers with and without palmitoylation in this study. Based on the MD simulations, the topological and electrostatic studies reveal how palmitoylation affects the E protein pentamer. Without the palmitoylation, the pentametric structure loses dynamic equilibrium and the pore radius was significantly decreased, and the pore collapsed. In the electrostatic studies, the luminal side of the E protein pentamer is highly negatively charged while the cytosolic side is highly negatively charged. The surface potential supports the attraction of cations on the luminal side and the repelling of cations on the cytosolic side. This finding is consistent with the finding of previous works that the viroporin prefers cation transfer. The palmitoylation on the E protein pentamer extends the positively charged area on the cytosolic side and increases the electric interaction with the membrane on the luminal side. Additionally, The PCA reveals that the palmitoylation may increase the motility of E protein pentamer. In the end, we simulated H+ ions approaching the E protein pentamer on the luminal side and leaving the E protein on the cytosolic side. The results indicate that the electrostatic force on palmitoylated E protein pentamer can provide a constant force along with a certain layer, helping cation transfer from the lumen to the cytoplasm. To go further and more accurately prove the ion transferring direction, more sophisticated biological experiments are required. This study reveals the importance of palmitoylation on the E protein pentamer, which is helpful for the treatment development of COVID-19 and other corona virus-related diseases.
Supplementary Material
Acknowledgement
This work is supported by Grant SC1GM132043 from the National Institutes of Health; Grant 5U54MD007592 from National Institutes on Minority Health and Health Disparities, a component of the NIH. The calculations and analyses were performed at the Texas Advanced Computing Center. We thank Mr.Jixuan Pan for the writing improvements.
Footnotes
Supporting Information
Additional details and results, including the supporting figures (Figure S1-S13) and tables (Table S1). This information is available free of charge via the Internet at https://nam10.safelinks.protection.outlook.com/?url=http%3A%2F%2Fpubs.acs.org%2F&data=04%7C01%7Cssun1%40miners.utep.edu%7C6df14369fd5a49f16c4008d961c2e374%7C857c21d21a1643a490cfd57f3fab9d2f%7C1%7C0%7C637648311938087061%7CUnknown%7CTWFpbGZsb3d8evJWIioiMC4wLiAwMDAiLCJQIioiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=eIrAFid613y%2BxZC2gre4c%2BYk6XBGq9c773HFI8%2F7v6g%3D&reserved=0
Reference
- 1.(a) Stadler K; Masignani V; Eickmann M; Becker S; Abrignani S; Klenk H-D; Rappuoli R J. N. R. M, SARS—beginning to understand a new virus. 2003, 1 (3), 209–218; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Wit E; Van Doremalen N; Falzarano D; Munster VJ J. N. R. M, SARS and MERS: recent insights into emerging coronaviruses. 2016, 14 (8), 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Park J-E; Jung S; Kim A J. B. p. h, MERS transmission and risk factors: a systematic review. 2018, 18 (1), 574; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Organization, W. H. WHO MERS global summary and assessment of risk, July 2019; World Health Organization: 2019. [Google Scholar]
- 3.(a) Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y J. N, A new coronavirus associated with human respiratory disease in China. 2020, 579 (7798), 265–269; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang P; Wang X J. C.; immunology, m, COVID-19: a new challenge for human beings. 2020, 17 (5), 555–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khudadad U; Safi N; Aftab W; Ali A; Siddiqi S, The COVID-19 pandemic: an opportunity to strengthen health systems in Afghanistan. 2021. [DOI] [PubMed] [Google Scholar]
- 5.Madjid M; Safavi-Naeini P; Solomon SD; Vardeny O J. J. c, Potential effects of coronaviruses on the cardiovascular system: a review. 2020, 5 (7), 831–840. [DOI] [PubMed] [Google Scholar]
- 6.Hall KS; Samari G; Garbers S; Casey SE; Diallo DD; Orcutt M; Moresky RT; Martinez ME; McGovern T J. T. I, Centring sexual and reproductive health and justice in the global COVID-19 response. 2020, 395 (10231), 1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao H; Chen J-H; Xu Y-F, Patients with mental health disorders in the COVID-19 epidemic. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moein ST; Hashemian SM; Mansourafshar B; Khorram-Tousi A; Tabarsi P; Doty RL In Smell dysfunction: a biomarker for COVID-19, International forum of allergy & rhinology, Wiley Online Library: 2020; pp 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramphul K; Mejias SG J. C, Coronavirus disease: a review of a new threat to public health. 2020, 12 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masters PS J. A. i. v. r, The molecular biology of coronaviruses. 2006, 66, 193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L J. N, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. 2020, 581 (7807), 215–220; [DOI] [PubMed] [Google Scholar]; (b) Xie Y; Karki CB; Du D; Li H; Wang J; Sobitan A; Teng S; Tang Q; Li L J. F. i. m. b, Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. 2020, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Haan CA; Rottier PJ J. A. i. v. r, Molecular interactions in the assembly of coronaviruses. 2005, 64, 165–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Schoeman D; Fielding BC J. V. j, Coronavirus envelope protein: current knowledge. 2019, 16 (1), 1–22; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neuman BW; Kiss G; Kunding AH; Bhella D; Baksh MF; Connelly S; Droese B; Klaus JP; Makino S; Sawicki SG J. J. o. s. b, A structural analysis of M protein in coronavirus assembly and morphology. 2011, 174 (1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatagopalan P; Daskalova SM; Lopez LA; Dolezal KA; Hogue BG J. V, Coronavirus envelope (E) protein remains at the site of assembly. 2015, 478, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Nieva JL; Madan V; Carrasco L J. N. R. M, Viroporins: structure and biological functions. 2012, 10 (8), 563–574; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nieto-Torres JL; DeDiego ML; Álvarez E; Jiménez-Guardeño JM; Regla-Nava JA; Llorente M; Kremer L; Shuo S; Enjuanes L J. V, Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. 2011, 415 (2), 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westerbeck JW; Machamer CE J. J. o. v, The infectious bronchitis coronavirus envelope protein alters Golgi pH to protect the spike protein and promote the release of infectious virus. 2019, 93 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) DeDiego ML; Álvarez E; Almazán F; Rejas MT; Lamirande E; Roberts A; Shieh W-J; Zaki SR; Subbarao K; Enjuanes L J. J. o. v, A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. 2007, 81 (4), 1701–1713; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortego J; Ceriani JE; Patiño C; Plana J; Enjuanes L J. V, Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. 2007, 368 (2), 296–308; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Curtis KM; Yount B; Baric RS J. J. o. v, Heterologous gene expression from transmissible gastroenteritis virus replicon particles. 2002, 76 (3), 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y; Surya W; Claudine S; Torres J J. J. o. B. C, Structure of a conserved Golgi complex-targeting signal in coronavirus envelope proteins. 2014, 289 (18), 12535–12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres J; Wang J; Parthasarathy K; Liu DX J. B. j, The transmembrane oligomers of coronavirus protein E. 2005, 88 (2), 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Wang C; Takeuchi K; Pinto L; Lamb RA J. J. o. v, Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. 1993, 67 (9), 5585–5594; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mould JA; Paterson RG; Takeda M; Ohigashi Y; Venkataraman P; Lamb RA; Pinto LH J. D. c, Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. 2003, 5 (1), 175–184; [DOI] [PubMed] [Google Scholar]; (c) Pham T; Perry JL; Dosey TL; Delcour AH; Hyser JM J. S. r, The rotavirus NSP4 viroporin domain is a calcium-conducting ion channel. 2017, 7 (1), 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nieto-Torres JL; Verdiá-Báguena C; Jimenez-Guardeño JM; Regla-Nava JA; Castaño-Rodriguez C; Fernandez-Delgado R; Torres J; Aguilella VM; Enjuanes L J. V, Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. 2015, 485, 330–339; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wilson L; Mckinlay C; Gage P; Ewart G J. V, SARS coronavirus E protein forms cation-selective ion channels. 2004, 330 (1), 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fung TS; Liu DX J. F. v, Post-translational modifications of coronavirus proteins: roles and function. 2018, 13 (6), 405–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Chen S-C; Lo S-Y; Ma H-C; Li H-C J. V. g, Expression and membrane integration of SARS-CoV E protein and its interaction with M protein. 2009, 38 (3), 365–371; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan Q; Liao Y; Torres J; Tam J; Liu D J. F. I, Biochemical evidence for the presence of mixed membrane topologies of the severe acute respiratory syndrome coronavirus envelope protein expressed in mammalian cells. 2006, 580 (13), 3192–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Y; Yuan Q; Torres J; Tam J; Liu D J. V, Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. 2006, 349 (2), 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Lopez LA; Riffle AJ; Pike SL; Gardner D; Hogue BG J. J. o. v, Importance of conserved cysteine residues in the coronavirus envelope protein. 2008, 82 (6), 3000–3010; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boscarino JA; Logan HL; Lacny JJ; Gallagher TM J. J. o. v, Envelope protein palmitoylations are crucial for murine coronavirus assembly. 2008, 82 (6), 2989–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Salaun C; Greaves J; Chamberlain LH J. J. o. C. B, The intracellular dynamic of protein palmitoylation. 2010, 191 (7), 1229–1238; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fujiwara Y; Kondo HX; Shirota M; Kobayashi M; Takeshita K; Nakagawa A; Okamura Y; Kinoshita K J. S. r, Structural basis for the membrane association of ankyrinG via palmitoylation. 2016, 6 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surya W; Li Y; Torres J J. B. e. B. A.-B, Structural model of the SARS coronavirus E channel in LMPG micelles. 2018, 1860 (6), 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DE; Chivian D; Baker D J. N. a. r, Protein structure prediction and analysis using the Robetta server. 2004, 32 (suppl_2), W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jo S; Kim T; Iyer VG; Im W J. J. o. c. c, CHARMM-GUI: a web-based graphical user interface for CHARMM. 2008, 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
- 29.Huang J; MacKerell AD Jr J. J. o. c. c, CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. 2013, 34 (25), 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Meer G; Voelker DR; Feigenson GW J. N. r. M. c. b, Membrane lipids: where they are and how they behave. 2008, 9 (2), 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackerell AD Jr; Feig M; Brooks CL III J. J. o. c. c, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. 2004, 25 (11), 1400–1415. [DOI] [PubMed] [Google Scholar]
- 32.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG J. T. J. o. c. p, A smooth particle mesh Ewald method. 1995, 103 (19), 8577–8593. [Google Scholar]
- 33.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K J. J. o. c. c, Scalable molecular dynamics with NAMD. 2005, 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W; Dalke A; Schulten K J. J. o. m. g, VMD: visual molecular dynamics. 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- 35.Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJ J. J. o. c. c, GROMACS: fast, flexible, and free. 2005, 26 (16), 1701–1718. [DOI] [PubMed] [Google Scholar]
- 36.Smart OS; Neduvelil JG; Wang X; Wallace B; Sansom MS J. J. o. m. g, HOLE: a program for the analysis of the pore dimensions of ion channel structural models. 1996, 14 (6), 354–360. [DOI] [PubMed] [Google Scholar]
- 37.Li L; Li C; Sarkar S; Zhang J; Witham S; Zhang Z; Wang L; Smith N; Petukh M; Alexov E J. B. b, DelPhi: a comprehensive suite for DelPhi software and associated resources. 2012, 5 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA J. N. a. r, PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. 2007, 35 (suppl_2), W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dias RP; Li L; Soares TA; Alexov E J. J. o. c. c, Modeling the electrostatic potential of asymmetric lipopolysaccharide membranes: The MEMPOT algorithm implemented in DelPhi. 2014, 35 (19), 1418–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE J. J. o. c. c, UCSF Chimera—a visualization system for exploratory research and analysis. 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- 41.Li L; Chakravorty A; Alexov E J. J. o. c. c, DelPhiForce, a tool for electrostatic force calculations: Applications to macromolecular binding. 2017, 38 (9), 584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ochsenbauer-Jambor C; Miller DC; Roberts CR; Rhee SS; Hunter E J. J. o. v, Palmitoylation of the Rous sarcoma virus transmembrane glycoprotein is required for protein stability and virus infectivity. 2001, 75 (23), 11544–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Percherancier Y; Planchenault T; Valenzuela-Fernandez A; Virelizier J-L; Arenzana-Seisdedos F; Bachelerie F J. J. o. B. C, Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. 2001, 276 (34), 31936–31944. [DOI] [PubMed] [Google Scholar]
- 44.Noland CL; Gierke S; Schnier PD; Murray J; Sandoval WN; Sagolla M; Dey A; Hannoush RN; Fairbrother WJ; Cunningham CN J. S, Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling. 2016, 24 (1), 179–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.