

Abstract
DNA-encoded library (DEL) technology features a time- and cost-effective interrogation format for the discovery of therapeutic candidates in the pharmaceutical industry. To develop DEL platforms, the implementation of water-compatible transformations that facilitate the incorporation of multifunctional building blocks (BBs) with high C(sp3) carbon counts is integral for success. In this report, a decarboxylative-based hydroalkylation of DNA-conjugated trifluoromethyl-substituted alkenes enabled by single-electron transfer (SET) and subsequent hydrogen atom termination through electron donor–acceptor (EDA) complex activation is detailed. In a further photoredox-catalyzed hydroarylation protocol, the coupling of functionalized, electronically unbiased olefins is achieved under air and within minutes of blue light irradiation through the intermediacy of reactive (hetero)aryl radical species with full retention of the DNA tag integrity. Notably, these processes operate under mild reaction conditions, furnishing complex structural scaffolds with a high density of pendant functional groups.
DNA-encoded library (DEL) technology facilitates the rapid identification of therapeutic candidates in pharmaceutical settings. Herein, the development of photoredox-mediated hydrocarbofunctionalization protocols of olefins is described.
Introduction
The identification of specific binding molecules remains a central theme in drug discovery efforts in academic and industrial laboratories.1 The global pharmaceutical industry invests over $186 billion annually on research and development to meet the ever-increasing demands for safe and improved therapeutics.2 In recent years, DNA-encoded library (DEL) technology (Fig. 1) has emerged as a novel interrogation platform to accelerate the advancement of small-molecule modulators of biomolecular targets.3 Conceptualized by Brenner and Lerner in 1992,4 DEL platforms confer unprecedented capabilities to overlap the versatility of chemical synthesis with the powerful features of genetic coding, allowing simultaneous testing of combinatorial libraries of exceptional magnitude (>106 to 1012 discrete members).3 Through “split and pool” synthetic cycles, diverse BBs are encoded by a unique DNA tag functioning as a molecular identifier.3 Following synthesis, DEL libraries are incubated with immobilized target proteins, after which non-binders are washed away. The chemical structures of the potent ligands are decoded using polymerase chain reaction (PCR) amplification and posterior next-generation DNA sequencing.3 In this vein, DELs feature a more effective and inexpensive discovery format ($0.0002 per library member) compared with conventional high-throughput screening (HTS) methods ($1000 per library member).5 To be successful, on-DNA chemistries are required to incorporate BBs from readily available chemicals bearing multifunctional handles for further diversification under mild, dilute, and aqueous conditions.6 In light of these considerations, the development of reliable transformations that operate through novel reactivity modes and employ an abundant, diverse set of starting materials would expedite progress in this field.
Fig. 1. Workflow of DEL technology.

As part of a program centered on the development of catalytic tools to yield novel structural scaffolds, we recently reported the synthesis of gem-difluoroalkenes,7,8 carbonyl mimics that display in vivo resistance toward metabolic processes,9 through photoinduced radical/polar crossover defluorinative alkylation.7,8,10 As a complementary approach to build chemical diversity, we became interested in pushing the limits of photochemical paradigms to access benzylically trifluoromethylated compounds, bioactive structural motifs in medicinal settings (Scheme 1A).11 Specifically, fluorine incorporation is a powerful strategy invoked by the pharmaceutical and agrochemical industries to alter a molecule's chemical, physical, and biological properties, such as its pKa, dipole moment, and molecular conformation.12,13 As a consequence of these factors, fluorinated scaffolds are prevalent in more than 25% of marketed drugs.11c As an important representative, the trifluoromethyl (–CF3) group renders increased metabolic stability, lipophilicity, and binding selectivity when embedded in therapeutic candidates.11,14 Typically, the trifluoromethyl group can be installed through nucleophilic, electrophilic, or radical routes.15 Although these strategies undoubtedly expand chemical space, these protocols remain elusive in the context of late-stage functionalization and the incorporation of sensitive functional groups in complex environments under DEL-like conditions. An underexplored opportunity to achieve C(sp)3 trifluoromethylation is the direct hydroalkylation of trifluoromethyl-substituted olefins. Specifically, the carbofunctionalization of these electrophilic unsaturated systems occurs readily at room temperature with exquisite functional group compatibility,16 thus rendering the incorporation of pharmaceutically relevant cores and complex alkyl fragments feasible in a library setting. However, given the established propensity of trifluoromethyl-substituted alkenes to undergo intramolecular E1cB-type fluoride elimination in metal-catalyzed cross-couplings that proceed through the intermediacy of α-CF3-metal species17via the nucleophilic addition of organometallic reagents18,19 or in the presence of traditional photoredox catalysts irrespective of the nature of the radical precursor (Scheme 1B),16,19a,20 hydrofunctionalization21 efforts remain challenging. In particular, the hydroalkylation of trifluoromethyl-substituted alkenes using unactivated alkyl counterparts presents a formidable, yet potentially powerful scenario to rapidly access unprecedented benzylically trifluoromethylated building blocks from commodity radical progenitors with a high content of C(sp3) carbons.
Scheme 1. (A) The trifluoromethyl group in medicinal chemistry. (B) Carbofunctionalization strategies of trifluoromethyl-substituted alkenes.

To address this challenge and unlock access to benzylically trifluoromethylated motifs from commodity chemicals in DEL synthesis, we report a decarboxylative-based, radical-mediated hydroalkylation of DNA-tagged trifluoromethyl-substituted alkenes enabled by the merger of electron donor–acceptor (EDA) complex photoactivation22,22j,22k and hydrogen atom transfer (HAT) chemistry (Scheme 2).23 Under blue light irradiation, a commercially available electron donor, Hantzsch ester (HE, diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate), functions as a strong photoreductant to induce C(sp3) radical generation from commercially available carboxylic acid derivatives.24 As part of its dual role, HE subsequently serves as a suitable hydrogen atom donor, impeding the formation of anionic intermediates upon radical addition as well as circumventing the necessity for alkylmetal complexes, species intrinsically primed to undergo β-F elimination in reactions with trifluoromethyl-substituted alkenes.17e,20,25 In this vein, the utility of this EDA paradigm is partially driven by its ability to deliver complex trifluoromethyl-substituted hydrofunctionalized products with high C(sp3) carbon counts selectively under mild and open-air conditions.
Scheme 2. Photoredox-mediated hydrocarbofunctionalization of olefins on DNA.

As a complement to the hydroalkylation protocol, a radical-mediated intermolecular hydroarylation of electronically unbiased olefins was developed (Scheme 2).26 Because alkenes are plentiful, structurally diverse, and versatile commodity feedstocks, readily available from petrochemical and renewable resources, they are ideal precursors for C–C bond formation in DELs, and the strategy developed is based on photoinduced reductive activation of DNA-conjugated (het)aryl halides to deliver reactive (het)aryl radical species that can be harnessed in useful synthetic operations followed by hydrogen atom termination.23
Discussion
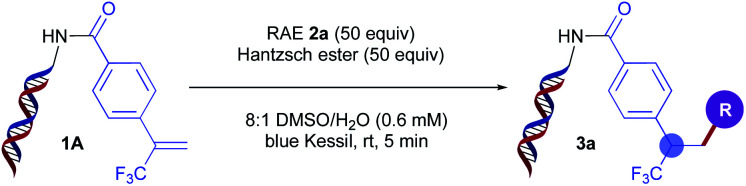
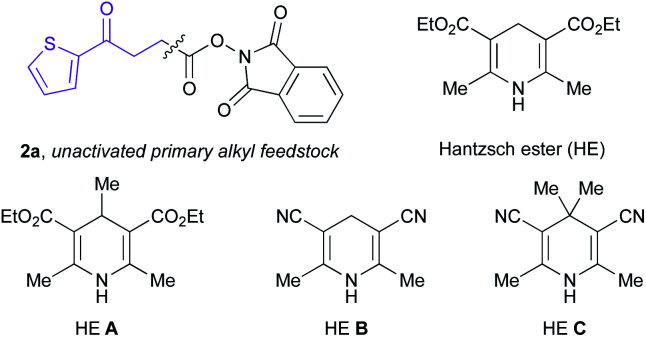
Recently, synthetic processes driven by EDA complex photochemistry have gained considerable traction, including protocols resulting in borylation, sulfonylation, and thioetherification.22,27 To harness the synthetic potential of EDA complex photoactivation toward DEL platforms, we examined the feasibility of the proposed decarboxylative hydroalkylation using on-DNA trifluoromethyl-substituted alkene 1A and unactivated primary redox-active ester (RAE) 2a as model substrates (Table 1). Under blue Kessil irradiation (λmax = 456 nm), efficient conversion to the desired benzylic trifluoromethyl-substituted product 3a was observed using 50 equivalents of the radical precursor under ambient reaction conditions within minutes of illumination. In contrast to radical-mediated alkylation promoted by metal reductants17e,28 or external photoredox catalysts,29 this open-to-air EDA paradigm provides an exceedingly low barrier to practical implementation in high-throughput settings and circumvents side reactivity stemming from singlet oxygen generation through triplet-energy transfer.29
On-DNA photoinduced decarboxylative alkylation: optimization of reaction conditionsa.
| ||
|---|---|---|
| Entry | Deviation from std conditions | 3ab (% conv.) |
| 1 | None | 84% |
| 2 | RAE (25 equiv.), HE (25 equiv.) | 65% |
| 3 | RAE (10 equiv.), HE (10 equiv.) | 55% |
| 4 | 0.5 mM | 84% |
| 5 | 0.4 mM | 80% |
| 6 | 0.3 mM | 77% |
| 7 | HE A | 0% |
| 8 | HE B | 20% |
| 9 | HE C | 0% |
| 10 | No HE | Trace |
| 11 | No light | Trace |
| ||
Reaction conditions: RAE 2a (50 equiv., 1.25 μmol), HE (50 equiv., 1.25 μmol), on-DNA trifluoromethyl-substituted alkene 1A (1.0 equiv., 25 nmol), 8 : 1 DMSO/H2O (0.6 mM), 5 min irradiation with blue Kessil lamps (λmax = 456 nm, 40 W).
Conversion to 3a as determined by LC/MS analysis.
To examine the influence of the dihydropyridine (DHP) backbone on the efficacy of this photochemical manifold, the reaction was conducted with four different DHP derivatives to gain a deeper understanding of their dual reactivity profile in EDA complex photoactivation and HAT catalysis. The C4-substituted DHP (HE A, entry 7) thus exhibited no reactivity under the reaction conditions, whereas cyano substitution at C3 and C5 of the DHP (HE B, entry 8) led to diminished product formation. As expected, 4,4′-dimethyl HE C (entry 9) failed to promote the reaction, presumably because of competitive back electron transfer (BET)22a that restores the ground-state EDA complex from its radical ion pair in the absence of a probable photooxidative aromatization event. Notably, commercially available and bench-stable HE30 displayed optimal performance (84% yield, entry 1), accommodating water as a co-solvent and high dilution conditions (0.3–0.6 mM), with only trace amounts of the corresponding gem-difluoroalkene detected. Using UV/vis absorption studies, a bathochromic shift of the reaction mixture in 8 : 1 DMSO/H2O (0.6 mM) was observed, with a wavelength band tailing to 500 nm (see ESI†). This is indicative of the formation of a new molecular aggregate between the electron-deficient aliphatic RAEs and the electron-rich HE. Using Job's method31 of continuous variation, we determined a molar donor–acceptor ratio of 1 : 1 for the colored EDA complex (see ESI†). Further spectrophotometric analysis at 450 nm revealed an association constant (KEDA) of 1.2 M−1 of HE with 1-methylcyclohexane-N-hydroxyphthalimide ester using the Benesi–Hildebrand method,32 highlighting a plausible association event of charge-transfer complexes prior to SET events under the conditions of the reaction. Finally, control experiments validated the necessity of all reaction parameters for effective C(sp3)–C(sp3) bond formation.
Next, we examined the scope of redox-active carboxylate derivatives using on-DNA trifluoromethylated alkene 1A (Scheme 3). In general, a broad palette of primary aliphatic systems that lack any radical stabilizing factors exhibited excellent reactivity. The method further benefits from broad functional group tolerance, facilitating the introduction of bifunctional handles including ketones (3a, 3f, 3q), aryl halides (3c, 3e, 3n), a terminal alkyne (3d), esters (3j, 3o), substituted alkenes (3k, 3l, 3m, 3o), free alcohols (3o, 3p), as well as medicinally-relevant heteroaromatic scaffolds (3a, 3n). In addition, Boc- and Fmoc-protected amines served as competent substrates. This is crucial in DEL settings, where library members should ideally bear multifunctional BBs that allow subsequent derivatization. The scope was further extended to the modification of biologically active molecules displaying a high density of pendant functional groups, including the herbicide 2,4-dichlorophenylacetic acid (3e), long-chain fatty acids (3k–3m), the anti-inflammatory agent indomethacin (3n), mycophenolic acid (3o), as well as various steroids (3p, 3q). In particular, this method provides a clear advantage in terms of scope over previously reported on-DNA photoinduced decarboxylative alkylation protocols, which are largely limited to α-heteroatom-stabilized radicals,7,33 or exclusively restricted to secondary and tertiary radicals.22k,34 More specifically, complementary decarboxylative methods employing zinc nanopowder as a reductant under strictly deoxygenated conditions fail to incorporate primary systems on DNA,28 presumably because of the higher reduction potentials associated with the radical precursor. Most importantly, these methods largely proceed through anionic intermediates, where in the case of the trifluoromethyl-substituted olefins, there is a predominant propensity for intramolecular E1cB-type fluoride elimination16a,19a,20 to afford the corresponding gem-difluoroalkenes (rather than trifluoromethyl-substituted alkanes) via radical/polar crossover pathways.10
Scheme 3. On-DNA photoinduced decarboxylative alkylation: Evaluation of aliphatic carboxylic acid derivatives.

In a similar manner, secondary and tertiary radical architectures are harnessed effectively to afford functionalized synthetic frameworks, including scaffolds derived from proteinogenic and non-proteinogenic amino acids (3u–3y), a glycoside (3z), and lipid-lowering agent gemfibrozil (3za) (Scheme 3). The reaction conditions proved general for both acyclic and cyclic carboxylate derivatives, including bridged bicyclics (3zf–3zh), as well as strained ring systems, such as cyclobutanes (3s, 3t) and a cyclopropane (3zc). Notably, trifluoromethyl-substituted bicyclo[1.1.1]pentane (BCP) product 3zg was obtained in good yield. These BCP derivatives serve as bioisosteres for arenes, internal alkynes, and tert-butyl groups in medicinal chemistry settings.35
With respect to the scope of trifluoromethyl-substituted alkenes, a diverse array of DNA headpieces (DNA-HPs) led to the desired benzylic trifluoromethyl-substituted products without compromising yields (Scheme 4). In general, both electron-withdrawing and electron-donating groups are well tolerated under the developed conditions. Substitution at the para-, meta-, and ortho-positions of the HPs' aryl moieties was explored, whereby efficient decarboxylative photocoupling took place. Furthermore, comparable reactivity was observed for unactivated primary, secondary, tertiary, as well as stabilized benzylic-, α-oxy-, and α-amino radical species, further underscoring the versatility of this photochemical EDA paradigm.
Scheme 4. On-DNA photoinduced decarboxylative alkylation: evaluation of trifluoromethyl-substituted alkenes.

The commercial availability and structural diversity of carboxylic acids render them particularly attractive for use as multifunctional BBs in DEL libraries. To validate the modularity of this approach further, we developed a telescoped, one-pot photoinduced decarboxylative alkylation protocol through in situ formation of aliphatic RAEs with N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (HITU),28 a bench-stable solid that can be readily prepared on kilogram scale (Scheme 5). This reagent features great versatility in reaction scope, accommodating a wide array of functional groups including ketone (3f), carbamate (3zi), aryl chloride (3zl), and Boc-protected amines (3zn, 3zo). Using 24-well plates, microdosing of the carboxylic acid, DIPEA, and HITU in DMSO is accomplished under air, followed by 3 h of activation time. The in situ formed RAEs can then be treated directly with a solution of HE and the corresponding DNA headpiece, reaching synthetically useful yields after 10 min of illumination (Scheme 5, workflow). Notably, this HITU-mediated alkylation performs equally well using unactivated- and α-heteroatom-stabilized radical progenitors, presenting a direct route toward C–C bond formation through oxidative quenching modes, an underexplored challenge in DEL-based environments.28
Scheme 5. On-DNA photoinduced decarboxylative alkylation: in situ activation of RAEs with N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (HITU).

As an extension of the hydroalkylation chemistry, an on-DNA multicomponent reaction (MCR) was developed. In recent years, MCRs36 have emerged as a powerful tool to furnish novel structural scaffolds with inherent molecular complexity from abundant feedstocks. Through sequential bond formation, MCRs enable the sampling of uncharted chemical space to accelerate drug discovery efforts.36 An underexplored realm in DEL synthesis is the development of olefin dicarbofunctionalization reactions.3a Specifically, alkenes serve as versatile BBs that possess functional group-rich handles for derivatization. However, in addition to chemo- and regio-selectivity concerns associated with DEL reactions that rely on high loadings of reagents, these processes are further complicated by the generation of undesired two-component coupling products. Keeping these considerations in mind and taking advantage of the electronically distinct nature of trifluoromethyl-substituted alkenes, a polarity-reversing radical cascade/trifluoromethylation of olefins has been developed through EDA complex photoactivation between HE and Umemoto's reagent,22a a commercially available trifluoromethylating agent (Scheme 6).
Scheme 6. On-DNA multicomponent trifluoromethylation promoted by photoactive EDA complex activation. Note: several diastereomers are expected to form under these conditions.

In particular, this open-to-air charge-transfer manifold harnesses electrophilic trifluoromethyl radicals for subsequent addition to electron-neutral or electron-rich alkenes, abundant yet currently underexplored partners in photoinduced DEL synthesis.3a,37 The resulting nucleophilic, open-shell radical intermediates may then engage in chemoselective coupling with on-DNA trifluoromethyl-substituted alkenes. As part of its dual role, the HE also functions as a hydrogen atom donor to furnish bis-trifluoromethylated products of significance in medicinal settings.11 Remarkably, the scope of the olefin partner proved general, tolerating diverse functional groups including a free alcohol (11a) and unprotected glycoside 11d. In this vein, we anticipate this mode of catalysis will help inform the design and implementation of unique synthetic disconnections toward complex, bioactive targets in DELs.
Having developed suitable conditions for the hydroalkylation of unsaturated DEL platforms, attention was turned toward hydroarylation transformations. Recently, research efforts have validated Ni/photoredox dual manifolds in DEL platforms using carboxylic acids,7,33b,33e 1,4-dihydropyridines (DHPs),7 α-silylamines,8 and alkyl bromides8,38 as radical precursors. Given our long-standing interest in the design of complex (hetero)aryl scaffolds with high C(sp3) carbon counts,39 we sought to expand reactivity in DEL synthesis through intermolecular radical-mediated hydroarylation of functionalized olefins to generate alkylarenes (Scheme 7). To develop a complementary approach toward C(sp3)–C(sp2) bond formation, we reasoned that single-electron reduction of DNA-bound, halogenated aryl subunits26 would grant access to reactive (het)aryl radical species in a regioselective fashion. Subsequent addition to alkenes followed by hydrogen atom termination would deliver unprecedented structures from readily available substrates. Inspired by pioneering work by Beckwith40 and related, precedented milestones,26 we hypothesized that photoinduced electron transfer from highly reducing transition-metal-based complexes would enable this strategy under mild reaction conditions. However, because of the high redox potentials associated with organohalides and the propensity of aryl radicals to undergo reduction through rapid HAT,26a the adaptation of this mechanistic proposal in DEL environments posed challenges. Importantly, aryl radicals have been shown to induce DNA strand damage,41 underscoring the requirement for a regulated generation of these high-energy intermediates and the necessity for well-orchestrated addition reactions. To achieve chemo- and regio-selectivity, the following criteria was considered: (i) the rate of (het)aryl radical addition to unsaturated systems must be competitive with C–X bond reduction stemming from undesired HAT pathways. (ii) The rate of hydrogen atom abstraction by the resulting alkyl radical must be competitive with its addition to another equivalent of the alkene. (iii) The rate of single-electron reduction of the aryl halide should take place preferentially over that of the alkyl radical intermediate. Specifically, the choice of both photocatalyst and hydrogen atom donor influences product distributions. We determined that 300 equiv. of the olefinic substrate and a 1 : 200 photocatalyst-to-HAT reagent ratio was optimal for reactivity. Toward this end, the combination of fac-Ir(ppy)3 and HE enabled the construction of alkylated arenes under air within minutes of blue light irradiation. Control experiments demonstrated that all reaction components are necessary for aryl radical generation.
Scheme 7. Employing alkene BBs for C(sp3)–C(sp2) bond formation in DEL synthesis: evaluation of olefins and aryl halides.

With optimized conditions established, we surveyed DNA-tagged (het)aryl halides with norbornene as the alkyl source (Scheme 7). Aryl iodide 13A bearing a chloride substituent afforded the desired product with the electrophilic cross-coupling handle intact, delivering linchpins for further functionalization. Electron-neutral iodobenzene 13B as well as derivatives bearing electron-donating groups (13C) or electron-withdrawing groups (13G) served as excellent substrates. Further extension to less activated aryl bromides was also possible (13D). Notably, electrophilic pyridyl radicals were employed as coupling partners, giving rise to functionalized heteroaromatics (19a, 19j–19n). Finally, in addition to the strained bicyclic norbornene, a broad array of functionalized alkenes was examined. In general, unactivated alkenes bearing unprotected alcohols (15b, 15c, 19j), an ester (19k), ketones (15i, 19l), and an epoxide (19n) were all accommodated. In addition, this photochemical paradigm was extended to the modification of activated styrene derivatives (15e, 15g) in synthetically useful yields. Even benzylically trifluoromethylated product 15g could be used as a substrate to afford product with complete retention of the bromide handle, presumably because of the high loading of alkene precursor compared to the photoredox catalyst, precluding an overreduction event of the halide. From the standpoint of DEL synthesis, which benefits from minimal reagent input (e.g., 10–25 nmol of HP per transformation), such equivalencies can be leveraged to achieve selectivity and unique reactivity trends that are otherwise untenable in traditional small molecule synthesis. In particular, these halogenated alkenes can further grow DEL libraries through transition metal-catalyzed cross-coupling efforts.
DNA compatibility with EDA complex photoactivation
Because the integrity of the DNA barcode is essential to a successful protein target selection, mock ligations and qPCR amplifications were performed to evaluate the ability of the RAE hydroalkylation conditions to be used in an actual library production. A model DNA conjugate composed of a representative headpiece ligated to a 4-cycle tag and equipped with a 2-base 5′ overhang was subjected to the standard hydroalkylation conditions. This model DNA conjugate was also subjected to control reactions where either Hantzsch ester or light was omitted. Post reactions, these DNA conjugates were further elongated by ligation to install the necessary PCR primers and quantified by qPCR. There was no significant difference in qPCR amplification across the various experiments, suggesting full DNA integrity (see ESI†). These findings further underscore the utility of EDA paradigms as a general blueprint toward selective on-DNA alkylation under open-to-air conditions.
DNA compatibility with aryl radical intermediates
Mindful of well-established precedent of DNA strand damage in the presence of reactive aryl radical species,41 the hydroarylation conditions were studied to evaluate the resulting DNA integrity. Again, a model DNA conjugate composed of a representative headpiece ligated to a 4-cycle tag and equipped with a 2-base 5′ overhang was reacted using the standard conditions. This model DNA conjugate was also subjected to control reactions where either Hantzsch ester, photocatalyst, or light was omitted. Post reactions, these DNA conjugates were further elongated by ligation to install the necessary PCR primers and quantified by qPCR. There was no significant difference in qPCR amplification across the various experiments, suggesting full DNA integrity (see ESI†). These results emphasize the mild nature of the developed photoredox paradigm, whereby the formation of reactive aryl radical intermediates in a regulated fashion facilitates productive on-DNA alkylation.
Conclusions
In summary, this report demonstrates a mechanistically-driven proof-of-concept for the implementation of charge-transfer complex activation as an enabling technology to introduce diverse C(sp3)-hybridized architectures from commodity chemicals in DEL platforms (including unactivated primary, secondary, tertiary, as well as stabilized benzylic, α-alkoxy, and α-amino systems). Specifically, this EDA paradigm was utilized to achieve the selective decarboxylative-based hydroalkylation of trifluoromethyl-substituted alkenes through radical/HAT crossover to access complex benzylic trifluoromethylated scaffolds, unlocking a complementary reactivity outcome to established carbodefluorinative protocols mediated by an external photoredox catalyst. Furthermore, a general intermolecular hydroarylation protocol of electronically unbiased olefins through selective C–X bond activation in DNA-tagged organohalides is reported. Remarkably, this photochemical paradigm delivers reactive (hetero)aryl radical species in a regulated fashion without compromising the DNA integrity. Notably, these open-to-air processes are chemoselective, operate under mild and dilute reaction conditions, and are completed within minutes, rendering them suitable for late-stage functionalization and high-throughput settings in the pharmaceutical industry. We anticipate these findings will expedite drug discovery research and provoke further development in radical-mediated DEL synthesis.
Data availability
All experimental procedures, characterization data, as well as copies of NMR and LCMS spectra supporting this article have been uploaded as part of the ESI.†
Author contributions
Dr Shorouk O. Badir conceived the topic. Dr Shorouk O. Badir and Dr Alexander Lipp contributed equally. Dr Shorouk O. Badir wrote the original draft of the manuscript with input from Professor Gary A. Molander and Dr Lisa A. Marcaurelle. All authors contributed to ideation, discussion of results, and writing of the paper.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G. M.). Dr Shorouk O. Badir is supported by the Bristol-Myers Squibb Graduate Fellowship for Synthetic Organic Chemistry. Dr Alexander Lipp is grateful to the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship (LI 3682/1-1). Dr María Jesús Cabrera-Afonso acknowledges the Fundación Ramón Areces for a postdoctoral fellowship. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr Katelyn Billings (formerly GSK, currently Relay Therapeutics), Dr Melissa C. Grenier-Davies (GSK), Dr Chris Dimitri (GSK), Dr Pradeep Bandarufor (GSK), and Haleh Kazemi (GSK) for stimulating discussions. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data. Access to a UV/vis spectrophotometer was provided by the Petersson laboratory (UPenn). Johnson Matthey is acknowledged for the donation of iridium(iii) chloride, and Kessil is thanked for the donation of lamps.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03191k
Notes and references
- (a) Blakemore D. C. Castro L. Churcher I. Rees D. C. Thomas A. W. Wilson D. M. Wood A. Nat. Chem. 2018;10:383–394. doi: 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]; (b) Erlanson D. A. Fesik S. W. Hubbard R. E. Jahnke W. Jhoti H. Nat. Rev. Drug Discovery. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]; (c) Schenone M. Dančík V. Wagner B. K. Clemons P. A. Nat. Chem. Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stockwell B. R. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statista, Research and Development worldwide, https://www.statista.com/study/70627/research-and-development-worldwide/, accessed June 2020
- For selected examples, see: ; (a) Patel S. Badir S. O. Molander G. A. Trends Chem. 2021;3:163–175. doi: 10.1016/j.trechm.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mullard A. Nature. 2016;530:367–369. doi: 10.1038/530367a. [DOI] [PubMed] [Google Scholar]; (c) Harris P. A. King B. W. Bandyopadhyay D. Berger S. B. Campobasso N. Capriotti C. A. Cox J. A. Dare L. Dong X. Finger J. N. Grady L. C. Hoffman S. J. Jeong J. U. Kang J. Kasparcova V. Lakdawala A. S. Lehr R. McNulty D. E. Nagilla R. Ouellette M. T. Pao C. S. Rendina A. R. Schaeffer M. C. Summerfield J. D. Swift B. A. Totoritis R. D. Ward P. Zhang A. Zhang D. Marquis R. W. Bertin J. Gough P. J. J. Med. Chem. 2016;59:2163–2178. doi: 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]; (d) Litovchick A. Dumelin C. E. Habeshian S. Gikunju D. Guié M.-A. Centrella P. Zhang Y. Sigel E. A. Cuozzo J. W. Keefe A. D. Clark M. A. Sci. Rep. 2015;5:10916–10923. doi: 10.1038/srep10916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Clark M. A. Acharya R. A. Arico-Muendel C. C. Belyanskaya S. L. Benjamin D. R. Carlson N. R. Centrella P. A. Chiu C. H. Creaser S. P. Cuozzo J. W. Davie C. P. Ding Y. Franklin G. J. Franzen K. D. Gefter M. L. Hale S. P. Hansen N. J. V. Israel D. I. Jiang J. Kavarana M. J. Kelley M. S. Kollmann C. S. Li F. Lind K. Mataruse S. Medeiros P. F. Messer J. A. Myers P. O'Keefe H. Oliff M. C. Rise C. E. Satz A. L. Skinner S. R. Svendsen J. L. Tang L. van Vloten K. Wagner R. W. Yao G. Zhao B. Morgan B. A. Nat. Chem. Biol. 2009;5:647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]; (f) Goodnow Jr R. A., A Handbook for DNA-Encoded Chemistry: Theory and Applications for Exploring Chemical Space and Drug Discovery, John Wiley & Sons, United States, 2014 [Google Scholar]; (g) Catalano M. Moroglu M. Balbi P. Mazzieri F. Clayton J. Andrews K. H. Bigatti M. Scheuermann J. Conway S. J. Neri D. ChemMedChem. 2020;15:1752–1756. doi: 10.1002/cmdc.202000528. [DOI] [PubMed] [Google Scholar]; (h) Franzini R. M. Ekblad T. Zhong N. Wichert M. Decurtins W. Nauer A. Zimmermann M. Samain F. Scheuermann J. Brown P. J. Hall J. Gräslund S. Schüler H. Neri D. Angew. Chem., Int. Ed. 2015;54:3927–3931. doi: 10.1002/anie.201410736. [DOI] [PubMed] [Google Scholar]; (i) Catalano M. Oehler S. Prati L. Favalli N. Bassi G. Scheuermann J. Neri D. Anal. Chem. 2020;92:10822–10829. doi: 10.1021/acs.analchem.0c02304. [DOI] [PubMed] [Google Scholar]; (j) Ottl J. Leder L. Schaefer J. V. Dumelin C. E. Molecules. 2019;24:1629–1651. doi: 10.3390/molecules24081629. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Martín A. Nicolaou C. A. Toledo M. A. Commun. Chem. 2020;3:1–9. doi: 10.1038/s42004-019-0249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Götte K. Chines S. Brunschweiger A. Tetrahedron Lett. 2020;61:151889–151899. doi: 10.1016/j.tetlet.2020.151889. [DOI] [Google Scholar]; (m) Flood D. T. Kingston C. Vantourout J. C. Dawson P. E. Baran P. S. Isr. J. Chem. 2020;60:268–280. doi: 10.1002/ijch.201900133. [DOI] [Google Scholar]
- Brenner S. Lerner R. A. Proc. Natl. Acad. Sci. U. S. A. 1992;89:5381–5383. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow R. A. Dumelin C. E. Keefe A. D. Nat. Rev. Drug Discovery. 2017;16:131–147. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Malone M. L. Paegel B. M. ACS Comb. Sci. 2016;18:182–187. doi: 10.1021/acscombsci.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan J. P. Lang S. B. Sim J. Berritt S. Peat A. J. Billings K. Fan L. Molander G. A. J. Am. Chem. Soc. 2019;141:3723–3732. doi: 10.1021/jacs.9b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badir S. O. Sim J. Billings K. Csakai A. Zhang X. Dong W. Molander G. A. Org. Lett. 2020;22:1046–1051. doi: 10.1021/acs.orglett.9b04568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Magueur G. Crousse B. Ourévitch M. Bonnet-Delpon D. Bégué J.-P. J. Fluorine Chem. 2006;127:637–642. doi: 10.1016/j.jfluchem.2005.12.013. [DOI] [Google Scholar]; (b) Leriche C. He X. Chang C.-W. T. Liu H.-W. J. Am. Chem. Soc. 2003;125:6348–6349. doi: 10.1021/ja021487+. [DOI] [PubMed] [Google Scholar]
- Wiles R. J. Molander G. A. Isr. J. Chem. 2020;60:281–293. doi: 10.1002/ijch.201900166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Purser S. Moore P. R. Swallow S. Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; (b) Harper D. B. O'Hagan D. Nat. Prod. Rep. 1994;11:123–133. doi: 10.1039/NP9941100123. [DOI] [PubMed] [Google Scholar]; (c) Wang J. Sánchez-Roselló M. Aceña J. L. del Pozo C. Sorochinsky A. E. Fustero S. Soloshonok V. A. Liu H. Chem. Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- (a) Patani G. A. LaVoie E. J. Chem. Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]; (b) Barrow J. C. Rittle K. E. Reger T. S. Yang Z.-Q. Bondiskey P. McGaughey G. B. Bock M. G. Hartman G. D. Tang C. Ballard J. ACS Med. Chem. Lett. 2010;1:75–79. doi: 10.1021/ml100004r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Meanwell N. A. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; (b) Gillis E. P. Eastman K. J. Hill M. D. Donnelly D. J. Meanwell N. A. J. Med. Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; (c) Hagmann W. K. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (d) Meanwell N. A. J. Med. Chem. 2018;61:5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Varenikov A. Gandelman M. Nat. Commun. 2018;9:3566–3572. doi: 10.1038/s41467-018-05946-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews, please see: ; (a) Xiao H. Zhang Z. Fang Y. Zhu L. Li C. Chem. Soc. Rev. 2021;50:6308–6319. doi: 10.1039/D1CS00200G. [DOI] [PubMed] [Google Scholar]; (b) Pan X. Xia H. Wu J. Org. Chem. Front. 2016;3:1163–1185. doi: 10.1039/C6QO00153J. [DOI] [Google Scholar]; (c) Bhaskaran R. P. Babu B. P. Adv. Synth. Catal. 2020;362:5219–5237. doi: 10.1002/adsc.202000996. [DOI] [Google Scholar]; (d) Li G.-B. Zhang C. Song C. Ma Y.-D. Beilstein J. Org. Chem. 2018;14:155–181. doi: 10.3762/bjoc.14.11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ma J.-A. Cahard D. J. Fluorine Chem. 2007;128:975–996. doi: 10.1016/j.jfluchem.2007.04.026. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Lang S. B. Wiles R. J. Kelly C. B. Molander G. A. Angew. Chem., Int. Ed. 2017;56:15073–15077. doi: 10.1002/anie.201709487. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yue W.-J. Day C. S. Martin R. J. Am. Chem. Soc. 2021;143:6395–6400. doi: 10.1021/jacs.1c03126. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Zhao X. Li C. Wang B. Cao S. Tetrahedron Lett. 2019;60:129–132. doi: 10.1016/j.tetlet.2018.11.073. [DOI] [Google Scholar]; (b) Liu Y. Zhou Y. Zhao Y. Qu J. Org. Lett. 2017;19:946–949. doi: 10.1021/acs.orglett.7b00168. [DOI] [PubMed] [Google Scholar]; (c) Miura T. Ito Y. Murakami M. Chem. Lett. 2008;37:1006–1007. doi: 10.1246/cl.2008.1006. [DOI] [Google Scholar]; (d) Gøgsig T. M. Søbjerg L. S. Lindhardt A. T. Jensen K. L. Skrydstrup T. J. Org. Chem. 2008;73:3404–3410. doi: 10.1021/jo7027097. [DOI] [PubMed] [Google Scholar]; (e) Lu X. Wang X.-X. Gong T.-J. Pi J.-J. He S.-J. Fu Y. Chem. Sci. 2019;10:809–814. doi: 10.1039/C8SC04335C. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang M. Pu X. Zhao Y. Wang P. Li Z. Zhu C. Shi Z. J. Am. Chem. Soc. 2018;140:9061–9065. doi: 10.1021/jacs.8b04902. [DOI] [PubMed] [Google Scholar]; (g) Yao C. Wang S. Norton J. Hammond M. J. Am. Chem. Soc. 2020;142:4793–4799. doi: 10.1021/jacs.9b13757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Chelucci G. Chem. Rev. 2012;112:1344–1462. doi: 10.1021/cr200165q. [DOI] [PubMed] [Google Scholar]; (b) Zhang X. Cao S. Tetrahedron Lett. 2017;58:375–392. doi: 10.1016/j.tetlet.2016.12.054. [DOI] [Google Scholar]; (c) Ji X. Liu Y. Shi H. Cao S. Tetrahedron. 2018;74:4155–4159. doi: 10.1016/j.tet.2018.06.018. [DOI] [Google Scholar]
- For selected examples using organolithium species, see: ; (a) Bégué J.-P. Bonnet-Delpon D. Rock M. H. Tetrahedron Lett. 1995;36:5003–5006. [Google Scholar]; (b) Bégué J.-P. Bonnet-Delpon D. Rock M. H. J. Chem. Soc., Perkin Trans. 1. 1996;1:1409–1413. doi: 10.1039/P19960001409. [DOI] [Google Scholar]
- (a) Xiao T. Li L. Zhou L. J. Org. Chem. 2016;81:7908–7916. doi: 10.1021/acs.joc.6b01620. [DOI] [PubMed] [Google Scholar]; (b) Wiles R. J. Phelan J. P. Molander G. A. Chem. Commun. 2019;55:7599–7602. doi: 10.1039/C9CC04265B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For hydroacylation protocols, see: ; (a) Fan P. Zhang C. Lan Y. Lin Z. Zhang L. Wang C. Chem. Commun. 2019;55:12691–12694. doi: 10.1039/C9CC07285C. [DOI] [PubMed] [Google Scholar]; (b) Zhang M. Xie J. Zhu C. Nat. Commun. 2018;9:3517–3527. doi: 10.1038/s41467-018-06019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Salaheldin A. M. Yi Z. Kitazume T. J. Fluorine Chem. 2004;125:1105–1110. doi: 10.1016/j.jfluchem.2004.01.028. [DOI] [Google Scholar]; ; For hydroarylation protocols, see: ; (d) Prakash G. K. S. Paknia F. Vaghoo H. Rasul G. Mathew T. Olah G. A. J. Org. Chem. 2010;75:2219–2226. doi: 10.1021/jo9026275. [DOI] [PubMed] [Google Scholar]; ; For hydroformylation protocols, see: ; (e) Fanfoni L. Diab L. Smejkal T. Breit B. Chimia. 2014;68:371–377. doi: 10.2533/chimia.2014.371. [DOI] [PubMed] [Google Scholar]; ; For hydroalkylation protocols limited to stabilized benzylic and aliphatic heteroatom-based radicals, see: ; (f) Bergstrom D. E. Ng M. W. Wong J. J. J. Chem. Soc., Perkin Trans. 1. 1983;1:741–745. doi: 10.1039/P19830000741. [DOI] [Google Scholar]; (g) Hosoya A. Umino Y. Narita T. Hamana H. J. Fluorine Chem. 2008;129:91–96. doi: 10.1016/j.jfluchem.2007.09.002. [DOI] [Google Scholar]; (h) Gu F. Huang W. Liu X. Chen W. Cheng X. Adv. Synth. Catal. 2018;360:925–931. doi: 10.1002/adsc.201701348. [DOI] [Google Scholar]; (i) Li Y. Miyazawa K. Koike T. Akita M. Org. Chem. Front. 2015;2:319–323. doi: 10.1039/C4QO00352G. [DOI] [Google Scholar]; (j) Alfonzo E. Hande S. M. ACS Catal. 2020;10:12590–12595. doi: 10.1021/acscatal.0c03851. [DOI] [Google Scholar]; ; For hydroalkylation protocols that proceed under elevated temperatures or exclusively with 2-aminomalonates, see: ; (k) Sánchez Merino A. Alcañiz F. R. Gaviña D. Delgado A. Sánchez Roselló M. del Pozo C. Eur. J. Org. Chem. 2019;2019:6606–6610. doi: 10.1002/ejoc.201901098. [DOI] [Google Scholar]; (l) Gao L. Wang G. Cao J. Yuan D. Xu C. Guo X. Li S. Chem. Commun. 2018;54:11534–11537. doi: 10.1039/C8CC06152A. [DOI] [PubMed] [Google Scholar]; (m) Wu L. H. Cheng J. K. Shen L. Shen Z. L. Loh T. P. Adv. Synth. Catal. 2018;360:3894–3899. doi: 10.1002/adsc.201800740. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Crisenza G. E. M. Mazzarella D. Melchiorre P. J. Am. Chem. Soc. 2020;142:5461–5476. doi: 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kammer L. M. Badir S. O. Hu R.-M. Molander G. A. Chem. Sci. 2021;12:5450–5457. doi: 10.1039/D1SC00943E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noble A. Mega R. S. Pflästerer D. Myers E. L. Aggarwal V. K. Angew. Chem., Int. Ed. 2018;57:2155–2159. doi: 10.1002/anie.201712186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu B. Lim C.-H. Miyake G. M. J. Am. Chem. Soc. 2017;139:13616–13619. doi: 10.1021/jacs.7b07390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wu J. Grant P. S. Li X. Noble A. Aggarwal V. K. Angew. Chem., Int. Ed. 2019;58:5697–5701. doi: 10.1002/anie.201814452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fawcett A. Pradeilles J. Wang Y. Mutsuga T. Myers E. L. Aggarwal V. K. Science. 2017;357:283–286. doi: 10.1126/science.aan3679. [DOI] [PubMed] [Google Scholar]; (g) Chen L. Liang J. Chen Z. y. Chen J. Yan M. Zhang X. j. Adv. Synth. Catal. 2019;361:956–960. [Google Scholar]; (h) Zhang J. Li Y. Xu R. Chen Y. Angew. Chem., Int. Ed. 2017;129:12793–12797. doi: 10.1002/ange.201707171. [DOI] [Google Scholar]; (i) Chen D. Xu L. Long T. Zhu S. Yang J. Chu L. Chem. Sci. 2018;9:9012–9017. doi: 10.1039/C8SC03493A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zheng C. Wang G.-Z. Shang R. Adv. Synth. Catal. 2019;361:4500–4505. doi: 10.1002/adsc.201900803. [DOI] [Google Scholar]; (k) Tu H.-Y. Zhu S. Qing F.-L. Chu L. Chem. Commun. 2018;54:12710–12713. doi: 10.1039/C8CC07344A. [DOI] [PubMed] [Google Scholar]; (l) Chowdhury R. Yu Z. Tong M. L. Kohlhepp S. V. Yin X. Mendoza A. J. Am. Chem. Soc. 2020;142:20143–20151. doi: 10.1021/jacs.0c09678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a selected review, please see: ; Capaldo L. Ravelli D. Eur. J. Org. Chem. 2017;2017:2056–2071. doi: 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murarka S. Adv. Synth. Catal. 2018;360:1735–1753. doi: 10.1002/adsc.201701615. [DOI] [Google Scholar]
- Lang S. B. Wiles R. J. Kelly C. B. Molander G. A. Angew. Chem., Int. Ed. 2017;56:15073–15077. doi: 10.1002/anie.201709487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples on (hetero)aryl radical generation off-DNA, please see: ; (a) Nguyen J. D. D'amato E. M. Narayanam J. M. Stephenson C. R. Nat. Chem. 2012;4:854–859. doi: 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]; (b) Devery J. J. Nguyen J. D. Dai C. Stephenson C. R. J. ACS Catal. 2016;6:5962–5967. doi: 10.1021/acscatal.6b01914. [DOI] [Google Scholar]; (c) Poelma S. O. Burnett G. L. Discekici E. H. Mattson K. M. Treat N. J. Luo Y. Hudson Z. M. Shankel S. L. Clark P. G. Kramer J. W. Hawker C. J. Read de Alaniz J. J. Org. Chem. 2016;81:7155–7160. doi: 10.1021/acs.joc.6b01034. [DOI] [PubMed] [Google Scholar]; (d) Arora A. Teegardin K. A. Weaver J. D. Org. Lett. 2015;17:3722–3725. doi: 10.1021/acs.orglett.5b01711. [DOI] [PubMed] [Google Scholar]; (e) Arora A. Weaver J. D. Org. Lett. 2016;18:3996–3999. doi: 10.1021/acs.orglett.6b01718. [DOI] [PubMed] [Google Scholar]; (f) Singh A. Kubik J. Weaver J. Chem. Sci. 2015;6:7206–7212. doi: 10.1039/C5SC03013G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Singh A. Fennell C. J. Weaver J. Chem. Sci. 2016;7:6796–6802. doi: 10.1039/C6SC02422J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Senaweera S. Weaver J. D. J. Am. Chem. Soc. 2016;138:2520–2523. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ghosh I. König B. Angew. Chem., Int. Ed. 2016;55:7676–7679. doi: 10.1002/anie.201602349. [DOI] [PubMed] [Google Scholar]; (j) Marzo L. Ghosh I. Esteban F. König B. ACS Catal. 2016;6:6780–6784. doi: 10.1021/acscatal.6b01452. [DOI] [Google Scholar]; (k) Discekici E. H. Treat N. J. Poelma S. O. Mattson K. M. Hudson Z. M. Luo Y. Hawker C. J. de Alaniz J. R. Chem. Commun. 2015;51:11705–11708. doi: 10.1039/C5CC04677G. [DOI] [PubMed] [Google Scholar]; (l) Ghosh I. Ghosh T. Bardagi J. I. König B. Science. 2014;346:725–728. doi: 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]; (m) Bardagi J. I. Ghosh I. Schmalzbauer M. Ghosh T. König B. Eur. J. Org. Chem. 2018;2018:34–40. doi: 10.1002/ejoc.201701461. [DOI] [Google Scholar]; (n) Aycock R. A. Wang H. Jui N. T. Chem. Sci. 2017;8:3121–3125. doi: 10.1039/C7SC00243B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Aycock R. A. Vogt D. B. Jui N. T. Chem. Sci. 2017;8:7998–8003. doi: 10.1039/C7SC03612D. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Boyington A. J. Riu M.-L. Y. Jui N. T. J. Am. Chem. Soc. 2017;139:6582–6585. doi: 10.1021/jacs.7b03262. [DOI] [PubMed] [Google Scholar]; (q) Seath C. P. Vogt D. B. Xu Z. Boyington A. J. Jui N. T. J. Am. Chem. Soc. 2018;140:15525–15534. doi: 10.1021/jacs.8b10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Shu C. Madhavachary R. Noble A. Aggarwal V. K. Org. Lett. 2020;22:7213–7218. doi: 10.1021/acs.orglett.0c02513. [DOI] [PubMed] [Google Scholar]; (b) Wu J. He L. Noble A. Aggarwal V. K. J. Am. Chem. Soc. 2018;140:10700–10704. doi: 10.1021/jacs.8b07103. [DOI] [PubMed] [Google Scholar]; (c) Wu J. Bär R. M. Guo L. Noble A. Aggarwal V. K. Angew. Chem., Int. Ed. 2019;58:18830–18834. doi: 10.1002/anie.201910051. [DOI] [PubMed] [Google Scholar]
- Wang J. Lundberg H. Asai S. Martín-Acosta P. Chen J. S. Brown S. Farrell W. Dushin R. G. O'Donnell C. J. Ratnayake A. S. Richardson P. Liu Z. Qin T. Blackmond D. G. Baran P. S. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E6404–E6410. doi: 10.1073/pnas.1806900115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K. Rankic D. A. MacMillan D. W. C. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.-Z. Chen J.-R. Xiao W.-J. Org. Biomol. Chem. 2019;17:6936–6951. doi: 10.1039/C9OB01289C. [DOI] [PubMed] [Google Scholar]
- Renny J. S. Tomasevich L. L. Tallmadge E. H. Collum D. B. Angew. Chem., Int. Ed. 2013;52:11998–12013. doi: 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesi H. A. Hildebrand J. J. Am. Chem. Soc. 1949;71:2703–2707. doi: 10.1021/ja01176a030. [DOI] [Google Scholar]
- (a) Kölmel D. K. Loach R. P. Knauber T. Flanagan M. E. ChemMedChem. 2018;13:2159–2165. doi: 10.1002/cmdc.201800492. [DOI] [PubMed] [Google Scholar]; (b) Ruff Y. Martinez R. Pellé X. Nimsgern P. Fille P. Ratnikov M. Berst F. ACS Comb. Sci. 2020;22:120–128. doi: 10.1021/acscombsci.9b00164. [DOI] [PubMed] [Google Scholar]; (c) Wu R. Gao S. Du T. Cai K. Cheng X. Fan J. Feng J. Shaginian A. Li J. Wan J. Liu G. Chem.–Asian J. 2020;15:4033–4037. doi: 10.1002/asia.202001105. [DOI] [PubMed] [Google Scholar]; (d) Wen H. Ge R. Qu Y. Sun J. Shi X. Cui W. Yan H. Zhang Q. An Y. Su W. Yang H. Kuai L. Satz A. L. Peng X. Org. Lett. 2020;22:9484–9489. doi: 10.1021/acs.orglett.0c03461. [DOI] [PubMed] [Google Scholar]; (e) Kölmel D. K. Meng J. Tsai M.-H. Que J. Loach R. P. Knauber T. Wan J. Flanagan M. E. ACS Comb. Sci. 2019;21:588–597. doi: 10.1021/acscombsci.9b00076. [DOI] [PubMed] [Google Scholar]
- Note: During the course of our studies, an independent report22k on the alkylation of a selected number of DNA-conjugated acrylamide and acrylate derivatives promoted by BuNAH and NADH was disclosed, albeit with low reaction rates that required prolonged blue light irradiation. The scope was limited to secondary and tertiary redox-active esters, in clear contrast with conditions developed herein (Schemes 3 and 4)
- Bauer M. R. Di Fruscia P. Lucas S. C. C. Michaelides I. N. Nelson J. E. Storer R. I. Whitehurst B. C. RSC Med. Chem. 2021;12:448–471. doi: 10.1039/D0MD00370K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobbe P. Ruijter E. Orru R. V. A. MedChemComm. 2012;3:1189–1218. doi: 10.1039/C2MD20089A. [DOI] [Google Scholar]
- Kölmel D. K. Ratnayake A. S. Flanagan M. E. Tsai M.-H. Duan C. Song C. Org. Lett. 2020;22:2908–2913. doi: 10.1021/acs.orglett.0c00574. [DOI] [PubMed] [Google Scholar]
- Kölmel D. K. Ratnayake A. S. Flanagan M. E. Biochem. Biophys. Res. Commun. 2020;533:201–208. doi: 10.1016/j.bbrc.2020.04.028. [DOI] [PubMed] [Google Scholar]
- (a) Milligan J. A. Phelan J. P. Badir S. O. Molander G. A. Angew. Chem., Int. Ed. 2019;58:6152–6163. doi: 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Badir S. O. Molander G. A. Chem. 2020;6:1327–1339. doi: 10.1016/j.chempr.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith A. L. Goh S. H. J. Chem. Soc., Chem. Commun. 1983:907. doi: 10.1039/C39830000907. [DOI] [Google Scholar]
- Ding H. Greenberg M. M. J. Org. Chem. 2010;75:535–544. doi: 10.1021/jo902071y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental procedures, characterization data, as well as copies of NMR and LCMS spectra supporting this article have been uploaded as part of the ESI.†