
Figure 4. Comparisons of intersubunit LB1 domains interfaces in the inactive and active states of CaSR.
(A) Left panel: The Cα trace of VFT module of CaSRfully inactive cryo-EM structure (cyan). The B-C Helix angle is 117°. Right panel: The Cα trace of VFT module of CaSRagonist+PAM cryo-EM structure (purple). The B-Helix angle is 89°. (B) Left panel: The Cα trace of VFT module of crystal structure of CaSR-ECDIoo (yellow) (PDB:5K5T). The B-Helix angle is 89°. Right panel: The Cα trace of VFT module of CaSR-ECDactive crystal structure (red) (PDB:5K5S). The B-Helix angle is 89°. (C) Left panel: The Cα trace of VFT module of CaSRIcc cryo-EM structure (brown) (PDB:7DTW). The B-Helix angle is 79°. Right panel: The Cα trace of VFT module of CaSRAcc cryo-EM structure (lavender) (PDB:7DTV). The B-Helix angle is 80°.
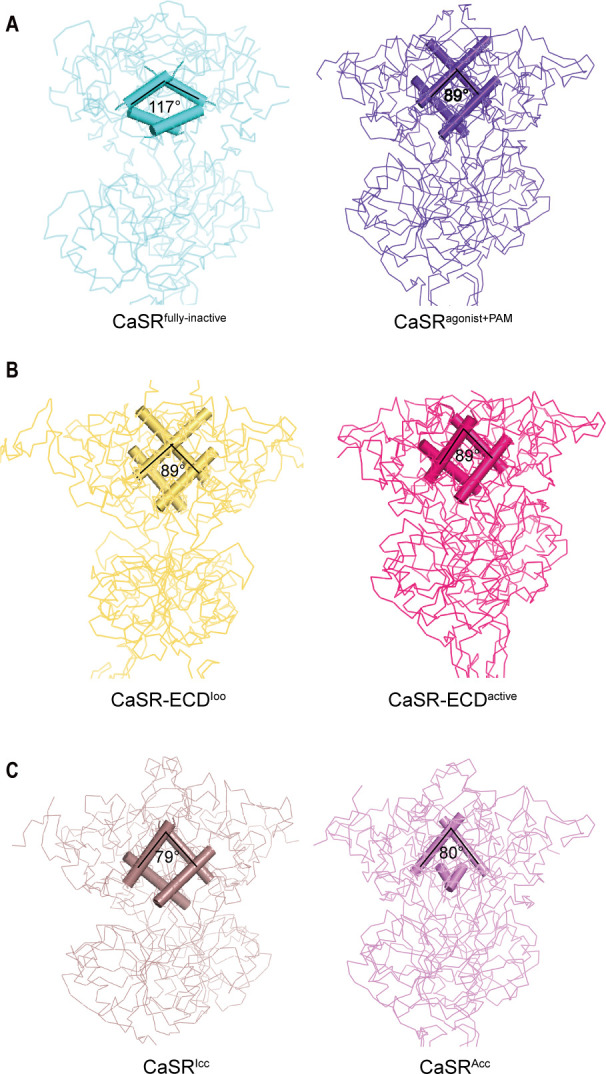
Figure 4—figure supplement 1. Comparisons of intersubunit LB1 domains interfaces in the inactive and active states of CaSR.

(A, B) The interface is a hydrophobic patch between residues on the B and C helices of each protomer. In the inactive conformation, it is an interface involving V115, V149, and L156 residues (A, cyan), whereas LB1 interface of the agonist+PAM bound state (B, purple) is packed with residues L112, L156, L159, and F160. (C) Left panel: The Cα trace of VFT module of inactive mGluR cryo-EM structure (raspberry) (PDB: 6N52). Right panel: The B-Helix angle is 125°. (D) Left panel: The Cα trace of VFT module of active mGluR cryo-EM structure (green) (PDB: 6N51). Right panel: The B-Helix angle is 66°. (E, F) Alignment of LB1 domain of CaSR in three conformations: fully inactive (cyan), intermediate (yellow, PDB:5K5T), and agonist+PAM bound (purple) states, ligand-binding residues (red). (E) Fully inactive and intermediate LB1 domain, the conformation of the ligand-binding region in LB1 domain is significantly different in two states. (F) intermediate and agonist+PAM LB1 domain, showing a well superposition.