

Summary
Transcription factors (TFs) are essential mediators of epigenetic regulation and modifiers of penetrance. Studies from the past decades have revealed a sub-class of TF that is capable of remodeling closed chromatin states through targeting nucleosomal motifs. This pioneer factor (PF) class of chromatin remodeler is ATP independent in its roles in epigenetic initiation, with nucleosome-motif recognition and association with repressive chromatin regions. Increasing evidence suggests that the fundamental properties of PFs can be coopted in human cancers. We explore the role of PFs in the larger context of tissue-specific epigenetic regulation. Moreover, we highlight an emerging class of chimeric PF derived from translocation partners in human disease and PFs associated with rare tumors. In the age of site-directed genome editing and targeted protein degradation, increasing our understanding of PFs will provide access to next-generation therapy for human disease driven from altered transcriptional circuitry.
Subject areas: Cancer systems biology, Epigenetics, Molecular biology, Systems biology
Graphical abstract
Cancer systems biology; Epigenetics; Molecular biology; Systems biology
Introduction
With approximately 3 billion DNA bp in the human genome and 38 million bp of coding sequence, we evolved precise keys to “unlock” regulatory motifs amidst long DNA sequences. The canonical transcription factors (TFs) evolved to recognize specific DNA motifs, providing precision, logic, and complexity in gene regulation. However, there is a general lack of sequence-specific nucleosome positioning in mammalian genomes. While TFs with relatively complex motifs can have million-fold specificity (an 11-bp OCT4-SOX2 motif has 411 specificity), even specific motifs occluded by nucleosomes may go unrecognized. “How do TFs establish or respond to cell fate changes, when a motif has variable accessibility in a cell of origin?” Pioneer factors (PFs) are capable of binding nucleosomal motifs and inducing DNA accessibility (Zaret and Carroll, 2011). The function of PFs has revealed fundamental properties for promoter selection (Hartley and Madhani, 2009) and core transcriptional circuitry (Michael et al., 2020), while little is known about chimeric- or oncogenic-fusion TFs in this context. This review aims to define central questions and fundamental advances in understanding the basic epigenetic functions of tissue-specific PFs and relate these findings to disease etiology.
PFs have the central features of (1) binding to closed chromatin states, (2) altering DNA accessibility at its targets, (3) the capability of reprograming cell fate decisions (Iwafuchi-Doi and Zaret, 2016), and increasing biophysical evidence has enabled new insights into the (4) direct molecular recognition of nucleosomal motifs (Donovan et al., 2019; Fernandez Garcia et al., 2019) (Figure 1). These central attributes are an aperture through which to view developmental changes and pathological cell fate decisions in human disease. We first focus on the mechanisms of binding to closed or repressive chromatin, highlighting transient site exposure, nucleosome motif recognition, histone mimicry, and genomics approaches for determining pioneer activity. Secondly, we focus on pioneer function and cell state, highlighting key PFs in each section, with examination of pluripotency, sarcomas, brain tumors, myogenesis, myogenic tumors, immune cell development, and leukemias. Key PFs we highlight are in the FOXO, FOXA, PAX, SOX, GATA, and CEBP families, among others. The attributes of PFs have implications for understanding altered transcription in human cancers (Bradner et al., 2017) and for defining the emergent transcriptional “circuitry” in response to a transforming event. The central four characteristics of PFs also provide exciting new context in the early changes in cancer genomes involving chromosomal translocations, resulting in PF chimeras. The unknown functions of translocation-induced PF chimerism motivate the evaluation of neomorphic pioneer characteristics in human tumors, linking DNA accessibility to human disease etiology. We examine the central attributes of PF function in a range of developmental and pathological processes and acknowledge PF chimeras with unknown function. There are several major implications for this analysis of the literature: PFs play essential roles in tissue-specific chromatin organization, and the molecular functions of PFs may stabilize maintenance of the epigenetic state across cell division (Figure 1).
Figure 1.

Pioneer function and epigenetic state changes
Evidence suggests that pioneer function is an early step in the establishment of an epigenetic state, with nucleosomal-motif recognition and binding in repressive chromatin regions. For distinct classes of pioneer factors, the recruitment of epigenetic machinery for chromatin activation might have distinct mechanisms.
Mechanisms of binding to closed chromatin
Transient site exposure
The nucleosome complex, comprising DNA and histone proteins, presents a steric challenge to canonical TFs in accessing their DNA sequence motifs: they are often buried against the surface of the histone octamer. A possible energetically neutral solution to this lies in the natural dynamics of the nucleosome itself. There is evidence that the DNA-nucleosome complex can fluctuate in equilibrium states between fully wrapped and transiently accessible states, which suggests a site exposure model (Anderson and Widom, 2000; Polach and Widom, 1995). Widom & colleagues observed that transient motif accessibility occurs at equilibrium, with 1–10% unwrapping at any given time (Figure 2). The relative abundance of accessible states varies such that motifs nearest to entry-exit sites may have increased exposure relative to motifs that are buried further into the nucleosome core (Battistini et al., 2012). Thus, context is important for considering the steric constraints of the nucleosome. These studies provide interesting insights into relative rates of natural DNA unwrapping from nucleosome particles, with rates nearly 20-times faster with enzymatic unwrapping (remodeling) than with equilibrium “site exposure”. For non-pioneer TFs that rely on accessible motifs, these observations lead to a model where opportunistic motif binding can occur with molecular recognition of transiently exposed nucleosomal sites. Nucleosome invasion can shift the equilibrium toward motif accessibility, increasing the stable binding equilibria of TFs (Li and Widom, 2004). A common energetic driver for this is that enthalpic costs of unwrapping are offset by increased local entropy and rotational freedom resulting from site exposure.
Figure 2.

Integration of the site exposure model with pioneer function at nucleosomal motifs
(A) Conceptual model for the kinetics of pioneer factors, which can associate with nucleosomes in wrapped or unwrapped states.
(B) Fitting our derived equation for fractional motif occupancy (Equation 3) with kinetic rate constants from Zaret and Poirier for pioneer factors and transcription factors in the context of fractional occupancy of TF-motif pairs.
The site exposure model also provides context for invasion from neighboring nucleosomes, where transient unwrapping has been shown to accommodate local nucleosome overlap (Engeholm et al., 2009). For equilibrium unwrapping, the relative free energy differences in accessibility can be calculated as follows:
| (Equation 1) |
where the free energy (ΔG) is a function of the equilibrium rate constant of DNA accessibility (Anderson et al., 2002). Clever experimental approaches to derive free energies of in vitro nucleosome positioning (Lowary and Widom, 1998) have measured relative restriction enzyme activities as a surrogate to approximate the unwrapping equilibrium constants. In reconstitution experiments and quantitative measurements of the site exposure Keq from nucleosome arrays of distinct lengths, the free energy differences relative to site exposure (ΔΔG) are not significantly impacted by lengthening of the nucleosome arrays. These results suggest that nucleosome translocation is a significantly slower process than equilibrium unwrapping. The activation barrier for nucleosome translocation is kinetically rate limiting, and the equilibrium process for unwrapping occurs rapidly on the order of seconds. The effects of ATP-dependent remodeling, nucleosome invasion by TFs, or acetylation of the histone tail increase the site exposure Keq, while distance of a motif inward toward the nucleosome core decreases the Keq (Anderson et al., 2001; Anderson and Widom, 2000). Studies with fluorescence resonance energy transfer (FRET) systems have also supported the model that local site exposure can lead to the establishment of accessible states locally, via TF binding (Poirier et al., 2009). While these observations suggest a possible mechanism of stabilizing TF motif accessibility, the question remains, “if the equilibrium state of the nucleosome-DNA complex is wrapped 90% of the time (Anderson and Widom, 2000; Polach and Widom, 1995), how can new chromatin states be rapidly established?”
Binding to nucleosomal motifs
The evolution of factors that have reduced off-rates with nucleosomal motifs perhaps is Nature's way of targeting the DNA-nucleosome complex independently of transient site exposure. Widom & colleagues predicted that factors binding to nucleosomal motifs may be negatively affected in their target search processes in proportion to the Keq of site exposure (Anderson et al., 2002; Polach and Widom, 1999), while there is evidence that PFs can also bind to transiently unwrapped states (Donovan et al., 2019) (Figure 2). Integrations of the site exposure model and pioneer function provide interesting questions about the equilibrium state of the chromatin fiber (Dudchenko et al., 2017; Sanborn et al., 2015). If the target- search and motif recognition properties of PFs are at equilibrium, then a slow could lead to transiently remodeled nucleosome structures that may relax into accessible states (Figure 2). However, with rapid signal flux or during the initiation of developmental or pathological cell reprograming changes, kinetic control may impose energetic constraints on the early targeting properties of pioneers.
Studies supporting the roles of chromatin remodeling in positioning of nucleosomes at promoters (Yen et al., 2012; Zhang et al., 2011) and the nucleosome-free region (NFR) (Hartley and Madhani, 2009) have motivated questions for how TFs and PFs might recognize motifs in either of these contexts. With nucleosome depletion at the NFR, it is predicted that TFs may have greater access to their motifs, while strongly positioned nucleosomes at promoters might be targeted with stronger binding by PFs. The target search function of non-pioneer TFs is likely facilitated by opportunistic binding to transiently unwrapped nucleosome-DNA complexes. In recent years, experiments at the single molecule and ensemble level have enabled the dissection of kinetic rate constants for this process. Under equilibrium conditions, TF binding to motifs may be facilitated by nucleosome-DNA unwrapping, with TF-motif dissociation constant:
| (Equation 2) |
The represents the off-rate from the TF-motif interface, while represents the on-rate. With single molecule studies, LexA (a non-pioneer) had 1000-fold faster from nucleosomal motifs relative to non-chromatinized DNA (Luo et al., 2014) (cf., Figure 2). Gal4 had a 100-fold increased from nucleosomal motifs relative to non-chromatinized DNA and 1000-fold weaker for nucleosomal motifs relative to naked DNA (Liang et al., 1996; Luo et al., 2014). One possible explanation for these results is that nucleosome re-wrapping can dramatically accelerate the for TFs (North et al., 2012) (Figure 2). For motifs proximal to the entry-exit sites of the nucleosome, a faster equilibrium can be established which may allow for rapid gene regulation. With several orders of magnitude slower dissociation constants for TFs with unwrapped motifs, the on-rates may influence the overall rates of gene activation.
At equilibrium, the fractional motif occupancy is dependent solely upon the and the concentration of TF, from the following derivation. The as a function of the concentration of TF and is derived from (Equation 2) and the equivalence of with . Moreover, the total abundance of motif is equivalent to the total sum of bound and unbound motif, which is the sum of and . This equivalence allows for the to be related directly to the ratio of and . This ratio simplifies to the expression in (Equation 3), as the ratio of and .
| (Equation 3) |
The binding dissociation constant for the TF and its motif is highly sensitive to for which nucleosome positioning is critical. A visual representation of this equivalence (Equation 3) is that for the pioneer FOXA1, where fractional occupancy at its motifs approaches saturation at much lower concentrations relative to non-pioneer LexA, which requires much higher concentrations to occupy its motifs when sterically occluded by nucleosomes (Figure 2). This visual representation is achieved merely by plotting as a function of with values from nucleosomal versus non-nucleosomal motifs obtained from kinetic studies (Iwafuchi-Doi et al., 2016; Luo et al., 2014) (Figure 2). These concepts are useful in describing TF-motif interactions at the “local” level, and extension of these principles to the genomic level leads to interesting hypotheses. Networks of TFs can regulate their own enhancer elements to integrate local fractional occupancy into what we think of as “core regulatory circuitry” (Boyer et al., 2005). Interestingly, the extension of local fractional occupancy into core circuitry then becomes dependent on TFs and PFs, nucleosome positioning, and chromatin remodeling. The locus-specific definition for becomes a single component of this circuitry, which is stabilized through TFs and PFs recognizing their motifs (e.g., enhancer accessibility) and inducing their own expression. An interesting extension of this concept is that core circuitry would be highly dependent only on the binding kinetics for TFs with their motifs and the expression level of TFs, without being a function of the intracellular abundance of motifs. We will discuss the implications of this concept for development and human disease for which cells are “locked” in an epigenetic state conferred by altered core circuitry.
An important caveat here is that the in vivo influences from the native milieu may invoke differences from predictions of in vitro TF binding models. These important differences can be evidenced from single-cell studies of transcriptional burst frequencies, which are highly sensitive to the local chromatin remodeling context (Kar et al., 2017). Evidence that local chromatin state influences transcriptional bursting is reinforced by the observation that dwell times of TFs in vitro would predict much longer transcriptional bursts than are observed in vivo. Thus, while the proximity of the motif with the nucleosome core may provide rapidly tunable and context-specific scenarios for gene regulation, further work will be important for understanding how TF residency times instruct transcriptional frequencies. These studies also provide insight into stably expressed TF networks that may require motif accessibility for maintenance.
The differences in behavior between TFs and PFs also have implications for the dynamics of transcriptional control. For classical TFs like Gal4, LexA, and c-Myc, the off-rates seem to be most sensitive to the nucleosomal context (Donovan et al., 2019; Luo et al., 2014; Soufi et al., 2015), essentially “tuning” the system for rapid gene control versus constitutive expression: (1) thermodynamically controlled with positioned nucleosomes, rapid , equilibrium can be achieved within the timescale of cell division, or (2) kinetically controlled with nucleosome depletion at motifs, equilibrium is achieved on the timescale approaching mitosis, with slow . For PFs, the nucleosomal motif context may actually reduce providing an alternative manifold for binding equilibria by reducing and stabilizing fractional occupancy.
Rap1, Reb1, Cbf1, and FOXA1: binding kinetics and histone mimicry
While classical TFs have relatively high dissociation rate constants from nucleosomal motifs, PFs are a class of TF with intrinsically slow off-rates from chromatinized motifs. Both Reb1 and Cbf1 preferentially remain associated with partially unwrapped DNA-nucleosome complexes in single-molecule experiments (Donovan et al., 2019). The approximately 100-fold decreases in for these factors recognizing nucleosomal DNA substrates are thought to represent kinetic trapping of nucleosomal motifs. Another TF with non-classical binding kinetics is the winged-helix TF, FOXA1, which has been shown to compete with, or mimic, histone H1 and exhibits slowed dissociation from nucleosomes (Cirillo et al., 1998, 2002; Shim et al., 1998). Nuclear mobility with fluorescence recovery after photobleaching (FRAP recovery) is several orders of magnitude slower for FOXA1 relative to c-Myc, NF-1, HMGB1, and GATA4 (Sekiya et al., 2009). The trends in mobility are inversely related with nucleosome-binding ability by EMSA, suggesting functional binding in vivo. On naked DNA, FOXA1 displays only a 2-fold enhanced binding affinity relative to MYC, while it has a ∼10-fold enhanced binding affinity relative to MYC on nucleosomes containing its motifs (Sekiya et al., 2009). In genomic localization studies, FOXA1 localizes to inactive chromatin and occupies its binding motifs independently of other TFs, which is consistent with pioneer function (Fu et al., 2019; Glont et al., 2019).
The yeast PF Rap1 has Myb-like domains that facilitate similar on rates for nucleosomal and non-nucleosomal motifs (Mivelaz et al., 2020). With single-molecule total internal reflection fluorescence (TIRF) imaging, binding interactions to compacted nucleosomal arrays encoding Rap1 motifs occur with a majority of binding events resulting in short residency times (< 1s) and additional interactions associated with longer residency. These studies support a model where Rap1 encounters higher-order chromatin structure but continually and rapidly “samples” motifs with short-lived binding interactions until stable motif recognition is achieved. Interestingly, rates were similar regardless of Rap1 motifs encoded in naked DNA, nucleosomal DNA, or compacted chromatin fibers. However, is markedly different (approximately 10-fold) when comparing Rap1 dissociation with nucleosomal vs free DNA motifs. Thus, rapid DNA-motif searching occurs at nucleosomal binding sites, with differences in dominating the kinetics until stable motif interactions can occur. In vitro, Rap1 can associate with chromatinized motifs to destabilize higher-order nucleosome structure without resulting in nucleosomal eviction. However, evidence suggests that stable Rap1 binding with nucleosomal motifs lowers the energy barrier for ATP-dependent nucleosome displacement. The pioneer function and domain structure of Rap1 provides an interesting contrast to the yeast pioneer Reb1, which also binds to chromatinized motifs, but inherently destabilizes its target nucleosomes in the process (Donovan et al., 2019).
The knockin of Reb1 motifs into nucleosomal substrates results in de novo formation of nucleosome-depleted regions (NDRs), which can be reversed with rapid degradation of Reb1 at the protein level (Hartley and Madhani, 2009). In an interesting comparison to Rap1, the yeast PF Reb1 has a 22-fold equilibrium binding preference for nucleosomes containing its specific binding site, with the serving as the determining factor to differentiate naked DNA from chromatinized motifs. For Reb1, the molecular recognition of its target on nucleosomes results in markedly slower off rates in comparison to non-nucleosomal motifs (cf., Figure 2). In genome-wide studies, Rap1 is enriched at the promoter-distal side of the −1 nucleosome, while Reb1 has been observed to associate with the promoter-proximal side of the −1 nucleosome at the NFR boarder (Koerber et al., 2009; Lee et al., 2007; Zhang et al., 2011).
Comparisons of Rap1 and Reb1 binding with MNase-ChIP have implicated the site exposure model in Reb1 function (Koerber et al., 2009). The observed nucleosomal-Reb1 DNA fragments released from MNase digestion were shorter than the expected length for an intact nucleosome, which is consistent with “invasion” through transient unwrapping. This provides interesting context for the site exposure model, which proposes that equilibrium binding of factors to nucleosomal DNA may not require additional energy (ATP), but rather the intrinsic, dynamic unwrapping and re-wrapping of nucleosomes may provide altered apparent values similar to naked DNA (Polach and Widom, 1996). Indeed, recent investigations of Rap1, Reb1, and Cbf1 provide examples where motif searching can occur rapidly, but dissociation rates are defining for pioneer function. Given the similar pattern for rates with nucleosomal motifs among these factors, it is possible that transient site exposure may influence the more than the on-rate. It will be of high interest to understand how target search and remodeling processes of pioneer function might be coupled to the thermodynamics of DNA unwrapping. Integrating these data with the model for fractional occupancy leads to questions about motif representation across the genome (cf., Equation 3): if the motif abundance matters less than the strength of binding of a TF-motif pair in a nucleosomal context, does it follow that the essentiality of TFs in a tissue type would be independent of their genomic motif representation?
Given the unique binding kinetics of PFs, which may have relatively slow dissociation rates from chromatinized motifs relative to TFs (Figure 2), it will be interesting to determine the extent to which these factors utilize conserved domains also with conserved functions. Recent structural comparisons suggest PFs often exhibit relatively short alpha-helical DNA recognition domains that sterically permit recognition of nucleosomal motifs (Fernandez Garcia et al., 2019). Yet, common predictive “rules” for how these structural domains influence PF dissociation rates will require further study.
While often considered the commonest transcriptional repressor, nucleosomes also have unique roles in regulated recruitment. Recent genome-wide proteomics screens have revealed that a highly conserved mode of interaction between chromatin regulatory factors and nucleosomes is the acidic patch region on the H2A-H2B interface (Kalashnikova et al., 2013; Skrajna et al., 2020). Recent studies to evaluate the roles of histone tails and nucleosome disk surface residues revealed that the nucleosome acidic patch is a major hub for regulated recruitment. The exciting progress in understanding nucleosome function in regulated recruitment leads to the questions, (1) do PFs target specific regions on the nucleosome in addition to their motifs, and (2) what are the individual influences of nucleosomal recognition and DNA binding on the ?
Early nucleosome protection assays have provided important clues for understanding how nucleosomes interact with PFs. Gal4 fails to alter MNase preferences in the presence of competitor DNA, while FOXA1 protects nucleosome fragments from nuclease digestion even with naked DNA encoding its motifs (Cirillo and Zaret, 1999). These findings have provided general analogy for similar nucleosomal binding preferences among winged-helix domain containing PFs (e.g., FOXA1, FOXO1) and for histone H1. In each case, the protein can engage through interactions with DNA at the nucleosome dyad and linker regions. These binding modes are distinct in comparison with the multi-zinc finger domain of Gal4, which is highly sensitive to the steric constraints of the nucleosome. Despite slower , the slower off-rates of FOXA1 with nucleosomal motifs result in stable positioning (Figure 2). These findings connect to more recent work on the single-cell level, showing nucleosome spacing is more stable at silent regions poised for gene activation, while 5′-positioning of nucleosomes is more stable at active regions (Lai et al., 2018). Future investigations will be important for determining the extent to which stable nucleosome spacing results from PF-nucleosome interactions, for which slow off-rates allow genomic association even in heterochromatic states. The slower dissociation kinetics for PFs from nucleosomes is also reflected in tandem chromatin immunoprecipitation (ChIP-reChIP) assays for TF histones which are effective for FOXA2, CEBPβ, and HNF4α (Iwafuchi-Doi et al., 2016). These results are consistent with PFs having slow dissociation rates from nucleosomal motifs.
Genomics approaches for determining pioneer activity
The synthetic biology approaches to “generate chromatin in a test tube” are powerful because they allow for complete control over the chromatin environment. The ability to uncouple the continuous biochemistry happening inside of the cell from functions of specific chromatin regulators can allow us to discern cause from consequence. However, in vitro reconstitution of nucleosome structures may yield different answers from cell biology approaches. The behavior of PFs including Rap1 and Reb1 at nucleosome-associated motifs in vitro deviates significantly from what is observed inside of the cell (Kaplan et al., 2009). This apparent discrepancy may be related to the off-rates of PFs with nucleosomes in the presence of active ATP-dependent chromatin remodeling (Figure 1) (Zhang et al., 2011). However, there have been impactful studies combining synthetic and cell biology approaches. The insertion of PF binding sites into stably positioned nucleosomes results in nucleosome displacing activity in vivo (Yan et al., 2018), whereas the eviction of nucleosomes is rare for the analogous in vitro experiments (Donovan et al., 2019). Despite differences in positioning in vivo, pioneer motifs have been found within nucleosomal binding sites in studies with distinct approaches to chromatin sequencing (Brogaard et al., 2012; Michael et al., 2020). Work with MNase-sequencing revealed that gene activation is highly predictive of strongly positioned nucleosomes near the promoter (Kaplan et al., 2010; Schones et al., 2008). The nucleosomal binding preferences for PFs can be challenging to measure by chromatin sequencing at steady state, and rapidly inducible control over PFs is important for their experimental characterization (Sunkel et al., 2021). Of note, the induced DNA unwrapping upon binding to Reb1 motifs near the entry/exit sites is supported by the predominance of sub-nucleosomal MNase fragments at its binding sites (Henikoff et al., 2011). These sites may result from partial nucleosome unwrapping or destabilization following PF binding inside of the cell. Observed PF occupancy at a given genomic locus in a cell may be the product of repetitive nucleosome recognition-eviction cycles and cooperative TF occupancy.
While the ability of PFs to recognize nucleosomes containing their motifs is a central attribute, a key question is, is this process limited by the local context in highly repressed regions of the genome? One powerful method to address this question relies on sequencing and proteomics analysis of regions of the genome that are refractory to endonuclease cleavage (sonication-resistant heterochromatin; srHC) (Becker et al., 2017; Fernandez Garcia et al., 2019; Sunkel et al., 2021). The conceptual basis for this is that the effects exerted by the mechanical force of sonication would be influenced by local chromatin repression. Sucrose gradients can been used to fractionate chromatin based on biophysical properties, prior to chromatin sequencing (Gilbert et al., 2004). By coupling sucrose gradient sedimentation of sonicated chromatin with genomics (Gradient-seq), it is possible to analyze PF binding sequences that are generally excluded from accessible regions. This addresses a common experimental bias in epigenetics, which leads to over-representation of accessible DNA in ChIP-seq, CUT&Tag, and MNase-seq experimental workflows (see Table 1). However, recent studies have enabled meta-analyses of ATAC-seq data with ChIP-seq data, computationally inferring nucleosome positioning from NucleoATAC (Beati and Chereji, 2020; Schep et al., 2015; Sunkel et al., 2021). Integrations of PF localization and nucleosome positioning will continue to illuminate the genomics of PFs.
Table 1.
Experimental approaches to define pioneer function
| Pioneera | Domain | Method | References |
|---|---|---|---|
| OCT4 (POU5F1) | POU, homeodomain | ChIP-seq, Cut-and-tag SeEN-seq, Cryo-EM, EMSA |
(Meers et al., 2019; Michael et al., 2020; Echigoya et al., 2020; Soufi et al., 2015) |
| SOX2 | HMG | ChIP-seq, Cut-and-tag SeEN-seq, EMSA |
(Meers et al., 2019; Michael et al., 2020; Soufi et al., 2015) |
| MYOD1 (MYF3) | bHLH | ChIP-seq, endonuclease assays | (Gerber et al., 1997; Bergstrom et al., 2002) |
| GATA3 | HMG | ChIP-seq, ATAC-seq, EMSA, Cryo-EM | (Tanaka et al., 2020) |
| GATA4 | HMG | Cut-and-tag, EMSA, nucleosome arrays | (Meers et al., 2019; Cirillo et al., 2002; Fernandez Garcia et al., 2019) |
| PU.1 (SPI1) | ETS, winged-helix | EMSA | (Feng et al., 2008; Fernandez Garcia et al., 2019) |
| EOMES | HMG | Cut-and-tag | (Meers et al., 2019) |
| PAX6 (FVH1) | Paired box, homeodomain | ChIP-seq, ectopic expression | (Sun et al., 2015; Halder et al., 1995) |
| PAX7 (HUP1) | Paired box, homeodomain | ATAC-seq, FAIRE-seq, ChIP-seq | (Budry et al., 2012; Mayran et al., 2018) |
| ASCL1 | bHLH | EMSA, ChIP-seq, DNase-seq | (Fernandez Garcia et al., 2019; Raposo et al., 2015) |
| FOXA1 (HNF3A) | Winged-helix | EMSA, nucleosome arrays | (Cirillo et al., 2002) |
| FOXA2 (HNF3B) | Winged-helix | Cut-and-tag | (Meers et al., 2019) |
| FOXD3 (HFH2) | Winged-helix | ChIP-seq | (Xu et al., 2009; Krishnakumar et al., 2016) |
| FOXO1 (FKHR) | Winged-helix | DNase1-footprinting, Nucleosome arrays | (Hatta and Cirillo, 2007) |
| CEBPα CEBPβ |
bZIP | ChIP-reChIP, EMSA | (Iwafuchi-Doi et al., 2016; Fernandez Garcia et al., 2019) |
| Rap1 | Myb-type | smTIRF | (Mivelaz et al., 2020) |
| Reb1 | Basic patch, HTH | PIFE, FRET, EMSA, degron alleles | (Hartley and Madhani, 2009; Donovan et al., 2019) |
| Cbf1 | bHLH | PIFE, FRET, EMSA | (Donovan et al., 2019) |
These factors are featured, with central attributes of pioneer function defined from the literature, along with domains and the relevant methods for characterization.
Indicates 2 or more of the 4 essential characteristics of pioneer function described in this manuscript.
The conceptual basis for Gradient-seq also relates to reports from the early 2000s where phenol-chloroform DNA purification was performed prior to reverse cross-linking but after chromatin shearing: the intrinsic physical properties of the DNA-nucleosome complexes in their domain contexts were shown to affect a partition coefficient across hydrophobic (chloroform-phenol: repressive chromatin) and hydrophilic (aqueous: accessible chromatin) phases. As this separation technique developed into FAIRE-seq, it allowed for the sequencing of repressive chromatin (Nagy et al., 2003). Conceptually, this has been further developed for defining the phasing properties of chromatin. The partitioning of chromatin regions into liquid-liquid “phase condensates” has revealed that H3K9me3-enriched domains may have the properties associated with hydrophobic phases, while H3K27ac regions have the properties associated with hydrophilic phases (Larson et al., 2017; Strom et al., 2017). Definition of mechanisms for formation of phase condensates will be important for understanding how DNA accessibility might regulate the local cellular environment.
The mechanistic insights from phase separation studies, FAIRE-seq, and srHC provided the framework for understanding sonication-resistant heterochromatin at both the genomics and proteomics levels (Becker et al., 2017). TFs expected to reside in the euchromatin are present in the heterochromatin gradient-seq fractions including EWSR1, commonly found translocated in Ewing sarcoma (Delattre et al., 1992). These advances raise pressing questions about the roles of PFs in srHC and the degree to which methods themselves are influencing our concept of chromatin structure, with (1) accessibility-based approaches (ATAC-seq, Cut&Tag) or (2) sequencing mono-nucleosomal fragment lengths (ChIP-seq, MNase-seq). As mutation rates are higher in srHC (Carone and Lawrence, 2013), it will be important to understand functional regulation in heterochromatin regions and whether one of the functions of srHC is for functional repression of mutations or repeat elements. With studies connecting the heterochromatin targeting of H4K20-methylated at H3K9me3-enriched regions, it will be interesting to further investigate unbiased proteomics approaches for srHC to understand how repressive histone marks and PFs may be interrelated (Sunkel et al., 2021).
The structural constraints of repression imposed by constitutive heterochromatin are coming into focus with recent imaging studies (Maiser et al., 2020; Ou et al., 2017). However, central questions remain for how PFs may be targeted to heterochromatin during interphase or in cycling cells. Job Dekker & colleagues recently observed that chromatin contact domains are cell cycle regulated, where CTCF and cohesin are released from chromatin during key stages in mitosis (Abramo et al., 2019). Cell cycle dependence of chromatin regulators has also been observed with “placeholder” nucleosomes in development (Dunleavy et al., 2011; Murphy et al., 2018; Tallan and Stanton, 2021). Understanding the bookmarking or placeholding function of PFs during the cell cycle will be critical for definition of epigenetic memory. How many PFs will also be bookmarking factors? Is bookmarking a central feature for pioneers, and will this distinction allow for parsing of sub-classes of PFs? These questions will be of interest for sub-classifying PFs and to define mechanisms of retention on mitotic chromatin for epigenetic inheritance.
With recent evidence that mitotic bookmarking is a function of key developmental PFs, we are able to gain new insight into epigenetic memory. PAX3, which contributes to myogenesis and neurogenesis, has intrinsic mitotic bookmarking capacity, which might contribute to tissue-specific memory (Wu et al., 2015). Patterns of histone marks and chromatin domains may also anticipate developmental changes in cell division. The de novo deposition of H4K20-methylation is highly associated with asymmetry in developmental chromatin replication (Ma et al., 2020). These findings connect the placement of histone marks associated with sonication-resistant heterochromatin and PFs in the context of epigenetic memory. Key insights about read-write mechanisms for heterochromatin inheritance have also provided clues for how pioneer function may also instruct inheritance of chromatin states, through associating with repressive regions (Reinberg and Vales, 2018). Characterizing bookmarking functions of PFs, modeled in experimental differentiation systems, will be of high interest for understanding both the maintenance and the loss of tissue-specific epigenetic inheritance.
Pioneer function and cell state
OCT4, SOX2, and ASCL1: pluripotency, connective tissue cancers, and brain tumors
The “core circuitry” or feed-forward transcriptional loops for OCT4-SOX2 implicate these PFs as binding to and stimulating their own regulatory elements (Boyer et al., 2005). These factors were found to primarily localize to their own promoters or enhancers or those of other TFs. The concerted autoregulatory capacity for OCT4-SOX2 targets scale with their expression levels. A model emerged where pluripotency master TFs could stabilize an epigenetic state defined by their own autoregulation. One of the central features of PFs as reprogramming factors in cell fate decisions (Figure 1) was revealed when pools of regulatory TFs were expressed in differentiated cells to isolate the master regulators that could direct pluripotency reprogramming. These studies revealed that OCT4-SOX2, NANOG, and KLF4 had sufficiency to reprogram transcriptional circuitry and induce a new epigenetically stable state (Takahashi and Yamanaka, 2006). Key questions emerged as to how this regulation occurred at the chromatin structural level.
In embryonic stem cells (ESCs), OCT4 and SOX2 bind as heterodimers to regulatory regions, primarily at DNaseI-resistant chromatin, within the initial stages of reprograming, which is consistent with pioneer function (Soufi et al., 2012; West et al., 2014). The regulatory targets of OCT4-SOX2 become nucleosome depleted during induced pluripotent stem cell (iPSC) generation, which also supports PF activity. Purified OCT4 and SOX2 display strong binding preferences for nucleosomal motifs relative to the TFs KLF4/MYC (Echigoya et al., 2020; Soufi et al., 2015). Investigations of direct versus indirect nucleosome recognition of OCT4-SOX2 revealed that OCT4-SOX2 binding can distort the nucleosomal DNA associated with H2A-H3 to remodel its binding sites (Michael et al., 2020). In these studies, Thomä and Schübeler developed a sequence-based approach to localize motifs in a nucleosomal context, selected engagement on nucleosomal sequencing (SeEN-seq), and defined the OCT4-SOX2 motif as residing near the entry-exit sites of nucleosomes, creating altered DNA-nucleosomal contacts, which has also been observed with cryo-electron microscopy (Cryo-EM) and chemical mapping (Echigoya et al., 2020). These PFs also have roles in human cancers.
In Ewing sarcoma, there is increasing evidence that anti-differentiation is driven from pioneer activity. Ewing sarcoma is a childhood bone cancer driven by a common translocation affecting the loci encoding EWSR1 and ETS family TFs, including FLI and ERG (Delattre et al., 1992; Ewing, 1972). Harboring less than 1 mutation per million bases of DNA, this malignancy has an exceedingly low mutational frequency (Table 2) (Crompton et al., 2014; Dharia et al., 2021; Pishas and Lessnick, 2018). The differentiation blockade in Ewing sarcoma is associated with transcriptional downregulation of lineage-specific genes (Kauer et al., 2009). The expression of SOX2 has been linked to EWS-FLI activity in some reports (Riggi et al., 2010, 2014). Excitingly, there is evidence that EWS-FLI engages its GGAA response elements even in repressed regions of the genome (Gangwal et al., 2008; Patel et al., 2012), though recent findings suggest that while the ETS domain is sufficient for DNA binding, a full-length fusion may be necessary for inaccessible chromatin binding (Boone et al., 2021). Understanding therapeutic mechanisms for destabilizing the motif recognition will continue to be highly impactful (Harlow et al., 2019). It will be of high interest to extend these fundamental principles to characterize essential roles of pioneer function in the establishment and maintenance of Ewing sarcoma epigenetics.
Table 2.
Pediatric tumors classified by alteration frequencies, classical translocations and deletions, in comparison with adult NSLC
| Tumor type | Alterations/Mb | Classical alterations |
|---|---|---|
| ATRT/MRT✧ | ∼3/whole exome <1 per Mb exome (Grobner et al., 2018) |
del(22q11.23) SNF5/SMARCB1 |
| Myeloid leukemias✧ | <1 per Mb exome 0.6 per Mb exome (Grobner et al., 2018) |
t(11q23) MLL-rearr. t(8; 21) AML-ETO inv(16) CBFb-MYH11 |
| Ewing sarcoma✧ | <1 per Mb exome 0.6 per Mb exome (Grobner et al., 2018) |
(11; 22) (q24; q12) EWS-FLI-1 |
| Diffuse Intrinsic Pontine Glioma✧ | 1.2 per Mb exome (Grobner et al., 2018) | H3.3K27M H3.1K27M |
| Synovial sarcoma✧ | 1.7 per Mb exome (Chalmers et al., 2017) | t(X; 18) SS18-SSX |
| Rhabdomyosarcoma✧ | 1.7 per Mb exome∗ (alveolar) 2.5 per Mb exome (Chalmers et al., 2017) (embryonal) 1.3 per Mb exome (Grobner et al., 2018) (embryonal) |
t(2; 13) (q35; q14) PAX3-FOXO1 |
| MPNST✧ | 2.5 per Mb exome (Chalmers et al., 2017) | del(17q11) NF1▵; SUZ12▵ del(11q14) EED▵ |
| NSCLC▵ | 8.1 per Mb exome (Chalmers et al., 2017) |
SMARCA4 mutations 19p13.2 |
| Osteosarcoma✧ | 14.6 per Mb exome (CNV) (Poos et al., 2015; Kovac et al., 2015) 1.8 per Mb exome (SNV) 1.3 per Mb exome (SNV) (Grobner et al., 2018) |
Complex karyotype/hyperdiploidy −10, -19 +4q, +8q, +17p |
The class of (✧) pediatric tumor and (▵) adult tumor are defined, as well as classical alterations, mutational burden per exome, and relevant pioneer factor.
Another childhood cancer with poor prognosis, diffuse intrinsic pontine glioma (DIPG), has transcriptional signatures that suggest the function of key PFs, including ASCL1 and SOX2. ASCL1 has strong nucleosome-motif binding capacity (Fernandez Garcia et al., 2019), is lineage restricted to the brain and pituitary, and is a vulnerability in neurogenic tumors (Figures 3A and 3B). Approximately, 80% of DIPG encodes a (H3F3A) histone H3 alteration in lysine-27 (Khuong-Quang et al., 2012). At the chromatin level, the loss of repressive histone marks including H3K27me3 is a common feature (Lewis et al., 2013). The genes that retain H3K27me3 are vulnerabilities in DIPG, including the INK4A locus (Mohammad et al., 2017). The PF, ASCL1, has been implicated in DIPG transcriptional circuitry (Anastas et al., 2019; Nagaraja et al., 2017). This neuronal TF is downregulated with in vitro drug treatments, including HDAC/LSD1 inhibition and BRD4 inhibition. A common feature in studies modeling new therapies for DIPG is the transcriptional downregulation of ASCL1 during loss of tumor proliferation. Enhancers regulating the SOX2 locus have also been shown to be important for DIPG transcriptional circuitry. Of note, SOX2 is a marker of neuronal stemness in DIPG (Anderson et al., 2017). While there has yet to be an established cell of origin for DIPG, the functional importance of SOX2 and ASCL1 may provide critical clues as to the neural precursor lineage and conceptually link the undifferentiated state of the tumor to pluripotent systems. It will be impactful to understand how H3.3K27M incorporates into nucleosome core particles and how would this affect nucleosomal-motif recognition and binding by PFs.
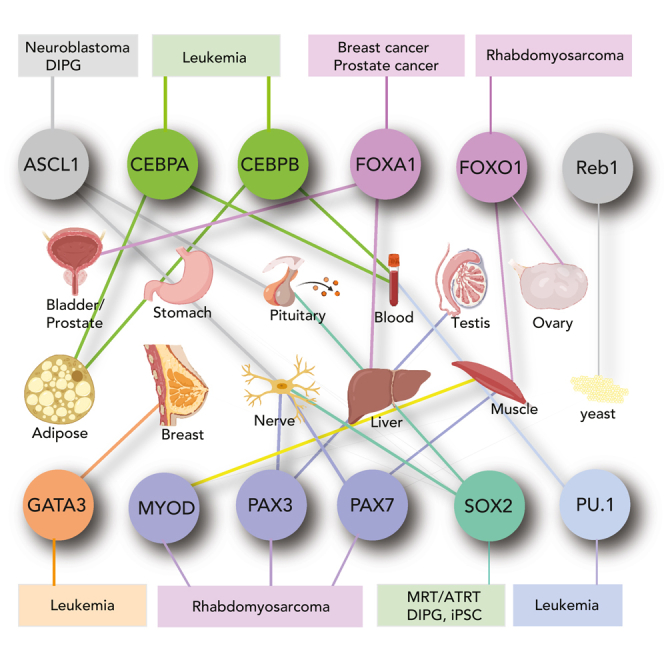
Figure 3.

Pioneer factors exhibit lineage-restricted expression and vulnerabilities
(A and B) Data analysis from the GTEx Consortium for expression levels of key pioneer factors in various tissues, with a focus on OCT4, SOX2, ASCL1, MYOD, PAX3, PAX7, FOXA1, FOXO1, FOXD3, FOXA2, GATA3, PU.1, CEBPα, β and (B) Examples of pioneer factors whose activity drives human cancers, with data analyses from DepMap, with a focus on SOX2, ASCL1, MYOD, PAX3, PAX7, FOXA1, FOXO1, PU.1, and CEBPα.
ASCL1 and SOX2 are also active in malignant rhabdoid tumors (MRTs) and atypical teratoid rhabdoid tumors (ATRTs), which are aggressive pediatric cancers that arise in soft tissues, CNS, or kidney, and have a poor prognosis (Sigauke et al., 2006). Recurrent deletions of chromosome 22 are characteristic of MRTs, and most of these alterations have been linked to loss of SMARCB1 (Table 2), a key subunit of BAF complexes (Biegel et al., 1999, 2002; Versteege et al., 1998). With SMARCB1 loss, these tumors can assemble non-canonical BAF (ncBAF) complexes, which are an acute vulnerability in MRTs and other SNF5-deficient cancers (Michel et al., 2018; Weissmiller et al., 2019). The strong dependence of SMARCB1-deficient MRTs on the continued expression and assembly of functional chromatin remodeling complexes (ncBAF) suggests they are under constant pressure to maintain an epigenetic program, perhaps instructed by TFs or PFs with transforming potential. In support of a rewired pioneer landscape driving MRT/ATRT phenotypes, a combined transcriptomic and epigenomic characterization of 191 ATRT tissues revealed subtypes with a distinct core transcriptional circuitry (Torchia et al., 2016). These include (1) tumors driven neurogenic factors ASCL1, MYCN, and SOX2, (2) tumors driven by OTX2 and MSX1, and (3) tumors driven by FOXC1, MYC, and various HOX family members (Figure 3; Table 2). The prevalence of pluripotency (SOX2) and neurogenic (ASCL1) PFs in these data suggest that neuronal stem cell-like features are present in ATRT and provide conceptual similarity to DIPG, which also has characteristic expression of these PFs. It is also of note that SOX2 and ASCL1 are each expressed in the brain, and SOX2 represents a vulnerability in ATRT (Figures 3A and 3B). In synovial sarcoma (SS), which also loses SMARCB1 assembly into BAF complexes, the pluripotency PFs, SOX2, and OCT4, is expressed at high levels, providing interesting connections between cancer stemness and sarcoma tumor initiation (Kadoch and Crabtree, 2013; Naka et al., 2010). Understanding mechanisms for how SOX2, OCT4, and ASCL1 interact with distinct SWI/SNF complexes to regulate epigenetic states in DIPG, MRT/ATRT, SS, Ewing sarcoma, and other childhood tumors will be of high interest.
In malignant peripheral nerve sheath tumors (MPNSTs), SOX-family pioneers have characteristic activities. MPNSTs are driven from loss of polycomb group genes (PcGs), with biallelic loss of SUZ12 or EED occurring together with alterations in Ras effectors. The dual loss of SUZ12 and NF1 has been reported in many cases (Kim et al., 2017; Miettinen et al., 2017). Null mutations of EED are also thought to drive malignant MPNSTs, resulting in a global defect in deposition of H3K27me3. Similar to MRT/ATRT, the deregulation of H3K27me3 can serve as an epigenetic driver, while MPNST has stereotypic losses, whereas ATRT has gains at CDKN2A/INK4A/ARF (Wilson et al., 2010). So how do MPNST epigenomes interact with transcription factors? MPNSTs are characterized by high expression of TWIST1 and SOX9, while SOX2 is often downregulated (Miller et al., 2009). SOX9 has been shown to bind to nucleosomal motifs in in vitro transcription assays (Coustry et al., 2010). While the binding preferences of SOX9 to DNA motifs have been characterized (Mertin et al., 1999), further studies will be important to understand if SOX9 satisfies the key criteria for pioneer function (Liu et al., 2018). Given global losses in PcG function, understanding the global accessibility in MPNST may require spike-in normalized sequencing of accessible DNA. Understanding pioneer function in MPNST will lead to a more complete understanding of how loss of H3K27me3 may alter motif binding preferences, with PFs that might not encounter nucleosomes as a steric deterrent. We hypothesize that either (1) a hyper-accessible MPNST genome would interact with TFs more broadly in this tumor, with less stringent requirements for srHC targeting or (2) compensatory H3K9me3 may provide an alternative pathway for the epigenetics of PFs in MPNST.
MYOD, PAX3, PAX7, and forkhead family: muscle development and myogenic tumors
MYOD functions as a master regulator in the conversion of embryonic fibroblast cells to a myoblast state (Davis et al., 1987). However, a recent report suggested the extended alpha-helical DNA binding domain of bHLH factors such as MYOD would prevent their recognition of nucleosomal motifs (Fernandez Garcia et al., 2019), potentially limiting intrinsic capacity for pioneer function. The basic patch of MYOD is essential for its instructive function in myogenesis (Figure 3) (Tapscott et al., 1988). Early observations connecting the molecular targeting of MYOD to chromatin acetylation emerged with mouse embryonic fibroblasts with inducible nuclear MYOD (Bergstrom et al., 2002; Hollenberg et al., 1993). In these studies, ChIP experiments were integrated with RNA expression analysis and revealed that MYOD binding to E-box motifs preceded histone acetylation and early gene expression. These early reports implicate MYOD as a PF, while direct nucleosomal binding remains to be systematically studied. While MYOD is predominantly expressed in the muscle and testis (Figure 3), it is also vulnerable in sarcomas. A recent study elegantly showed MYOD as a key dependency in a highly aggressive pediatric muscle tumor, rhabdomyosarcoma (RMS) (Dharia et al., 2021). Mechanistically, there is evidence that this MYOD dependency in RMS is linked to activation of SNAI2 transcription (Pomella et al., 2021). While wild-type MYOD can serve as a tumor driver in RMS (Dharia et al., 2021; Gryder et al., 2019), mutant MYOD has been shown as a driver in a subtype of highly aggressive rhabdomyosarcomas with poorest clinical outcomes (Kohsaka et al., 2014; Shern et al., 2014, 2021). Connecting nucleosome binding kinetics, MYOD's role in establishing core circuitry and its roles in childhood sarcoma will be impactful. Given that MYOD is a master regulator for muscle development but also plays instructive roles in tumors with a muscle differentiation blockade (RMS), understanding fractional occupancy and dosage sensitivity of MYOD could provide essential clues.
Forkhead TFs with winged-helix domains are critical for the initial phases of lineage commitment during establishment of endoderm development (Ang et al., 1993; Genga et al., 2019). FOXA1 has essential roles in liver development (Lee et al., 2005) and binds strongly to nucleosomal motifs, with a KD in the low nanomolar range (Fernandez Garcia et al., 2019) (Figures 2B and 3A). A related forkhead family member PF FOXD3 has been shown as essential for early development, while it has remained challenging to understand the mechanistic interdependence of FOXD3 and FOXA1 at the chromatin interface (Gualdi et al., 1996; Krishnakumar et al., 2016). There have also been interesting connections between FOXD3 function and CpG methylation states (Xu et al., 2009), which presents possibilities for integrating a direct pioneer function in opposing de novo DNA methyltransferase activity (Domcke et al., 2015). It is important to note that PFs from different families might not have identical properties at distinct phases of the cell cycle or retention on DNA motifs within mitotic chromatin. While the forkhead family member FOXO1 has pioneer function (Hatta and Cirillo, 2007), there is no current evidence that it can associate with mitotic chromosomes, while SOX2 remains associated through mitosis (Teves et al., 2016). It is of note that FOXO1 ranks as vulnerability in RMS, likely because of its role in the fusion PF oncoprotein PAX3-FOXO1 (Figure 3B) (Dharia et al., 2021).
The PFs PAX3 and PAX7 are capable of inducing the fibroblast-to-myoblast transition (Ito et al., 2017). PAX3 has been shown to function as a PF and mitotic bookmarking factor and to induce accessibility during myogenesis (Magli et al., 2019; Wu et al., 2015). PAX7 functions as a PF with nucleosomal motif occupancy (Mayran et al., 2018; Pelletier et al., 2021). Conversion of mouse ESCs into muscle progenitor cells has been reported with exogenous expression of PAX3, which enables cell fate transition presumably through initiating myoblast-like core circuitry (Darabi et al., 2008). The PAX3 paralog PAX7 is also critical for myogenesis and has been studied in the context of its ability to induce a myogenic transcriptional circuitry in iPSCs (Darabi et al., 2011). Induced PAX7 can target H3K27me3- and H3K9me3-enriched chromatin and influence DNA accessibility within hours at its binding sites (Lilja et al., 2017). PAX7-dependent accessibility induction is linked with the placement of H3K4me1 and H3K27ac. These findings are consistent with a model where PAX7 functions as a PF to interact with repressive chromatin (Figure 1). It will be important for the field in the coming years to define the precise maintenance interplay between MYOD, PAX3, PAX7, and MYOG in myogenic development. Future investigations to understand the establishment and maintenance of PF function in myogenesis (Figure 1) will have implications for definition of mirrored processes in myogenic tumors and dystrophic diseases.
RMS is a rare pediatric tumor with a poor prognosis (Shern et al., 2021). The more aggressive alveolar RMS subtype is driven from translocations between the forkhead (FOXO1) activator, with the PAX3/7 DNA binding domains, resulting in expression of t(2;13) or t(1;13) fusion oncoproteins PAX3-FOXO1 or PAX7-FOXO1 (Barr et al., 1993; Galili et al., 1993). The fusion oncoprotein PAX3-FOXO1 has been shown to induce transformation but also cell cycle arrest, depending on “oncogene dosage” and co-expression of other TFs (Pandey et al., 2017). The genetic drivers of the fusion-negative RMS (FN-RMS) embryonal subtype are fundamentally distinct from the alveolar subtype, including RAS mutations (Stratton et al., 1989) and mutations in NF1 and FGFR4 (Chen et al., 2015; Taylor et al., 2009). The classical markers of skeletal muscle development from myoblast-to-myotube are repressed in fusion-positive RMS (FP-RMS), resulting in loss of myosin heavy chain and wt-PAX3/7 expression, and upregulation of MYCN, MYOD, MYOG, and IGF2 (Keller and Guttridge, 2013; Khan et al., 1998). The major translocations PAX3/7-FOXO1 comprise fusion partners PAX3 and PAX7 that have individually demonstrated pioneer function (Hatta and Cirillo, 2007; Magli et al., 2019; Mayran et al., 2018). Recent evidence suggests that PAX3-FOXO1 has intrinsic pioneer activity to bind to repressive chromatin and nucleosomal motifs (Sunkel et al., 2021). PAX3 also ranks highly as a vulnerability in RMS (Figure 3) (Dharia et al., 2021).
There are strong neurogenic signatures in RMS, including the aforementioned ASCL1, PAX3, and PAX7 genes (Mansouri and Gruss, 1998). In experimental models of neuronal development, ASCL1 binds to repressed regions of chromatin, which subsequently become accessible at distinct stages of differentiation (Raposo et al., 2015). The predominance of motifs for the PFs ASCL1 and MYOD in chromatin regions bound by mammalian SWI/SNF complexes (B.D.S. and B.Z.S., unpublished data) provides insight into the steady-state coregulation of accessibility with PFs and ATP-dependent chromatin remodeling (Figure 1). Future efforts will be important for understanding the role of neuronal chromatin signatures in RMS, related to the concerted action of TF networks. While the predominant developmental model for RMS is a blockade of myogenesis, PF chromatin signatures may provide motivation to explore hypotheses of a neuronal cell of origin for this tumor.
GATA3, PU.1, and CEBPα, β: immune cell development and leukemias
Cascading expression and function of TFs orchestrates immune cell development. While the concerted activities of cell-type-specific TFs can facilitate lineage specification in the innate and acquired branches of the immune system, we focus on the master regulators GATA3, PU.1, and CEBP. Hematopoietic stem cells (HSCs) develop in the bone marrow and migrate to the thymus during T cell maturation before reaching the periphery. During commitment stages (DN1, DN2), selection stages (DN3, DN4), and lineage specification for CD4+ helper T cells (DP to CD4), the function of the PF GATA3 (NF-E1c) is required (Yamamoto et al., 1990; Zheng and Flavell, 1997). Connection between T cell development and the PF activity of GATA3 has been observed in the context of binding to nucleosomes, and its activity linked to induction of accessibility (Tanaka et al., 2020). GATA3 is essential for DN2 stage entry (Hosoya et al., 2009) and establishment of the DN4 stage (Pai et al., 2003) implicating this PF in multiple steps of early T cell maturation. GATA3 is also important for development of CD4+ T helper cells (Farrar et al., 2001; Nawijn et al., 2001). Thus, evidence supports an instructive pioneer function for GATA3 being necessary and sufficient for T cell development in both early and later stages.
Motif accessibility can serve as a gateway to understanding the distinct pathways in hematopoietic development. Hematopoietic PFs GATA3, PU.1, and CEBP often have accessible motifs in immune cell development (Corces et al., 2016). In studies of B cell and myeloid development, PU.1 (SPI1) is highlighted with motif enrichments in human multipotential progenitor (MPP) cells (Nerlov and Graf, 1998), and its expression can lead to opening of chromatin at tissue-specific enhancers (Heinz et al., 2010). While PU.1 motifs are enriched in B cells (Corces et al., 2016), pioneer function of PU.1 was revealed in early work on B cell development, where it regulates chromatin accessibility at IgG heavy chain enhancers (Nikolajczyk et al., 1999). PU.1 alters MNase cutting activity, presumably through its binding to nucleosomal motifs, and it associates with chromatinized motifs over IgG heavy chain μ-enhancer. PU.1 is instructive for accessibility changes and remodeling of enhancer chromatin structure. Indeed, it has been recently reported that PU.1, CEBP, and GATA family PFs can bind to nucleosomal motifs, consistent with pioneer activity (Fernandez Garcia et al., 2019). However, CEBPα has comparatively weak nucleosome-motif binding, relative to other PFs.
PU.1 is also a master regulator of myeloid development (Nerlov and Graf, 1998; Scott et al., 1994). To model the transition of multi-potent myeloid progenitors into myeloblasts, Graf & colleagues used an in vitro differentiation system with induced expression of PU.1. The activation domain and the ETS domain were both necessary for this instructive role in myeloid differentiation. These results are not restricted to MPPs, and PU.1 expression can induce the expression of macrophage markers in fibroblasts as well (Feng et al., 2008). CEBPα,β regulate enhancers in macrophage development (Corces et al., 2016; Heinz et al., 2010). The myeloid marker, macrophage-1 antigen (Mac-1), is induced approximately 100-fold upon PU.1 expression in NIH3T3 cells, an effect that is augmented with co-expression of CEBPα. In macrophages stimulated with lipopolysaccharide (LPS), PU.1 motifs are among the most highly enriched in P300 ChIP-seq (Ghisletti et al., 2010). This suggests that enhancer establishment during macrophage activation is highly coupled to PU.1 motif accessibility, indicating a role in mature macrophages in addition to earlier steps in development. Strikingly, many of the LPS-induced P300 sites are pre-associated with PU.1, which is consistent with a pioneer function in inactive chromatin binding prior to accessibility and deposition of H3K27ac. The roles of PU.1 in enhancer regulation have been highlighted in B cells as well as macrophages (Heinz et al., 2010). PU.1 emerges as the most highly enriched motif correlated with enhancer-associated chromatin modifications in B cells and macrophages, and its binding precedes enhancer activation. PU.1 motifs can be occupied in gene deserts or inaccessible regions in macrophages, which is consistent with pioneer function (Pham et al., 2013). In surface plasmon resonance (SPR) studies, PU.1 associates with its motif slower than non-pioneer ETS-1, while PU.1 dissociates on timescales of minutes in comparison to seconds for ETS-1 (Wang et al., 2014). Thus, similar to Reb1, PU.1 has kinetics defined by a slow (Figure 2). Combining MNase-seq with ChIP-seq revealed that PU.1 binding is correlated with nucleosome depletion in myeloid cells, while it has the capability of intrinsically binding to nucleosomal motifs (Barozzi et al., 2014). Thus, PU.1 pioneer function is essential for multiple branches of innate and acquired immune cell development.
The PFs PU.1 and CEBPα have unique activity in acute myeloid leukemia (AML), an aggressive blood cancer that derives from myeloid cells that proliferate and fail to differentiate. AML has among the lowest mutational frequencies of sequenced cancers (Cancer Genome Atlas Research et al., 2013), with fewer than 1 mutation per Mb in adult AML (Alexandrov et al., 2013), approximately 0.17 mutations per Mb in pediatric AML (Ma et al., 2018), and fewer than 0.1 coding mutations per Mb (Grobner et al., 2018) (Table 2). To support the role of PFs in the core circuitry of AML, the master regulatory GATA family members and CEBPβ are highly associated with DNA accessibility in leukemic blasts (Corces et al., 2016). Childhood AML has similar genetic drivers to adult AML (Grobner et al., 2018), with the notable absence of the commonest alterations occurring in adult clonal hematopoiesis (DNMT3A, IDH2, TET2) (Busque et al., 2012; Quek et al., 2018). The major childhood AML subtypes include (1) core binding factor AML t(8;21) RUNX1-RUNX1T1 (AML1-ETO) (Faber et al., 2016) and inv(16) (CBFb-MYH11) (Cancer Genome Atlas Research et al., 2013; Liu et al., 1993) and (2) KMT2A/MLL-rearranged (11q23) AML (Gu et al., 1992; Meyer et al., 2006; Taki et al., 1996). The insights into the genetic drivers of AML lead to the question do the driving alterations affect pioneer activity? A pattern emerges supporting a model where genetic drivers in AML systematically alter the function of several key PFs.
The fusion TF AML1-ETO (RUNX1-RUNX1T1) has been shown to disrupt the normal function of the PF, PU.1, in myeloid development (Vangala et al., 2003). Mutations in PU.1 are mutually exclusive with AML-ETO alterations in leukemia, suggesting a converging epigenetic pathway. The AML-ETO chimeric fusion oncoprotein alters the function of CEBPα (Pabst et al., 2001; Vangala et al., 2003). Evidence suggests that core circuitry established by CEBPα is suppressed at the chromatin level by AML-ETO fusions. Interestingly, CEBPα deletion is toxic in AML, suggesting that maintaining the right balance of pioneer activity is essential, as has been evidenced in sarcomas (Seong et al., 2021) (Figure 3). Expression of the CBFb-MYH11 chimera during early development restricts myeloid lineage specification (Castilla et al., 1996). The CBFb-MYH11 fusion binds strongly to gene promoters and is associated with high levels of RNAP2, including E-box and GATA motifs (Mandoli et al., 2014). Strikingly, CBFb-MYH11 genomic binding sites overlap strongly with PU.1. This provides an interesting insight into the co-opting of core regulatory machinery for anti-differentiation programs.
In MLL-rearranged leukemias, translocation partners co-opt the H3K79-methyltransferase activity of DOT1L complexes, retargeting this histone mark. While inhibition of H3K79 methylation in MLL-rearranged AML results in DNA accessibility reductions, inhibition of H3K4-demethylase activity (LSD1) results in leukemic differentiation through accessibility changes at PU.1 and CEBPα motifs (Cusan et al., 2018). These findings motivate further efforts to understand the interactions of histone marks and PFs in AML. Taken together, across AML-ETO, CBF-MYH11, and MLL-rearranged AML, altered interactions with hematopoietic PFs PU.1 and CEBPα result in distinct epigenetic states.
The inhibition of pioneer function or the co-opting of pioneer function by chimeric translocations represents common epigenetic features of leukemogenesis, with striking parallels to other translocation-driven tumors (Boone et al., 2021; Gryder et al., 2019; Sunkel et al., 2021). Future studies will be critical to examine the four definitions of pioneer function in the context of AML translocations and the precise mechanisms by which these chimeric oncogenes interfere with PFs in myeloid differentiation blockade. The pattern of systematically altered PF activity in AML leads to the question of whether the transcriptional regulation of these PFs might serve as a vulnerability (cf., Figure 3). Further studies of the precise epigenetic requirements for regulation of PU.1 and CEBPα and their precise chromatin targeting will be important for defining therapeutic opportunities for AML differentiation.
Summary and outlook
In cancers bearing high rates of somatic mutations, integrative analyses of have revealed high frequency alterations in TF binding sites. Yet these cancers often retain transcriptional regulatory circuitry resembling their tissue of origin. PFs may be central to reconciling these observations, in establishing the tumor epigenome and transcriptome. Selective pressure for conservation of coding sequences may be less stringent outside of the exome. Indeed, a potential driver of regulatory diversity may be the sequence heterogeneity within TF binding sites (Gerhart and Kirschner, 2007; Vierstra et al., 2020; Villar et al., 2015). Thus, “motif stability” for TFs and PFs may be under distinct evolutionary requirements compared with coding regions of regulatory factors. How is gene regulatory circuitry maintained for a cell to preserve its identity in the face of mutability of cis-regulatory elements?
Fractional occupancy for TF-motif pairs (Equation 3) depends on both the expression level and the affinity of the TF for its cognate motif but lacks strict requirements for motif's abundance. Thus, there is a “buffer” where PFs may retain the capability to establish core circuitry despite varying degrees of (1) accessibility of their motifs and (2) the relative abundance of wild-type motifs. This also raises the possibility of core circuitry establishment being relatively stable even in the presence of non-coding genetic variation in a certain organism or at the population level. Perhaps there are severe implications for this conceptual framework as well, such that an oncogene with pioneer function may be independent of the motif abundance or the cell of origin. Increasing evidence suggests that low mutational burden tumors are driven from epigenetic alterations (Boone et al., 2021; Lin et al., 2019; Pomella et al., 2021; Sunkel et al., 2021). It will be important to understand how these chromatin-level changes are translated into stable transcriptional states. PF function shapes the epigenetic accessibility landscape in development and also provides clues for the establishment of chromatin states that prevent differentiation in cancers. Do low-mutational burden tumors result from altered pioneer function? Studies of differentiation blockades may encode essential clues.
With the recent studies of srHC, our view is that new methods would allow us to better understand the fraction of the inaccessible genome that may be bound by bookmarking factors, PFs, and other factors, through distinct phases of the cell cycle. The Tn5 transposase does not function as a PF, making kinetic resolution in vivo challenging in terms of (1) defining partially unwrapped nucleosomes in the context of transient site exposure and (2) measurement of off-rates for PFs. If a TF induces DNA accessibility, the adage post hoc ergo propter hoc is a good reminder that definitions of “PF” require key attributes independent from merely inducing open chromatin. Rigorous examination of nucleosomal targeting and association with repressive chromatin are important. Finally, defining the shift from epigenetic establishment to maintenance requires an understanding of when equilibrium has been reached on chromatin, which is quite challenging.
Future studies will be important for providing empirical evidence to replace a priori definitions: with new translocations sequenced in rare disease, it may not be possible to predict pioneer function as a “gestalt phenomenon”, where the whole is more than the sum of its parts, despite component parts being derived from PF pairs (e.g., PAX3-FOXO1). New insights into conserved function versus conserved structure will be impactful. Understanding if there is a pioneer for each tissue type, and by extension, a pioneer driving circuitry and epigenetic memory in each human cancer will be important questions to address. Furthermore, targeted protein degradation of PFs (Tallan and Stanton, 2021) may allow nucleosomes to productively occlude non-pioneer TFs to alter genome structure in human cancer. Our analysis of the literature strengthens the view that PFs, and TFs more broadly, should be directly included in our conception of epigenetics. While ATP-dependent chromatin remodeling complexes lack specific consensus binding motifs, pioneer function is likely to establish targeting of the accessible genome in human tissues.
Acknowledgments
We apologize to the authors whom we could not include due to space constraints. We gratefully acknowledge all members of the Stanton Lab for discussions and motivations and edits from B. Donovan and M.G. Poirier. We thank M. Parthun, S.L. Lessnick, and T. Cripe for discussions related to this work. We acknowledge the St. Baldrick's Foundation (B.Z.S.), The Mark Foundation for Cancer Research (B.Z.S.), the Andrew McDonough B+ Foundation (B.Z.S.), CancerFree Kids Foundation (B.D.S.), and Nationwide Children's Hospital for support and inspiration to understand historical etiology of childhood cancer as chromatin-driven diseases.
Declaration of interests
The authors declare no competing interests.
References
- Abramo K., Valton A.L., Venev S.V., Ozadam H., Fox A.N., Dekker J. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat. Cell Biol. 2019;21:1393–1402. doi: 10.1038/s41556-019-0406-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas J.N., Zee B.M., Kalin J.H., Kim M., Guo R., Alexandrescu S., Blanco M.A., Giera S., Gillespie S.M., Das J. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell. 2019;36:528–544 e510. doi: 10.1016/j.ccell.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Anderson J.D., Lowary P.T., Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- Anderson J.D., Thastrom A., Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol. Cell Biol. 2002;22:7147–7157. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J.D., Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- Anderson J.L., Muraleedharan R., Oatman N., Klotter A., Sengupta S., Waclaw R.R., Wu J., Drissi R., Miles L., Raabe E.H. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro Oncol. 2017;19:1068–1078. doi: 10.1093/neuonc/now299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang S.L., Wierda A., Wong D., Stevens K.A., Cascio S., Rossant J., Zaret K.S. The formation and maintenance of the definitive endoderm lineage in the mouse: involvement of HNF3/forkhead proteins. Development. 1993;119:1301–1315. doi: 10.1242/dev.119.4.1301. [DOI] [PubMed] [Google Scholar]
- Barozzi I., Simonatto M., Bonifacio S., Yang L., Rohs R., Ghisletti S., Natoli G. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell. 2014;54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr F.G., Galili N., Holick J., Biegel J.A., Rovera G., Emanuel B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993;3:113–117. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- Battistini F., Hunter C.A., Moore I.K., Widom J. Structure-based identification of new high-affinity nucleosome binding sequences. J. Mol. Biol. 2012;420:8–16. doi: 10.1016/j.jmb.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Beati P., Chereji R.V. Creating 2D occupancy plots using plot2DO. Methods Mol. Biol. 2020;2117:93–108. doi: 10.1007/978-1-0716-0301-7_5. [DOI] [PubMed] [Google Scholar]
- Becker J.S., McCarthy R.L., Sidoli S., Donahue G., Kaeding K.E., He Z., Lin S., Garcia B.A., Zaret K.S. Genomic and proteomic resolution of heterochromatin and its restriction of alternate fate genes. Mol. Cell. 2017;68:1023–1037.e1015. doi: 10.1016/j.molcel.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom D.A., Penn B.H., Strand A., Perry R.L., Rudnicki M.A., Tapscott S.J. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell. 2002;9:587–600. doi: 10.1016/s1097-2765(02)00481-1. [DOI] [PubMed] [Google Scholar]
- Biegel J.A., Tan L., Zhang F., Wainwright L., Russo P., Rorke L.B. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002;8:3461–3467. [PubMed] [Google Scholar]
- Biegel J.A., Zhou J.Y., Rorke L.B., Stenstrom C., Wainwright L.M., Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- Boone M.A., Taslim C., Crow J.C., Selich-Anderson J., Byrum A.K., Showpnil I.A., Sunkel B.D., Wang M., Stanton B.Z., Theisen E.R. The FLI portion of EWS/FLI contributes a transcriptional regulatory function that is distinct and separable from its DNA-binding function in Ewing sarcoma. Oncogene. 2021;40:4759–4769. doi: 10.1038/s41388-021-01876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J.E., Hnisz D., Young R.A. Transcriptional addiction in cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K., Xi L., Wang J.P., Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budry L., Balsalobre A., Gauthier Y., Khetchoumian K., L’honoré A., Vallette S., Brue T., Figarella-Branger D., Meij B., Drouin J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012;26:2299–2310. doi: 10.1101/gad.200436.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busque L., Patel J.P., Figueroa M.E., Vasanthakumar A., Provost S., Hamilou Z., Mollica L., Li J., Viale A., Heguy A. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N., Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A., Hoadley K., Triche T.J., Jr., Laird P.W. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone D.M., Lawrence J.B. Heterochromatin instability in cancer: from the Barr body to satellites and the nuclear periphery. Semin. Cancer Biol. 2013;23:99–108. doi: 10.1016/j.semcancer.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla L.H., Wijmenga C., Wang Q., Stacy T., Speck N.A., Eckhaus M., Marin-Padilla M., Collins F.S., Wynshaw-Boris A., Liu P.P. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87:687–696. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- Chalmers Z.R., Connelly C.F., Fabrizio D., Gay L., Ali S.M., Ennis R., Schrock A., Campbell B., Shlien A., Chmielecki J. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9 doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Shern J.F., Wei J.S., Yohe M.E., Song Y.K., Hurd L., Liao H., Catchpoole D., Skapek S.X., Barr F.G. Clonality and evolutionary history of rhabdomyosarcoma. Plos Genet. 2015;11:e1005075. doi: 10.1371/journal.pgen.1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M., Zaret K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Cirillo L.A., McPherson C.E., Bossard P., Stevens K., Cherian S., Shim E.Y., Clark K.L., Burley S.K., Zaret K.S. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998;17:244–254. doi: 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo L.A., Zaret K.S. An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol. Cell. 1999;4:961–969. doi: 10.1016/s1097-2765(00)80225-7. [DOI] [PubMed] [Google Scholar]
- Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustry F., Oh C.D., Hattori T., Maity S.N., de Crombrugghe B., Yasuda H. The dimerization domain of SOX9 is required for transcription activation of a chondrocyte-specific chromatin DNA template. Nucl. Acids Res. 2010;38:6018–6028. doi: 10.1093/nar/gkq417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton B.D., Stewart C., Taylor-Weiner A., Alexe G., Kurek K.C., Calicchio M.L., Kiezun A., Carter S.L., Shukla S.A., Mehta S.S. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4:1326–1341. doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- Cusan M., Cai S.F., Mohammad H.P., Krivtsov A., Chramiec A., Loizou E., Witkin M.D., Smitheman K.N., Tenen D.G., Ye M. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPalpha-dependent enhancers in AML. Blood. 2018;131:1730–1742. doi: 10.1182/blood-2017-09-807024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabi R., Gehlbach K., Bachoo R.M., Kamath S., Osawa M., Kamm K.E., Kyba M., Perlingeiro R.C. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat. Med. 2008;14:134–143. doi: 10.1038/nm1705. [DOI] [PubMed] [Google Scholar]
- Darabi R., Pan W., Bosnakovski D., Baik J., Kyba M., Perlingeiro R.C. Functional myogenic engraftment from mouse iPS cells. Stem Cell Rev Rep. 2011;7:948–957. doi: 10.1007/s12015-011-9258-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.L., Weintraub H., Lassar A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Delattre O., Zucman J., Plougastel B., Desmaze C., Melot T., Peter M., Kovar H., Joubert I., de Jong P., Rouleau G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- Dharia N.V., Kugener G., Guenther L.M., Malone C.F., Durbin A.D., Hong A.L., Howard T.P., Bandopadhayay P., Wechsler C.S., Fung I. A first-generation pediatric cancer dependency map. Nat. Genet. 2021;53:529–538. doi: 10.1038/s41588-021-00819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S., Bardet A.F., Adrian Ginno P., Hartl D., Burger L., Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–579. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- Donovan B.T., Chen H., Jipa C., Bai L., Poirier M.G. Dissociation rate compensation mechanism for budding yeast pioneer transcription factors. Elife. 2019;8:e43008. doi: 10.7554/eLife.43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudchenko O., Batra S.S., Omer A.D., Nyquist S.K., Hoeger M., Durand N.C., Shamim M.S., Machol I., Lander E.S., Aiden A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy E.M., Almouzni G., Karpen G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus. 2011;2:146–157. doi: 10.4161/nucl.2.2.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echigoya K., Koyama M., Negishi L., Takizawa Y., Mizukami Y., Shimabayashi H., Kuroda A., Kurumizaka H. Nucleosome binding by the pioneer transcription factor OCT4. Sci. Rep. 2020;10:11832. doi: 10.1038/s41598-020-68850-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeholm M., de Jager M., Flaus A., Brenk R., van Noort J., Owen-Hughes T. Nucleosomes can invade DNA territories occupied by their neighbors. Nat. Struct. Mol. Biol. 2009;16:151–158. doi: 10.1038/nsmb.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York pathological society, 1921. CA Cancer J. Clin. 1972;22:95–98. doi: 10.3322/canjclin.22.2.95. [DOI] [PubMed] [Google Scholar]
- Faber Z.J., Chen X., Gedman A.L., Boggs K., Cheng J., Ma J., Radtke I., Chao J.R., Walsh M.P., Song G. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016;48:1551–1556. doi: 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar J.D., Ouyang W., Lohning M., Assenmacher M., Radbruch A., Kanagawa O., Murphy K.M. An instructive component in T helper cell type 2 (Th2) development mediated by GATA-3. J. Exp. Med. 2001;193:643–650. doi: 10.1084/jem.193.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R., Desbordes S.C., Xie H., Tillo E.S., Pixley F., Stanley E.R., Graf T. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc. Natl. Acad. Sci. U S A. 2008;105:6057–6062. doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez Garcia M., Moore C.D., Schulz K.N., Alberto O., Donague G., Harrison M.M., Zhu H., Zaret K.S. Structural features of transcription factors associating with nucleosome binding. Mol. Cell. 2019;75:921–932.e926. doi: 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Pereira R., De Angelis C., Veeraraghavan J., Nanda S., Qin L., Cataldo M.L., Sethunath V., Mehravaran S., Gutierrez C. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc. Natl. Acad. Sci. U S A. 2019;116:26823–26834. doi: 10.1073/pnas.1911584116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili N., Davis R.J., Fredericks W.J., Mukhopadhyay S., Rauscher F.J., 3rd, Emanuel B.S., Rovera G., Barr F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993;5:230–235. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- Gangwal K., Sankar S., Hollenhorst P.C., Kinsey M., Haroldsen S.C., Shah A.A., Boucher K.M., Watkins W.S., Jorde L.B., Graves B.J. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proc. Natl. Acad. Sci. U S A. 2008;105:10149–10154. doi: 10.1073/pnas.0801073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genga R.M.J., Kernfeld E.M., Parsi K.M., Parsons T.J., Ziller M.J., Maehr R. Single-cell RNA-sequencing-based CRISPRi screening resolves molecular drivers of early human endoderm development. Cell Rep. 2019;27:708–718.e710. doi: 10.1016/j.celrep.2019.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber A.N., Klesert T.R., Bergstrom D.A., Tapscott S.J. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. 1997;11:436–450. doi: 10.1101/gad.11.4.436. [DOI] [PubMed] [Google Scholar]
- Gerhart J., Kirschner M. The theory of facilitated variation. Proc. Natl. Acad. Sci. U S A. 2007;104:8582–8589. doi: 10.1073/pnas.0701035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Gilbert N., Boyle S., Fiegler H., Woodfine K., Carter N.P., Bickmore W.A. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Glont S.E., Chernukhin I., Carroll J.S. Comprehensive genomic analysis reveals that the pioneering function of FOXA1 is independent of hormonal signaling. Cell Rep. 2019;26:2558–2565.e2553. doi: 10.1016/j.celrep.2019.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobner S.N., Worst B.C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V.A., Johann P.D., Balasubramanian G.P., Segura-Wang M., Brabetz S. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- Gryder B.E., Pomella S., Sayers C., Wu X.S., Song Y., Chiarella A.M., Bagchi S., Chou H.C., Sinniah R.S., Walton A. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019;51:1714–1722. doi: 10.1038/s41588-019-0534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Nakamura T., Alder H., Prasad R., Canaani O., Cimino G., Croce C.M., Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- Gualdi R., Bossard P., Zheng M., Hamada Y., Coleman J.R., Zaret K.S. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev. 1996;10:1670–1682. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- Halder G., Callaerts P., Gehring W.J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science. 1995;267:1788–1792. doi: 10.1126/science.7892602. [DOI] [PubMed] [Google Scholar]
- Harlow M.L., Chasse M.H., Boguslawski E.A., Sorensen K.M., Gedminas J.M., Kitchen-Goosen S.M., Rothbart S.B., Taslim C., Lessnick S.L., Peck A.S. Trabectedin inhibits EWS-FLI1 and Evicts SWI/SNF from chromatin in a schedule-dependent manner. Clin. Cancer Res. 2019;25:3417–3429. doi: 10.1158/1078-0432.CCR-18-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley P.D., Madhani H.D. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137:445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta M., Cirillo L.A. Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J. Biol. Chem. 2007;282:35583–35593. doi: 10.1074/jbc.M704735200. [DOI] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff J.G., Belsky J.A., Krassovsky K., MacAlpine D.M., Henikoff S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. U S A. 2011;108:18318–18323. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg S.M., Cheng P.F., Weintraub H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc. Natl. Acad. Sci. U S A. 1993;90:8028–8032. doi: 10.1073/pnas.90.17.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya T., Kuroha T., Moriguchi T., Cummings D., Maillard I., Lim K.C., Engel J.D. GATA-3 is required for early T lineage progenitor development. J. Exp. Med. 2009;206:2987–3000. doi: 10.1084/jem.20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N., Kii I., Shimizu N., Tanaka H., Takeda S. Direct reprogramming of fibroblasts into skeletal muscle progenitor cells by transcription factors enriched in undifferentiated subpopulation of satellite cells. Sci. Rep. 2017;7:8097. doi: 10.1038/s41598-017-08232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M., Donahue G., Kakumanu A., Watts J.A., Mahony S., Pugh B.F., Lee D., Kaestner K.H., Zaret K.S. The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol. Cell. 2016;62:79–91. doi: 10.1016/j.molcel.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M., Zaret K.S. Cell fate control by pioneer transcription factors. Development. 2016;143:1833–1837. doi: 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C., Crabtree G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova A.A., Porter-Goff M.E., Muthurajan U.M., Luger K., Hansen J.C. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interf. 2013;10:20121022. doi: 10.1098/rsif.2012.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Moore I., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., Hughes T.R., Lieb J.D., Widom J., Segal E. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat. Struct. Mol. Biol. 2010;17:918–920. doi: 10.1038/nsmb0810-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., LeProust E.M., Hughes T.R., Lieb J.D., Widom J. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar G., Kim J.K., Kolodziejczyk A.A., Natarajan K.N., Torlai Triglia E., Mifsud B., Elderkin S., Marioni J.C., Pombo A., Teichmann S.A. Flipping between Polycomb repressed and active transcriptional states introduces noise in gene expression. Nat. Commun. 2017;8:36. doi: 10.1038/s41467-017-00052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer M., Ban J., Kofler R., Walker B., Davis S., Meltzer P., Kovar H. A molecular function map of Ewing's sarcoma. PLoS ONE. 2009;4:e5415. doi: 10.1371/journal.pone.0005415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C., Guttridge D.C. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 2013;280:4323–4334. doi: 10.1111/febs.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J., Simon R., Bittner M., Chen Y., Leighton S.B., Pohida T., Smith P.D., Jiang Y., Gooden G.C., Trent J.M. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res. 1998;58:5009–5013. [PubMed] [Google Scholar]
- Khuong-Quang D.A., Buczkowicz P., Rakopoulos P., Liu X.Y., Fontebasso A.M., Bouffet E., Bartels U., Albrecht S., Schwartzentruber J., Letourneau L. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A., Stewart D.R., Reilly K.M., Viskochil D., Miettinen M.M., Widemann B.C. Malignant peripheral nerve sheath tumors state of the science: leveraging clinical and biological insights into effective therapies. Sarcoma. 2017;2017:7429697. doi: 10.1155/2017/7429697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerber R.T., Rhee H.S., Jiang C., Pugh B.F. Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol. Cell. 2009;35:889–902. doi: 10.1016/j.molcel.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka S., Shukla N., Ameur N., Ito T., Ng C.K., Wang L., Lim D., Marchetti A., Viale A., Pirun M. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat. Genet. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac M., Blattmann C., Ribi S., Smida J., Mueller N.S., Engert F., Castro-Giner F., Weischenfeldt J., Kovacova M., Krieg A. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015;6 doi: 10.1038/ncomms9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R., Chen A.F., Pantovich M.G., Danial M., Parchem R.J., Labosky P.A., Blelloch R. FOXD3 regulates pluripotent stem cell potential by simultaneously initiating and repressing enhancer activity. Cell Stem Cell. 2016;18:104–117. doi: 10.1016/j.stem.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B., Gao W., Cui K., Xie W., Tang Q., Jin W., Hu G., Ni B., Zhao K. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature. 2018;562:281–285. doi: 10.1038/s41586-018-0567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–240. doi: 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.S., Friedman J.R., Fulmer J.T., Kaestner K.H. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- Lee W., Tillo D., Bray N., Morse R.H., Davis R.W., Hughes T.R., Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- Lewis P.W., Muller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Widom J. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- Liang S.D., Marmorstein R., Harrison S.C., Ptashne M. DNA sequence preferences of GAL4 and PPR1: how a subset of Zn2 Cys6 binuclear cluster proteins recognizes DNA. Mol. Cell Biol. 1996;16:3773–3780. doi: 10.1128/mcb.16.7.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja K.C., Zhang N., Magli A., Gunduz V., Bowman C.J., Arpke R.W., Darabi R., Kyba M., Perlingeiro R., Dynlacht B.D. Pax7 remodels the chromatin landscape in skeletal muscle stem cells. PLoS ONE. 2017;12:e0176190. doi: 10.1371/journal.pone.0176190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.L., Wilson K.M., Ceribelli M., Stanton B.Z., Woo P.J., Kreimer S., Qin E.Y., Zhang X., Lennon J., Nagaraja S. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.F., Angelozzi M., Haseeb A., Lefebvre V. SOX9 is dispensable for the initiation of epigenetic remodeling and the activation of marker genes at the onset of chondrogenesis. Development. 2018;145:dev164459. doi: 10.1242/dev.164459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Tarle S.A., Hajra A., Claxton D.F., Marlton P., Freedman M., Siciliano M.J., Collins F.S. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luo Y., North J.A., Rose S.D., Poirier M.G. Nucleosomes accelerate transcription factor dissociation. Nucl. Acids Res. 2014;42:3017–3027. doi: 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B., Trieu T.J., Cheng J., Zhou S., Tang Q., Xie J., Liu J.L., Zhao K., Habib S.J., Chen X. Differential histone distribution patterns in induced asymmetrically dividing mouse embryonic stem cells. Cell Rep. 2020;32:108003. doi: 10.1016/j.celrep.2020.108003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Liu Y., Liu Y., Alexandrov L.B., Edmonson M.N., Gawad C., Zhou X., Li Y., Rusch M.C., Easton J. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli A., Baik J., Mills L.J., Kwak I.Y., Dillon B.S., Mondragon Gonzalez R., Stafford D.A., Swanson S.A., Stewart R., Thomson J.A. Time-dependent Pax3-mediated chromatin remodeling and cooperation with Six4 and Tead2 specify the skeletal myogenic lineage in developing mesoderm. PLoS Biol. 2019;17:e3000153. doi: 10.1371/journal.pbio.3000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiser A., Dillinger S., Langst G., Schermelleh L., Leonhardt H., Nemeth A. Super-resolution in situ analysis of active ribosomal DNA chromatin organization in the nucleolus. Sci. Rep. 2020;10:7462. doi: 10.1038/s41598-020-64589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandoli A., Singh A.A., Jansen P.W., Wierenga A.T., Riahi H., Franci G., Prange K., Saeed S., Vellenga E., Vermeulen M. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia. 2014;28:770–778. doi: 10.1038/leu.2013.257. [DOI] [PubMed] [Google Scholar]
- Mansouri A., Gruss P. Pax3 and Pax7 are expressed in commissural neurons and restrict ventral neuronal identity in the spinal cord. Mech. Dev. 1998;78:171–178. doi: 10.1016/s0925-4773(98)00168-3. [DOI] [PubMed] [Google Scholar]
- Mayran A., Khetchoumian K., Hariri F., Pastinen T., Gauthier Y., Balsalobre A., Drouin J. Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat. Genet. 2018;50:259–269. doi: 10.1038/s41588-017-0035-2. [DOI] [PubMed] [Google Scholar]
- Meers M., Janssens D.H., Henikoff S. Pioneer Factor-Nucleosome Binding Events during Differentiation Are Motif Encoded. Mol. Cell. 2019;75:562–575. doi: 10.1016/j.molcel.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertin S., McDowall S.G., Harley V.R. The DNA-binding specificity of SOX9 and other SOX proteins. Nucl. Acids Res. 1999;27:1359–1364. doi: 10.1093/nar/27.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C., Schneider B., Jakob S., Strehl S., Attarbaschi A., Schnittger S., Schoch C., Jansen M.W., van Dongen J.J., den Boer M.L. The MLL recombinome of acute leukemias. Leukemia. 2006;20:777–784. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- Michael A.K., Grand R.S., Isbel L., Cavadini S., Kozicka Z., Kempf G., Bunker R.D., Schenk A.D., Graff-Meyer A., Pathare G.R. Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science. 2020;368:1460–1465. doi: 10.1126/science.abb0074. [DOI] [PubMed] [Google Scholar]
- Michel B.C., D'Avino A.R., Cassel S.H., Mashtalir N., McKenzie Z.M., McBride M.J., Valencia A.M., Zhou Q., Bocker M., Soares L.M.M. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018;20:1410–1420. doi: 10.1038/s41556-018-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen M.M., Antonescu C.R., Fletcher C.D.M., Kim A., Lazar A.J., Quezado M.M., Reilly K.M., Stemmer-Rachamimov A., Stewart D.R., Viskochil D. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017;67:1–10. doi: 10.1016/j.humpath.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.J., Jessen W.J., Mehta T., Hardiman A., Sites E., Kaiser S., Jegga A.G., Li H., Upadhyaya M., Giovannini M. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol. Med. 2009;1:236–248. doi: 10.1002/emmm.200900027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mivelaz M., Cao A.M., Kubik S., Zencir S., Hovius R., Boichenko I., Stachowicz A.M., Kurat C.F., Shore D., Fierz B. Chromatin fiber invasion and nucleosome displacement by the Rap1 transcription factor. Mol. Cell. 2020;77:488–500.e489. doi: 10.1016/j.molcel.2019.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F., Weissmann S., Leblanc B., Pandey D.P., Hojfeldt J.W., Comet I., Zheng C., Johansen J.V., Rapin N., Porse B.T. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017;23:483–492. doi: 10.1038/nm.4293. [DOI] [PubMed] [Google Scholar]
- Murphy P.J., Wu S.F., James C.R., Wike C.L., Cairns B.R. Placeholder nucleosomes underlie Germline-to-Embryo DNA methylation reprogramming. Cell. 2018;172:993–1006.e1013. doi: 10.1016/j.cell.2018.01.022. [DOI] [PubMed] [Google Scholar]
- Nagaraja S., Vitanza N.A., Woo P.J., Taylor K.R., Liu F., Zhang L., Li M., Meng W., Ponnuswami A., Sun W. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31:635–652.e636. doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P.L., Cleary M.L., Brown P.O., Lieb J.D. Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc. Natl. Acad. Sci. U S A. 2003;100:6364–6369. doi: 10.1073/pnas.1131966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka N., Takenaka S., Araki N., Miwa T., Hashimoto N., Yoshioka K., Joyama S., Hamada K., Tsukamoto Y., Tomita Y. Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28:1119–1131. doi: 10.1002/stem.452. [DOI] [PubMed] [Google Scholar]
- Nawijn M.C., Dingjan G.M., Ferreira R., Lambrecht B.N., Karis A., Grosveld F., Savelkoul H., Hendriks R.W. Enforced expression of GATA-3 in transgenic mice inhibits Th1 differentiation and induces the formation of a T1/ST2-expressing Th2-committed T cell compartment in vivo. J. Immunol. 2001;167:724–732. doi: 10.4049/jimmunol.167.2.724. [DOI] [PubMed] [Google Scholar]
- Nerlov C., Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998;12:2403–2412. doi: 10.1101/gad.12.15.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolajczyk B.S., Sanchez J.A., Sen R. ETS protein-dependent accessibility changes at the immunoglobulin mu heavy chain enhancer. Immunity. 1999;11:11–20. doi: 10.1016/s1074-7613(00)80077-1. [DOI] [PubMed] [Google Scholar]
- North J.A., Shimko J.C., Javaid S., Mooney A.M., Shoffner M.A., Rose S.D., Bundschuh R., Fishel R., Ottesen J.J., Poirier M.G. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 2012;40:10215–10227. doi: 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O’Shea C.C. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357 doi: 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst T., Mueller B.U., Harakawa N., Schoch C., Haferlach T., Behre G., Hiddemann W., Zhang D.E., Tenen D.G. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat. Med. 2001;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- Pai S.Y., Truitt M.L., Ting C.N., Leiden J.M., Glimcher L.H., Ho I.C. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity. 2003;19:863–875. doi: 10.1016/s1074-7613(03)00328-5. [DOI] [PubMed] [Google Scholar]
- Pandey P.R., Chatterjee B., Olanich M.E., Khan J., Miettinen M.M., Hewitt S.M., Barr F.G. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 2017;241:626–637. doi: 10.1002/path.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M., Simon J.M., Iglesia M.D., Wu S.B., McFadden A.W., Lieb J.D., Davis I.J. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012;22:259–270. doi: 10.1101/gr.125666.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier A., Mayran A., Gouhier A., Omichinski J.G., Balsalobre A., Drouin J. Pax7 pioneer factor action requires both paired and homeo DNA binding domains. Nucl. Acids Res. 2021;49:7424–7436. doi: 10.1093/nar/gkab561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T.H., Minderjahn J., Schmidl C., Hoffmeister H., Schmidhofer S., Chen W., Langst G., Benner C., Rehli M. Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucl. Acids Res. 2013;41:6391–6402. doi: 10.1093/nar/gkt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishas K.I., Lessnick S.L. Ewing sarcoma resistance to SP-2509 is not mediated through KDM1A/LSD1 mutation. Oncotarget. 2018;9:36413–36429. doi: 10.18632/oncotarget.26326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier M.G., Oh E., Tims H.S., Widom J. Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol. 2009;16:938–944. doi: 10.1038/nsmb.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach K.J., Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- Polach K.J., Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- Polach K.J., Widom J. Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol. 1999;304:278–298. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- Pomella S., Sreenivas P., Gryder B.E., Wang L., Milewski D., Cassandri M., Baxi K., Hensch N.R., Carcarino E., Song Y. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in Fusion Negative Rhabdomyosarcoma. Nat. Commun. 2021;12:192. doi: 10.1038/s41467-020-20386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poos K., Smida J., Maugg D., Eckstein G., Baumhoer D., Nathrath M., Korsching E. Genomic Heterogeneity of Osteosarcoma - Shift from Single Candidates to Functional Modules. PLoS One. 2015;10 doi: 10.1371/journal.pone.0123082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek L., David M.D., Kennedy A., Metzner M., Amatangelo M., Shih A., Stoilova B., Quivoron C., Heiblig M., Willekens C. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018;24:1167–1177. doi: 10.1038/s41591-018-0115-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo A., Vasconcelos F.F., Drechsel D., Marie C., Johnston C., Dolle D., Bithell A., Gillotin S., van den Berg D.L.C., Ettwiller L. Ascl1 coordinately regulates gene expression and the chromatin landscape during neurogenesis. Cell Rep. 2015;10:1544–1556. doi: 10.1016/j.celrep.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinberg D., Vales L.D. Chromatin domains rich in inheritance. Science. 2018;361:33–34. doi: 10.1126/science.aat7871. [DOI] [PubMed] [Google Scholar]
- Riggi N., Knoechel B., Gillespie S.M., Rheinbay E., Boulay G., Suva M.L., Rossetti N.E., Boonseng W.E., Oksuz O., Cook E.B. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26:668–681. doi: 10.1016/j.ccell.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi N., Suva M.L., De Vito C., Provero P., Stehle J.C., Baumer K., Cironi L., Janiszewska M., Petricevic T., Suva D. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010;24:916–932. doi: 10.1101/gad.1899710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn A.L., Rao S.S., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U S A. 2015;112:E6456–E6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep A.N., Buenrostro J.D., Denny S.K., Schwartz K., Sherlock G., Greenleaf W.J. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25:1757–1770. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott E.W., Simon M.C., Anastasi J., Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- Sekiya T., Muthurajan U.M., Luger K., Tulin A.V., Zaret K.S. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong B.K.A., Dharia N.V., Lin S., Donovan K.A., Chong S., Robichaud A., Conway A., Hamze A., Ross L., Alexe G. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell. 2021;39:1262. doi: 10.1016/j.ccell.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shern J.F., Chen L., Chmielecki J., Wei J.S., Patidar R., Rosenberg M., Ambrogio L., Auclair D., Wang J., Song Y.K. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shern J.F., Selfe J., Izquierdo E., Patidar R., Chou H.C., Song Y.K., Yohe M.E., Sindiri S., Wei J., Wen X. Genomic classification and clinical outcome in rhabdomyosarcoma: a report from an international Consortium. J. Clin. Oncol. 2021;39:JCO2003060. doi: 10.1200/JCO.20.03060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim E.Y., Woodcock C., Zaret K.S. Nucleosome positioning by the winged helix transcription factor HNF3. Genes Dev. 1998;12:5–10. doi: 10.1101/gad.12.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigauke E., Rakheja D., Maddox D.L., Hladik C.L., White C.L., Timmons C.F., Raisanen J. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod. Pathol. 2006;19:717–725. doi: 10.1038/modpathol.3800581. [DOI] [PubMed] [Google Scholar]
- Skrajna A., Goldfarb D., Kedziora K.M., Cousins E.M., Grant G.D., Spangler C.J., Barbour E.H., Yan X., Hathaway N.A., Brown N.G. Comprehensive nucleosome interactome screen establishes fundamental principles of nucleosome binding. Nucleic Acids Res. 2020;48:9415–9432. doi: 10.1093/nar/gkaa544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Donahue G., Zaret K.S. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Garcia M.F., Jaroszewicz A., Osman N., Pellegrini M., Zaret K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015;161:555–568. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M.R., Fisher C., Gusterson B.A., Cooper C.S. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989;49:6324–6327. [PubMed] [Google Scholar]
- Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–245. doi: 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Rockowitz S., Xie Q., Ashery-Padan R., Zheng D., Cvekl A. Identification of in vivo DNA-binding mechanisms of Pax6 and reconstruction of Pax6-dependent gene regulatory networks during forebrain and lens development. Nucleic Acids Res. 2015;43:6827–6846. doi: 10.1093/nar/gkv589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkel B.D., Wang M., LaHaye S., Kelly B.J., Fitch J.R., Barr F.G., White P., Stanton B.Z. Evidence of pioneer factor activity of an oncogenic fusion transcription factor. iScience. 2021;24:102867. doi: 10.1016/j.isci.2021.102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Taki T., Ida K., Bessho F., Hanada R., Kikuchi A., Yamamoto K., Sako M., Tsuchida M., Seto M., Ueda R. Frequency and clinical significance of the MLL gene rearrangements in infant acute leukemia. Leukemia. 1996;10:1303–1307. [PubMed] [Google Scholar]
- Tallan A., Stanton B.Z. Inducible protein degradation to understand genome architecture. Biochemistry. 2021;60:2387–2396. doi: 10.1021/acs.biochem.1c00306. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Takizawa Y., Takaku M., Kato D., Kumagawa Y., Grimm S.A., Wade P.A., Kurumizaka H. Interaction of the pioneer transcription factor GATA3 with nucleosomes. Nat. Commun. 2020;11:4136. doi: 10.1038/s41467-020-17959-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott S.J., Davis R.L., Thayer M.J., Cheng P.F., Weintraub H., Lassar A.B. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- Taylor J.G.t., Cheuk A.T., Tsang P.S., Chung J.Y., Song Y.K., Desai K., Yu Y., Chen Q.R., Shah K., Youngblood V. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves S.S., An L., Hansen A.S., Xie L., Darzacq X., Tjian R. A dynamic mode of mitotic bookmarking by transcription factors. Elife. 2016;5:e22280. doi: 10.7554/eLife.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchia J., Golbourn B., Feng S., Ho K.C., Sin-Chan P., Vasiljevic A., Norman J.D., Guilhamon P., Garzia L., Agamez N.R. Integrated (epi)-Genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 2016;30:891–908. doi: 10.1016/j.ccell.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangala R.K., Heiss-Neumann M.S., Rangatia J.S., Singh S.M., Schoch C., Tenen D.G., Hiddemann W., Behre G. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood. 2003;101:270–277. doi: 10.1182/blood-2002-04-1288. [DOI] [PubMed] [Google Scholar]
- Versteege I., Sevenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E. Global reference mapping of human transcription factor footprints. Nature. 2020;583:729–736. doi: 10.1038/s41586-020-2528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D., Berthelot C., Aldridge S., Rayner T.F., Lukk M., Pignatelli M., Park T.J., Deaville R., Erichsen J.T., Jasinska A.J. Enhancer evolution across 20 mammalian species. Cell. 2015;160:554–566. doi: 10.1016/j.cell.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Linde M.H., Munde M., Carvalho V.D., Wilson W.D., Poon G.M. Mechanistic heterogeneity in site recognition by the structurally homologous DNA-binding domains of the ETS family transcription factors Ets-1 and PU.1. J. Biol. Chem. 2014;289:21605–21616. doi: 10.1074/jbc.M114.575340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmiller A.M., Wang J., Lorey S.L., Howard G.C., Martinez E., Liu Q., Tansey W.P. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat. Commun. 2019;10:2014. doi: 10.1038/s41467-019-10022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J.A., Cook A., Alver B.H., Stadtfeld M., Deaton A.M., Hochedlinger K., Park P.J., Tolstorukov M.Y., Kingston R.E. Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat. Commun. 2014;5:4719. doi: 10.1038/ncomms5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B.G., Wang X., Shen X., McKenna E.S., Lemieux M.E., Cho Y.J., Koellhoffer E.C., Pomeroy S.L., Orkin S.H., Roberts C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.F., Yao Y.L., Lai I.L., Lai C.C., Lin P.L., Yang W.M. Loading of PAX3 to mitotic chromosomes is mediated by arginine methylation and associated with Waardenburg syndrome. J. Biol. Chem. 2015;290:20556–20564. doi: 10.1074/jbc.M114.607713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Watts J.A., Pope S.D., Gadue P., Kamps M., Plath K., Zaret K.S., Smale S.T. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23:2824–2838. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Ko L.J., Leonard M.W., Beug H., Orkin S.H., Engel J.D. Activity and tissue-specific expression of the transcription factor NF-E1 multigene family. Genes Dev. 1990;4:1650–1662. doi: 10.1101/gad.4.10.1650. [DOI] [PubMed] [Google Scholar]
- Yan C., Chen H., Bai L. Systematic study of nucleosome-displacing factors in budding yeast. Mol. Cell. 2018;71:294–305.e294. doi: 10.1016/j.molcel.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K., Vinayachandran V., Batta K., Koerber R.T., Pugh B.F. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret K.S., Carroll J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Wippo C.J., Wal M., Ward E., Korber P., Pugh B.F. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., Flavell R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]