

Abstract
Strong epidemiological evidence demonstrates an association between chronic arsenic exposure and anemia. We recently found that As+3 impairs erythropoiesis by disrupting the function of GATA-1; however the downstream pathways impacted by the loss of GATA-1 function have not been evaluated. Additionally, our previous findings indicate that the predominant arsenical in the bone marrow of mice exposed to As+3 in their drinking water for 30 days was MMA+3, but the impacts of this arsenical on erythorpoisis also remain largely unknown. The goal of this study was to address these critical knowledge gaps by evaluating the comparative effects of arsenite (As+3) and the As+3 metabolite, monomethyarsonous acid (MMA+3) on two critical regulatory pathways that control the differentiation and survival of early erythroid progenitor cells. We found that 500 nM As+3 and 100 and 500 nM MMA+3 suppress erythropoiesis by impairing the differentiation of early stage erythroid progenitors. The suppression of early erythroid progenitor cell development was attributed to combined effects on differentiation and survival pathways mediated by disruption of GATA-1 and STAT5. Our results show that As+3 primarily disrupted GATA-1 function; whereas, MMA+3 suppressed both GATA-1 and STAT5 activity. Collectively, these findings provide novel mechanistic insights into arsenic-induced dyserythropoiesis and suggest that MMA+3 may be more toxic than As+3 to early developing erythroid cells.
Keywords: Arsenic, Arsenite (As+3), Monomethylarsonous acid (MMA+3), Erythropoiesis, STAT5, GATA-1
1. INTRODUCTION
Chronic exposure to arsenic in drinking water has been linked to a multitude of detrimental health outcomes, including cancer, immunosuppression, cardiovascular disease, neurological disorders, and anemia (Hughes, 2002; Heck et al., 2008; Naujokas et al., 2013; Tyler and Allan, 2014; Ferrario et al., 2016). The primary form of arsenic in drinking water is the trivalent inorganic arsenical, arsenite (As+3). As+3 is metabolized through a series of oxidative methylation and reduction reactions catalyzed by the arsenic (+3 oxidation state) methyltransferase (As3MT) enzyme to monomethylarsonic acid (MMA+5), monomethylarsonous acid (MMA+3), dimethylarsinic acid (DMA+5), and dimethylarsinous acid (DMA+3) (Thomas et al., 2001; Vahter, 2002). Numerous in vitro and in vivo studies report MMA+3 to be one of the most toxic arsenicals (Styblo et al., 2000; Petrick et al., 2001). In addition, a large body of evidence from human populations chronically exposed to arsenic underscores the relationship between arsenic metabolism (i.e. methylation capacity) with adverse disease outcomes (Yu et al., 2000; Chen et al., 2003; Ahsan et al., 2007; Huang et al., 2008; Chen et al., 2009; Lin et al., 2018; Luo et al., 2018).
Anemia is a hematological disorder that adversely affects the health of millions of people worldwide (Koury, 2014; WHO, 2015). The etiology of anemia is complex and often associated with numerous factors, including nutritional deficiencies, inflammation, genetic disorders, and cancer (Chaparro and Suchdev, 2019); however, strong epidemiological evidence show that environmental arsenic exposures may contribute to the onset or exacerbation of this condition (Hopenhayn et al., 2006; Heck et al., 2008; Surdu et al., 2015; Kile et al., 2016). In a cross-sectional study of clinical indicators of anemia, we found strong inverse relationships between measures of arsenic exposure (water and urine) and urinary arsenic metabolites (MMA and DMA) with red blood cell counts and hematocrits among a subset of male Health Effects of Arsenic Longitudinal Study (HEALS) cohort volunteers from rural Bangladesh (Parvez et al., 2017). Follow-up studies in mice, revealed that 60-day exposures to As+3 via drinking water resulted in anemia due to the impaired development of early erythroid progenitor cells in the bone marrow (Medina et al., 2017). Therefore, we hypothesize that the inhibition of erythropoiesis may be a plausible mechanism underlying arsenic-associated anemias.
Red blood cells develop in the bone marrow in response to the hormone, erythropoietin (EPO) (Supplementary Fig. S1A). EPO acts on early erythroid progenitors (burst-forming-unit erythroid (BFU-E) and colony-forming-unit erythroid (CFU-E) to promote survival, proliferation, and differentiation (Hattangadi et al., 2011; Dzierzak and Philipsen, 2013). Erythropoiesis is a dynamic and highly regulated process that relies on coordination of multiple pathways for normal differentiation of erythroid progenitors (Tsiftsoglou et al., 2009; Hattangadi et al., 2011; Dzierzak and Philipsen, 2013). GATA-1 is an essential transcriptional regulator of erythropoiesis and is responsible for inducing and repressing many genes critical for differentiation (Ferreira et al., 2005). Another important regulatory mechanism in early erythroid progenitors is the EPO-activated Signal Transducer and Activator of Transcription 5 (STAT5) prosurvival pathway (Socolovsky et al., 1999; Socolovsky et al., 2001; Liu et al., 2006).
EPO-activated STAT5 signaling utilizes both GATA-1 and STAT5 to regulate and maintain the expression of the prosurvival factor, B-cell lymphoma-extra-large (Bcl-xL), thereby preventing caspase-3 mediated cell death of early erythroid progenitors (Supplementary Fig. S1B) (Gregory et al., 1999). Under basal EPO conditions, erythroid progenitors mature to the CFU-E stage, but most (~60%) succumb to cell death via this mechanism (Wu et al., 1995; Dzierzak and Philipsen, 2013). During hypoxic conditions, EPO levels are elevated and this prosurvival mechanism is fully activated, thereby allowing for the survival and rapid expansion of the early erythroid progenitor pool (Dzierzak and Philipsen, 2013). This survival mechanism represents a critical regulatory pathway that directly links EPO levels to the control of erythropoietic rate by regulating the survival and differentiation of early erythroid progenitors (Koulnis et al., 2012).
We recently found that As+3 interacts with the zinc finger motifs of GATA-1 impairing both the DNA binding and protein-protein interaction activities of this critical transcription factor (Zhou et al., 2020). Despite our understanding of As+3 effects on GATA-1, the downstream pathways impacted by the loss of GATA-1 function have not been evaluated. Additionally, very little is known on the impacts of MMA+3 exposure on early developing erythroid progenitor cells. Our previous findings indicate that the predominant arsenical in the bone marrow of mice exposed to As+3 in their drinking water for 30 days was MMA+3, which reached concentrations similar to the low dose used in this study (80 vs. 100 nM, respectively) (Xu et al., 2016b). Interestingly, there was no detectable As+3 in the bone marrow of these mice and only minimal levels of other arsenic species (Xu et al., 2016b), suggesting that cells at this site are at an increased risk of MMA+3 exposure and toxicity. However, to our knowledge, the relative contribution of MMA+3 vs. As+3 to arsenic-induced hematotoxicity has also not been investigated.
Considering the importance of GATA-1 function in regulating erythroid differentiation, along with the importance of this factor in combination with STAT5 for promoting the survival of early erythroid progenitors, we hypothesized that impairment of these pathways may be responsible for arsenic-induced suppression of erythropoiesis. We further postulated that MMA+3 would produce stronger suppressive effects on the development of early erythroid progenitors than As+3. Taking into account the potential for As+3 metabolites to drive toxicity in vivo, we performed a comparative analysis of As+3 and MMA+3-induced toxicity in early erythroid progenitor cells. In this study, the effects of As+3 and MMA+3 on GATA-1-regulated differentiation of erythroid progenitors were evaluated. We also investigated the effects of As+3 and MMA+3 on STAT5 and GATA-1 mediated survival of early erythroid progenitor cells.
2. METHODS
2.1. Chemicals and reagents
Sodium meta-arsenite (NaAsO2, ≥95% purity, CAS 774-46-5, Cat. No. S7400) was purchased from Sigma Aldrich. Monomethylarsonous acid (CH5AsO2, ≥95% purity, CAS 25400-23-1, Cat. No. M565100) was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). A complete list of reagents utilized in this study can be found in supplementary materials.
As+3 and MMA+3 stock solutions were prepared fresh at the onset of each experiment using cell culture grade water and cell culture medium. The As+3 concentrations used in this study, 100 nM (7.5 ppb) and 500 nM (37.5 ppb) exceed the WHO and US EPA drinking water standard of 10 ppb, but are within the range of environmentally relevant arsenic levels that many human populations around the world are exposed to in drinking water (Hopenhayn et al., 2006; Heck et al., 2008; Naujokas et al., 2013; Surdu et al., 2015; Kile et al., 2016; Parvez et al., 2017). The low concentration of MMA+3 used in this study (100 nM) was selected in accordance with our previous work showing that in vivo exposures of mice to 500 ppb As+3 in their drinking water results in concentrations of MMA+3 in the bone marrow of ~80 nM (Xu et al., 2016b). The higher MMA+3 dose used in this study (500 nM) served primarily for comparisons of equimolar doses of As+3.
2.2. Primary bone marrow cell isolation
All experiments were performed in accordance with protocols approved by the Institutional Animal Use and Care Committee at the University of New Mexico Health Sciences Center. Male C57BL/6J mice were purchased at 11 weeks of age from Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate in our animal facility for one week. All experiments were performed using male C57BL/6J mice between 12-16 weeks of age.
Bone marrow cells from each femur set of three mice were pooled together for all experiments. Primary mouse bone marrow cells were isolated as previously described (Ezeh et al., 2016b). Briefly, bone marrow cells were flushed from both femurs of each mouse by passing approximately 6-9 mL of media through the marrow shaft using a 1cc syringe and 25-G needle. The cell suspension was transferred using a 9 in Pasteur pipette to a 15 mL centrifuge tube, centrifuged at 200×g for 10 min, and resuspended in 20 mL of Isocove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (HI FBS), 2 mM L-glutamine, and 100 mg/ml streptomycin and 100 units/ml penicillin). Cell viabilities and concentrations of isolated cells was then determined using acridine orange/propidium iodide (AO/PI) staining and a Nexcelom Cellometer Auto 2000.
2.3. Hematopoietic progenitor cell isolation and in vitro erythropoiesis model
Hematopoietic progenitor cells (HPC) were isolated from total bone marrow cells using the EasySep mouse HPC isolation kit according to manufacturer’s instructions and as described previously (Zhou et al., 2020). Briefly, bone marrow cells were concentrated to 1×108 cells/mL in Easy Sep Buffer (DPBS without calcium and magnesium (DPBS−), 2% HI FBS, and 1 mM EDTA) and stained with a cocktail of biotinylated lineage specific antibodies (CD5, CD11b, CD19, CD45R/B220, Ly6G/C (Gr-1), TER119) for 15 min at 4°C. After 15 min, streptavidincoated magnetic particles were added to each sample and incubated for an additional 10 min. Samples were brought up to 2.5 mL with EasySep buffer and placed into the EasySep™ magnet (STEMCELL Technologies, Cambridge, MA) for 3 min. After two rounds of isolation, supernatant containing purified HPCs (~>75% Lin−, >45% cKit+) were utilized in an in vitro model of erythropoiesis as described previously by Shuga et al. (2007) and Zhou et al., (2020). HPC were cultured in the presence of 0, 100, and 500 nM As+3 or MMA+3 in SF StemSpan hematopoietic progenitor expansion media supplemented with 100 ng/mL murine SCF and 5 IU/mL human recombinant EPO (31.25 ng/mL) to stimulate erythroid lineage commitment and differentiation (Shuga et al. 2007; Zhou et al., 2020). Growth of erythroid progenitor cells was determined based on the concentration of viable cells after 24 and 48 h of erythroid differentiation. The total number of viable cells was measured using AO/PI staining and a Nexcelom Cellometer.
2.4. RNA isolation and quantitative real-time PCR
RNA was isolated from 0, 100, and 500 nM As+3 or MMA+3 exposed erythroid progenitor cells using the QIAshredder and RNeasy kit according to manufacturer’s instructions. RNA concentrations were determined using an Agilent Nanodrop Spectrophotometer. A total of 100 ng of RNA was utilized for cDNA synthesis using the High Capacity Reverse Transcription Kit. Samples were diluted 1:4 in RNase/DNase free water and stored at −80°C. Quantitative real-time PCR (qPCR) was performed in 10 μL reactions consisting of RNase/DNase free water, TaqMan universal PCR master mix, and TaqMan gene expression assay probes for Gapdh, Gata-1, Gata-2, EpoR, Klf1, Alas2, Alad, or Bcl-xL using a BioRad CFX384 Touch Real-Time PCR Detection System (BioRad, Hercules, CA). The comparative CT method was used for relative quantification of gene expression using Gapdh as an endogenous control.
2.5. Flow cytometry
Early Erythroid progenitor subsets (i.e., BFU-E, CFU-E, and Pro-erythroblast) were classified as Lin-, cKit+, SCA-1−, CD16/32−, CD150+,−, CD105+ based on cell surface marker phenotypes previously described by Pronk and Bryder (2011) and Grover et al. (2014) (Supplementary Fig. S2A). Late-stages of erythroblast differentiation were measured based on Ter119 surface marker expression as previously described by Socolovsky et al. (2001); Koulnis et al. (2011).
For surface marker analysis, 0.5-1×106 cells were collected from each treatment group (0, 100, and 500 nM As+3 or MMA+3) and stained with 0.5 μg of monoclonal antibodies with the following antigen and fluorochrome conjugations: CD117 (APC-R700), SCA-1 (BV605), CD16/32 (BV510), CD150 (BV421), CD105 (BB515), TER119 (FITC) in 50 μL BD Horizon Brilliant Stain Buffer at room temperature in the dark for 30 min. Samples were washed twice and resuspended in 0.5 mL wash/stain buffer (DPBS− with 2% FBS and 0.09% sodium azide) prior to analysis using a BD Accuri™ C6 flow cytometer or BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA). For assessment of cell death, erythroid progenitors were stained as above, followed by staining with propidium iodide in DPBS− (0.25 μg/100 μL sample) according to manufacturer’s instructions (BD Biosciences, San Jose, CA).
Intracellular staining was performed using the BD Transcription Factor buffer kit according to manufacturer’s instructions. Following As+3 or MMA+3 exposure, 0.5-1×106 cells were stained with surface markers for 30 min at 4°C in the dark (as described above). Cells were washed and resuspended in 1 mL of 1X fixation/permeabilization buffer and incubated at 4°C for 45 min. After 45 min, 1 mL of 1X permeabilization/wash reagent was added to each tube and the samples are centrifuged at 350×g for 6 min. Cells were resuspended in 50 μL of 1X permeabilization/wash buffer containing the manufacturer recommended test volume of GATA-1 (PE), BCL-xL (Alexa Fluor® 647), cleaved caspase-3 (Asp175; Alexa Fluor® 647 ), phosphoSTAT5 (pY694; PE), or concentration, fluorochrome, and isotype matched controls and incubated at room temperature for 1 h. Cells were washed twice with 1X permeabilization/wash buffer and resuspended in 0.3 mL wash/stain buffer prior to analysis using a BD LSRFortessa flow cytometer.
2.6. Data analysis and statistics
All flow cytometry data was processed using FlowJo version 10 software (FlowJo LLC, Ashland, OR). Data were analyzed with Excel 2010 and GraphPad Prism version 9 (GraphPad Software, San Diego, CA). Differences between control and treatment groups and between As+3 and MMA+3 groups were determined using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Statistically significant changes were considered at significance levels of *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Three replicates were performed and analyzed for each group (control, As+3, and MMA+3) and at least three independent experiments were conducted and comparable results attained.
3. RESULTS
3.1. As+3 and MMA+3 impair erythroid differentiation of primary mouse bone marrow hematopoietic progenitor cells
To determine the effects of As+3 and MMA+3 on erythroblast differentiation, we utilized an in vitro model of erythropoiesis using primary mouse bone marrow HPC stimulated with EPO and SCF to promote erythroid lineage commitment and differentiation (Shuga et al., 2007). Utilizing this in vitro model of erythropoiesis, we assessed erythroblast differentiation based on cell surface marker phenotype using multi-parameter flow cytometry (Pronk and Bryder, 2011; Grover et al., 2014) after 48 h As+3 and MMA+3 exposure (Fig. 1A-B).
Fig. 1.
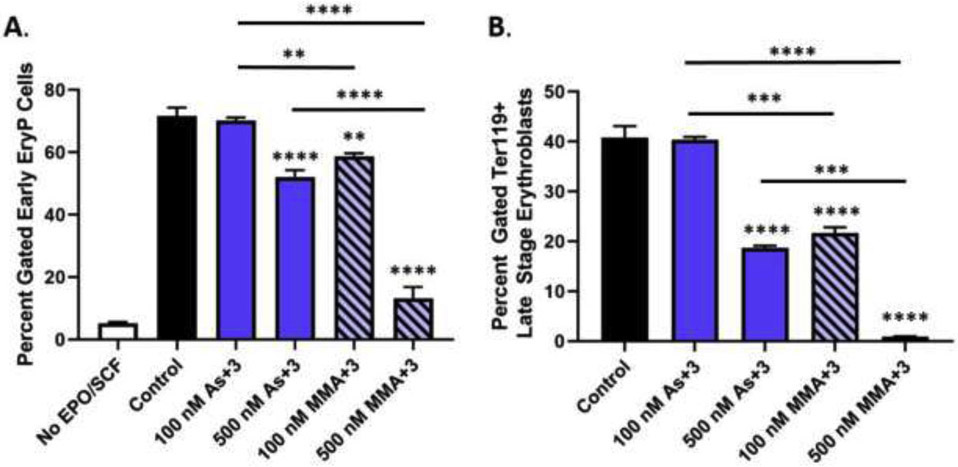
As+3 and MMA+3 suppress the differentiation of erythroid progenitor cells. Erythroid differentiation of primary mouse bone marrow hematopoietic progenitors (HPC) was induced in vitro with erythropoietin and stem cell factor in the presence of 100 and 500 nM As+3 or MMA+3 for 48 h. (A) Percentage of early erythroid cells after treatment with As+3 or MMA+3 (see Supplementary Fig. S2A for gating strategy used to identify early erythroid progenitors). (B) Percentage of late-stage erythroblasts (TER119+) following exposure to As+3 or MMA+3. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
A significant reduction in the percentage of early erythroid progenitors (i.e., BFU-E, CFU-E, and Proerythroblasts) was found with 500 nM As+3 (Fig. 1A). A similar suppression of early erythroid progenitors was found with MMA+3 at 100 nM; however, early erythroid differentiation was nearly abolished with 500 nM MMA+3 (Fig. 1A). Interestingly, the suppressive effects of As+3 and MMA+3 were selective for erythroid progenitors, as no significant decreases were observed in other cell lineages (i.e., lymphoid progenitors and myeloid progenitors; Supplementary Fig. S3). These findings were confirmed by assessing the BFU-E colony-forming ability of primary mouse bone marrow HPC cultured ex vivo (in the presence of As+3 or MMA+3) in serum free methylcellulose based medium supplemented with EPO to promote the differentiation and expansion of erythroid progenitor cells (Supplementary Fig. S4).
To determine if the reduction of early erythroid progenitors persisted throughout erythroblast differentiation, we evaluated later-stage erythroblasts based on TER119 cell surface marker expression using flow cytometry (Socolovsky et al., 2001; Koulnis et al., 2011) (Fig. 1B). Similar to the effects observed on early erythroid progenitors, the percentage of later-stage erythroblasts (TER119+) was strongly reduced by 500 nM As+3 and 100 nM MMA+3, respectively and very few erythroblasts reached later stages of differentiation with 500 nM MMA+3 (<5 % of cells) (Fig. 1B).
3.2. Suppression of erythroid progenitor cell growth by As+3 and MMA+3 exposures
The differentiation of erythroid progenitors is highly dependent on proliferation (Kinross et al., 2006; Tallack et al., 2007; Pop et al., 2010). As such, we utilized AO/PI staining and a Nexcelom Cellometer to evaluate cell growth after 24 and 48 h exposure to As+3 and MMA+3. A significant suppression of cell proliferation (as measured by the total number of viable cells) was found after 24 and 48 h exposure to both 100 nM and 500 nM MMA+3, whereas 500 nM As+3 only significantly disrupted growth after 48 h of exposure (Fig. 2 A-B). Similar to the effects on differentiation, the magnitude of cell growth inhibition produced by 500 nM As+3 was comparable to 100 nM MMA+3 (Fig. 2B).
Fig. 2.
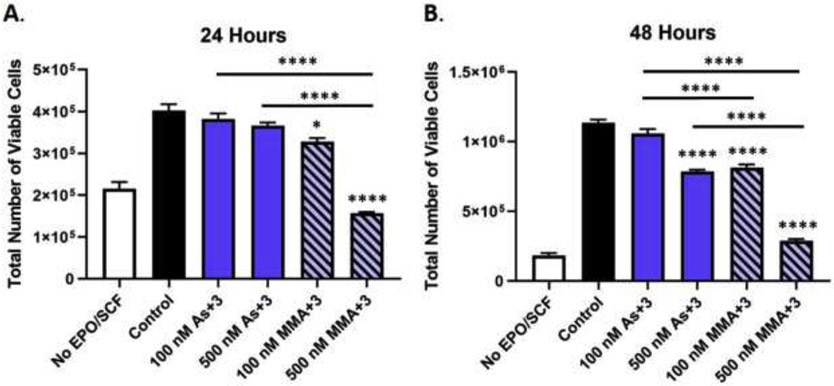
Erythroid progenitor cell growth is suppressed by As+3 and MMA+3 exposure. Erythroid differentiation of primary mouse bone marrow hematopoietic progenitors (HPC) was induced in vitro with erythropoietin and stem cell factor and the total number of viable cells was measured by AO/PI staining using a Nexcelom Cellometer following (A) 24 h and (B) 48 h exposure to 0, 100, and 500 nM As+3 or MMA+3. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; *p<0.05, ****p<0.0001).
Collectively, these results demonstrate that As+3 and MMA+3 impair erythropoiesis by suppressing very early stages of erythroid development, likely through combined effects on proliferation, differentiation, and cell viability. Our findings also show that MMA+3 produced more potent suppression of erythropoiesis than As+3.
3.3. Differential suppressive effects of As+3 and MMA+3 on GATA-1 expression and activation
We recently showed that the master regulator of erythropoiesis, GATA-1, is a sensitive molecular target of As+3. As+3 was found to selectively interact with the zinc finger motifs of GATA-1 disrupting DNA binding and protein-protein interactions (Zhou et al., 2020). In an effort to determine whether disruption of GATA-1 is responsible for the suppressed differentiation of early erythroid progenitors following arsenical exposure, we measured the expression of GATA-1-regulated genes important for erythroid differentiation using qPCR following 24 h As+3 or MMA+3 exposure. We also assessed GATA-1 protein and mRNA levels in early erythroid progenitors using intracellular flow cytometry and qPCR, respectively.
The expression of several GATA-1-regulated genes critical for erythroid differentiation were significantly reduced following 500 nM As+3 exposure, including Gata-2, Krüppel-like factor-1 (Klf-1), and Epo receptor (EpoR) (Fig. 3A-G). The effects of MMA+3 on GATA-1-regulated gene expression were more pronounced. All genes measured (Gata-2, Klf-1, EpoR, 5- aminolevulinate synthase 2 (Alas2), 5-aminolevulinate dehydratase (Alad), ATP binding cassette mitochondrial erythroid (Abcb10) were significantly reduced with 100 and 500 nM MMA+3 (Fig. 3A-G). However, The percentage of GATA-1+ early erythroblasts, the fluorescence intensity of GATA-1 in early erythroblasts and Gata-1 mRNA expression were significantly reduced after 24 h exposure to 100 nM and 500 nM MMA+3 (Fig. 3A, Fig. 4A-B, and Supplementary Fig. S2B). Interestingly, As+3 significantly reduced Gata-1 mRNA expression (Fig. 3A), but did not significantly modulate GATA-1 protein in early erythroid progenitors (Fig. 4A-B).
Fig. 3.
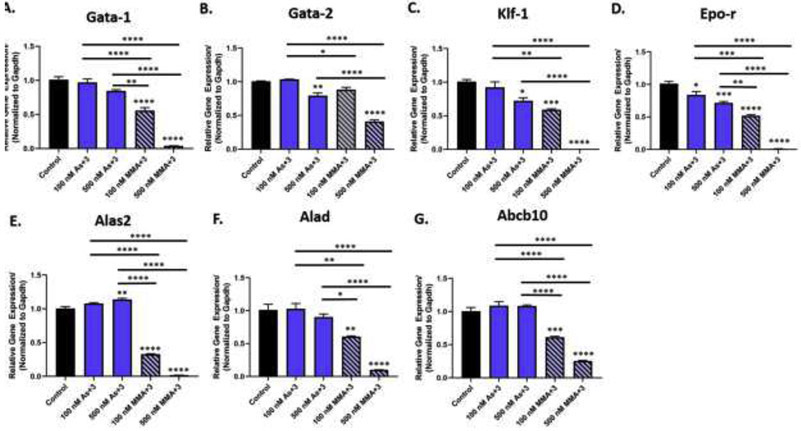
As+3 and MMA+3 produce differential suppressive effects on GATA-1-regulated gene expression in early erythroid progenitor cells. Erythroid progenitor cells were treated in vitro with 100 and 500 nM As+3 or MMA+3 for 24 h and the expression of GATA-1-regulated genes was measured by qPCR. Relative expression of GATA-1-regulated genes (normalized to Gapdh) (A) Gata-1, (B) Gata-2, (C) Klf-1, (D) Epo-r, (E) Alas2, (F) Alad, (G) Abcb10 in erythroid progenitor cells after As+3 or MMA+3 exposure. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Fig. 4.
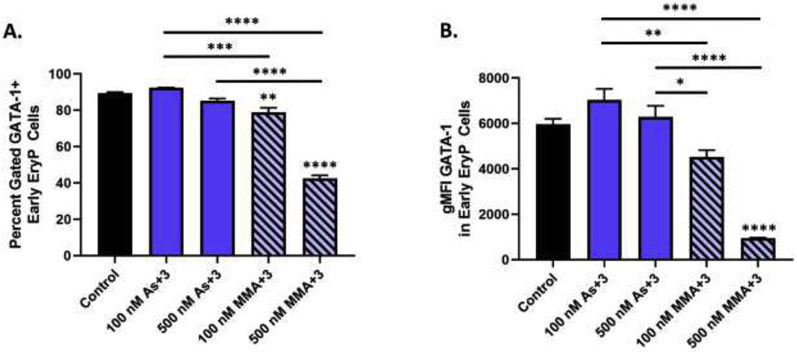
As+3 and MMA+3 produce differential suppressive effects on GATA-1 protein in surface marker defined early erythroid progenitor cells. (A) Percentage of GATA-1+ early erythroid progenitors following 24 h exposure to As+3 or MMA+3. (B) GATA-1 geometric mean fluorescence intensity in early erythroid progenitors. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Taken together, these findings show differential effects of As+3 and MMA+3 on GATA-1 in early erythroid progenitor cells. Whereas As+3 primarily disrupts GATA-1 function, MMA+3 disrupts both expression and function of GATA-1, as evidenced by decreased GATA-1 protein and mRNA levels in early erythroid progenitors as well as reductions of GATA-1-regulated gene expression.
3.4. MMA+3 also suppresses STAT5 phosphorylation in early erythroid progenitors
A downstream pathway regulated by GATA-1 in coordination with EPO-activated STAT5 is a critical prosurvival pathway in early erythroid progenitors. As+3 and MMA+3 have been reported to inhibit the development of early B cells in the bone marrow and early T cells in the thymus by reducing STAT5 activation (Ezeh et al., 2016a; Xu et al., 2016a). Since the survival and subsequent maturation of erythroid progenitors is dependent on STAT5 signaling (Socolovsky et al., 1999; Socolovsky et al., 2001), and taking into account that this pathway is partially regulated by GATA-1, we hypothesized that the suppression of early erythroid progenitor cell development may also be influenced by inhibition of STAT5 signaling.
To determine if As+3 or MMA+3 alter the activation of STAT5, we evaluated the phosphorylation of STAT5 (pSTAT5) in early erythroid progenitors using intracellular flow cytometry, a method that we have previously validated by western blot (Ezeh et al., 2016a; Xu et al., 2016a). A significant reduction in the percentage of pSTAT5+ early erythroid progenitors and the fluorescence intensity of pSTAT5 in early erythroid progenitors was detected following 24 h exposure to 100 and 500 nM MMA+3 (Fig. 5A-B and Supplementary Fig. S2C). A slight, albeit non statistically significant reduction of pSTAT5 was also observed with 500 nM As+3 (p = 0.053; Fig. 5A-B and Supplementary Fig. S2C).
Fig. 5.
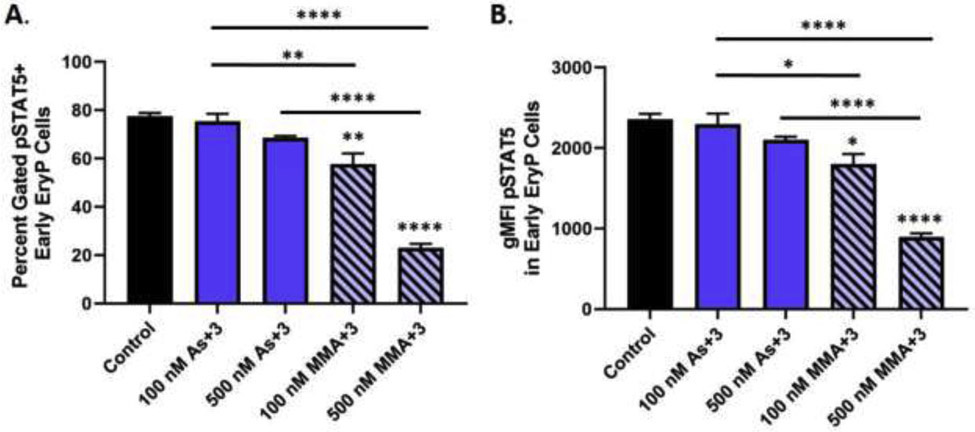
MMA+3, but not As+3 suppresses STAT5 phosphorylation in early erythroid progenitor cells. STAT5 phosphorylation (pY694) (pSTAT5) was measured by flow cytometry in surface marker defined early erythroid progenitors following in vitro exposure to 100 and 500 nM As+3 or MMA+3 for 24 h. (A) Percentage of pSTAT5+ early erythroid progenitors following exposure to As+3 or MMA+3. (B) pSTAT5 geometric mean fluorescence intensity in early erythroid progenitors. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; *p<0.05, **p<0.01, ****p<0.0001.
These findings show that MMA+3, but not As+3 significantly disrupted the activation of STAT5 in early erythroid progenitor cells. These findings suggest that; whereas, As+3 acts dominantly through the inhibition of GATA-1 function, MMA+3 likely impairs multiple pathways (GATA-1 and pSTAT5) important for erythroid progenitor differentiation.
3.5. As+3 and MMA+3 impaired the GATA-1 and pSTAT5 regulated survival of early erythroid progenitors
To determine if arsenical-mediated suppression of GATA-1 and STAT5 activation resulted in disruption of STAT5 and GATA-1 regulated survival of early erythroid progenitors, we measured two downstream factors in the prosurvival pathway (i.e., Bcl-xL and cleaved caspase-3) using intracellular flow cytometry. MMA+3, but not As+3, significantly decreased Bcl-xL gene expression after 24 h of exposure (Fig. 6A). Additionally, a significant reduction of the percentage of Bcl-xL+ erythroid progenitors was found after 24 h exposure to 500 nM As+3 and 100 and 500 nM MMA+3 (Fig. 6B and Supplementary Fig. S2D).
Fig. 6.

Effects of As+3 or MMA+3 on Bcl-xL in surface marker defined early erythroid progenitor cells. Erythroid progenitors were treated in vitro with 100 and 500 nM As+3 or MMA+3 for 24 h and Bcl-xL was measured in early erythroid progenitor cells by flow cytometry. (A) Relative Bcl-xL mRNA expression (normalized to Gapdh) in erythroid progenitor cells. (B) Percentage of Bcl-xL+ early erythroid progenitors following treatment with As+3 or MMA+3. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; **p<0.01, ***p<0.001, ****p<0.0001).
Cleaved caspase-3, the terminal factor downstream of Bcl-xL in the STAT5 and GATA-1 regulated survival pathway, was significantly increased in early erythroid progenitors treated with 500 nM As+3, and 100 and 500 nM MMA+3 for 24 h (Fig. 7A and Supplementary Fig. S1E). In further support of the As+3 and MMA+3-induced impairment of the GATA-1 and STAT5 mediated survival pathway, we measured cell death using PI staining in surface marker defined early erythroid progenitors by flow cytometry. The percentage of cell death (PI+) among early erythroid progenitors was significantly increased by approximately 2-fold with 500 nM As+3 and 100 nM MMA+3 and approximately 20-fold with 500 nM MMA+3 (Fig. 7B and Supplementary Fig. S2F).
Fig. 7.
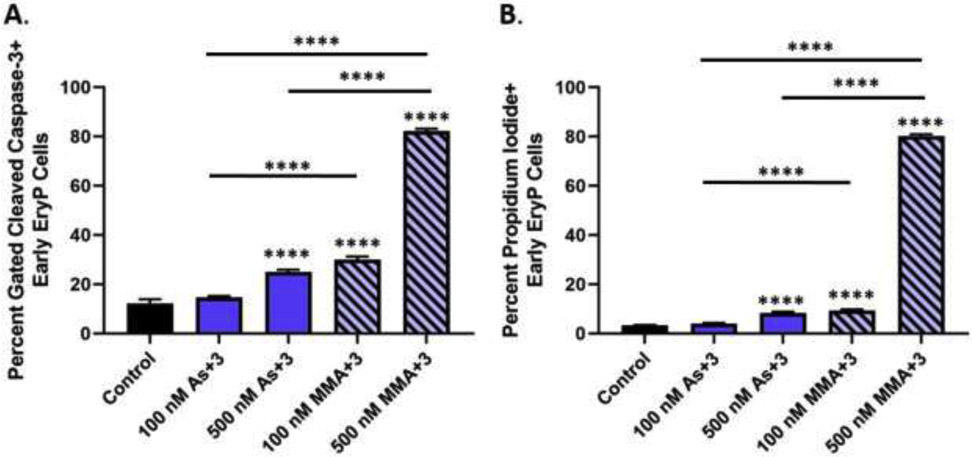
As+3 and MMA+3 increase cleaved caspase-3 mediated cell death of early erythroid progenitor cells. Erythroid progenitor cells were treated in vitro with 100 and 500 nM As+3 or MMA+3 for 24 h and cleaved caspase-3 and cell death (PI+) were measured in surface marker defined early erythroid progenitors by flow cytometry. (A) Percentage of cleaved-caspase 3 in early erythroid progenitor cells. (B) Percentage of cell death (PI+) in early erythroid progenitors. Data are expressed as mean ± SEM. Statistically significant differences compared to control or between exposure groups (as indicated by the bars) in one-way ANOVA followed by Tukey’s post hoc test (n = 3; ****p<0.0001).
Taken together, these findings provide support that As+3 and MMA+3 impair GATA-1 and STAT5 regulated survival of early erythroid progenitors, as indicated by the loss of Bcl-xL, increased caspase-3 activation, and increased death of this population. Additionally, as observed in our previous experiments, the effects of MMA+3 on early erythroid progenitor cell survival were greater compared to As+3.
4. DISCUSSION
Erythropoiesis is a dynamic and highly controlled process that requires the coordination of GATA-1 and STAT5 regulated signaling pathways for normal development of erythroid progenitors (Dzierzak and Philipsen, 2013). In this study, we report that both As+3 and MMA+3 impair erythropoiesis by disrupting the differentiation and survival of early erythroid progenitor cells. As+3 and MMA+3 were found to suppress GATA-1-regulated differentiation and GATA-1 and STAT5-regulated survival of early erythroid progenitor cells. We previously reported that the development of early erythroid progenitor cells is sensitive to As+3 exposure in vivo and in vitro, likely resulting from downstream consequences caused by the loss of GATA-1 function (Medina et al., 2017; Zhou et al., 2020).
Importantly, As+3 and MMA+3 appear to produce differential effects on GATA-1. As+3 primarily impaired GATA-1 function; whereas, MMA+3 also impaired GATA-1 regulation, as evidenced by decreased protein expression. As a result, MMA+3 produced stronger suppressive effects on the expression of erythroid-specific differentiation target genes and subsequent erythroid progenitor cell differentiation than equimolar doses of As+3. The decrease of GATA-1-regulated genes with As+3 is consistent with our previous findings demonstrating that As+3 disrupts the zinc finger motifs of GATA-1, thereby inhibiting DNA binding activity and interactions with the transcriptional coactivator, Friend of GATA-1 (FOG-1) both of which are essential in the regulation of downstream gene expression (Pevny et al., 1991; Pevny et al., 1995; Welch et al., 2004; Ferreira et al., 2005; Zhou et al., 2020). This suggests that the reduction of GATA-1 responsive gene expression is a contributing factor to the suppression of early erythroid progenitor cell differentiation and subsequent impairment of erythropoiesis observed here and in previous studies (Medina et al., 2017; Zhou et al., 2020). Whether MMA+3 also disrupts GATA-1 function through zinc finger interactions or alternate mechanisms has yet to be determined and will be the topic of our future investigations.
Early erythroid cells are dependent on EPO for survival and differentiation (Wu et al., 1995; Dzierzak and Philipsen, 2013). The EPO receptor stimulates activation of STAT5, which in combination with GATA-1, functions to regulate the expression of the prosurvival factor, Bcl-xL (Gregory et al., 1999). STAT5 knockout studies in mice and EPO deprivation studies in erythroid progenitor cell models show that loss of STAT5 corresponds to reduced Bcl-xL, increased cell death, and inhibition of erythropoiesis (Silva et al., 1996; Gregory et al., 1999; Socolovsky et al., 1999; Socolovsky et al., 2001). Similarly, GATA-1 knockout studies in erythroid progenitor cell models have also demonstrated that erythroid cells lacking this factor undergo maturation arrest and apoptosis (Weiss et al., 1994; Weiss and Orkin, 1995; Fujiwara et al., 1996).
In this study, we found that As+3 and MMA+3 reduced EPO receptor mRNA expression, the upstream regulator of STAT5. Additionally, we found that MMA+3, but not As+3 significantly disrupted STAT5 activation. The suppression of EPO-activated STAT5 phosphorylation, along with the disruption of GATA-1 expression/function reduced Bcl-xL in early erythroid progenitors. The loss of Bcl-xL in early erythroid progenitors resulted in increased activation of caspase-3 mediated cell death. The greater cell death response observed with MMA+3 likely resulted from the combined loss of pSTAT5 and GATA-1 regulation of Bcl-xL.
Several studies have reported the phosphorylation of STAT5 to be impaired by As+3 and MMA+3 exposures (Cheng et al., 2004; Ezeh et al., 2016a; Xu et al., 2016a). We previously found that As+3 and MMA+3 inhibit the development of pre-B cells in the bone marrow and pre-T cells in the thymus by reducing STAT5 activation (Ezeh et al., 2016a; Xu et al., 2016a). In both of the aforementioned studies, As+3 had a limited ability to disrupt STAT5 phosphorylation, in comparison to MMA+3 (Ezeh et al., 2016a; Xu et al., 2016a). Similarly, in the present study, phosphorylation of STAT5 was only significantly reduced by MMA+3. Considering that the impairment of STAT5 activation has been observed in two separate progenitor cell lineages (i.e., lymphoid and erythroid), it may represent a common mechanism of arsenic-induced toxicity among HPC.
Our results show that MMA+3 is more toxic than As+3, which is consistent with many previous studies (Styblo et al., 2000; Petrick et al., 2001; Ezeh et al., 2016a; Xu et al., 2017). In comparison to As+3, MMA+3 has a greater affinity for interactions with sulfhydryl groups in proteins, exerts greater inhibitory effects on enzymes, and is more cytotoxic than As+3 (Styblo et al., 1995; Styblo et al., 1997; Lin et al., 1999; Petrick et al., 2000; Shen et al., 2013). These differences in toxicity may account for the differential effects of As+3 and MMA+3 observed in this study, and suggests that MMA+3 disrupts a broader spectrum of factors important for erythroid development than As+3.
In addition, arsenic metabolism may have a significant role in the inhibition of erythropoiesis and anemia observed in vivo (Medina et al., 2017; Zhou et al., 2020). This is significant because our previous studies revealed inverse associations between urinary inorganic arsenicals (As+5 + As+3), monomethylated arsenical species (MMA+5 + MMA+3), and demethylated arsenical (DMA+5 + DMA+3) with red blood cell counts in a cohort of men from rural Bangladesh (Parvez et al., 2017). This highlights the importance of arsenic metabolism in arsenic-associated anemias, and when coupled with findings from the present study, suggests that people with variants of As3MT which modulate As+3 methylation efficiency may be at an elevated risk of anemia resulting from As+3 and MMA+3-induced suppression of erythropoiesis.
It is important to keep in mind that findings from the present study were based solely on an ex vivo primary mouse cell model system that does not accurately reflect the in vivo metabolism of arsenic, nor the resultant tissue and arsenical specific exposures. Additionally, although in the present study we exposed cells directly to As+3 or MMA+3, we cannot completely rule out the possibility that under our ex vivo experimental conditions that As+3 is metabolized into MMA+3 or that MMA+3 is further metabolized to DMA. Further in vivo studies evaluating the comparative hematotoxicity of As+3 vs. MMA+3 are needed, and will be a major focus of future investigations.
In summary, As+3 and MMA+3 were found to impair erythroid differentiation at environmentally relevant and physiologically achievable concentrations, respectively. We identified that two important erythropoiesis regulatory pathways are compromised by As+3 and MMA+3 (Fig. 8 and Supplementary Fig. S1B). As+3 and MMA+3 were found to suppress the expression of GATA-1-regulated genes important for promoting the differentiation of early erythroid progenitors. Additionally, As+3 and MMA+3 decreased the Bcl-xL-mediated survival of early erythroid progenitor cells through impairments of GATA-1/STAT5 and GATA-1, respectively. These findings provide evidence that arsenic disrupts both differentiation and survival of early erythroid progenitors, resulting in dyserythropoiesis. Collectively, results from the present study provide novel mechanistic insights by which arsenic exposures can contribute to the development and/or exacerbation of anemia.
Figure 8.
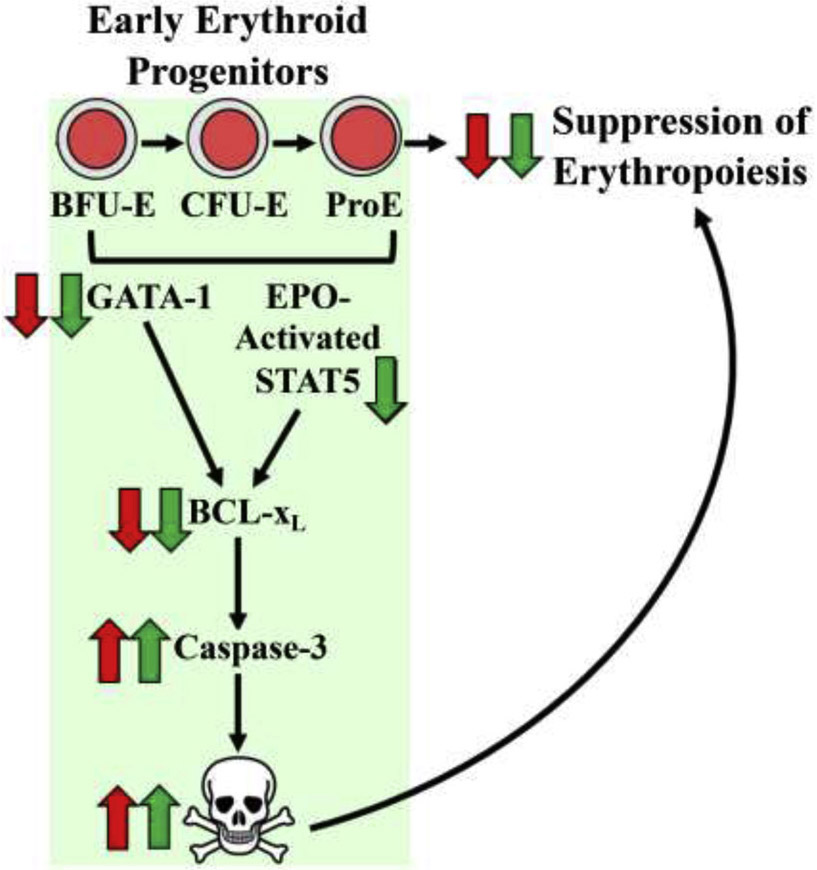
As+3 and MMA+3 impair erythropoiesis by disrupting two critical regulatory pathways controlling the differentiation and survival of early erythroid progenitor cells. As+3 and MMA+3 suppress the development of early erythroid progenitors likely through combined effects on GATA-1 and EPO-activated STAT5 regulated differentiation and survival pathways. The effects of As+3 were primarily mediated through effects on GATA-1, whereas, the MMA+3 impaired both GATA-1 and EPO-activated STAT5. Effects of As+3 or MMA+3 are indicated with red or green arrows, respectively.
Supplementary Material
Highlights.
Development of early-stage erythroid progenitors is impaired by both As+3 and MMA+3.
As+3 and MMA+3 disrupt the differentiation and survival of early erythroid progenitors.
Differentiation and survival were impaired by combined effects on GATA-1 and STAT5.
MMA+3 is more toxic in vitro than As+3 to early developing erythroid cells.
FUNDING
This work was supported by the National Institutes of Environmental Health Sciences RO1 ES029369, R01 ES029369-03S1 (K.J.L); RO1 ES019968 (S.W.B.); the UNM Clinical and Translational Science Center Grant Number UL1TR001449 (S.W.B.); the National Institute of Environmental Health Sciences and UNM METALS Superfund Research Program Grant Number P42 ES025589 (S.W.B. and K.J.L.); and the National Institute of General Medical Sciences Grant Number P20GM130422 (A.M.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Ahsan H, Chen Y, Kibriya MG, Slavkovich V, Parvez F, Jasmine F, Gamble MV, Graziano JH, 2007. Arsenic metabolism, genetic susceptibility, and risk of premalignant skin lesions in Bangladesh. Cancer Epidemiol Biomarkers Prev 16, 1270–1278. [DOI] [PubMed] [Google Scholar]
- Chaparro CM, Suchdev PS, 2019. Anemia epidemiology, pathophysiology, and etiology in low- and middle-income countries. Ann N Y Acad Sci 1450, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, Graziano JH, Ahsan H, 2009. Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol Appl Pharmacol 239, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Guo YL, Su HJ, Hsueh YM, Smith TJ, Ryan LM, Lee MS, Chao SC, Lee JY, Christiani DC, 2003. Arsenic methylation and skin cancer risk in southwestern Taiwan. J Occup Environ Med 45, 241–248. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Li P, David M, Smithgall TE, Feng L, Lieberman MW, 2004. Arsenic inhibition of the JAK-STAT pathway. Oncogene 23, 3603–3612. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, Philipsen S, 2013. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med 3, a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh PC, Xu H, Lauer FT, Liu KJ, Hudson LG, Burchiel SW, 2016a. Monomethylarsonous acid (MMA+3) Inhibits IL-7 Signaling in Mouse Pre-B Cells. Toxicol Sci 149, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh PC, Xu H, Wang SC, Medina S, Burchiel SW, 2016b. Evaluation of Toxicity in Mouse Bone Marrow Progenitor Cells. Curr Protoc Toxicol 67, 18 19 11–18 19 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario D, Gribaldo L, Hartung T, 2016. Arsenic Exposure and Immunotoxicity: a Review Including the Possible Influence of Age and Sex. Curr Environ Health Rep 3, 1–12. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Ohneda K, Yamamoto M, Philipsen S, 2005. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol 25, 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH, 1996. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci U S A 93, 12355–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ, 1999. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94, 87–96. [PubMed] [Google Scholar]
- Grover A, Mancini E, Moore S, Mead AJ, Atkinson D, Rasmussen KD, O'Carroll D, Jacobsen SE, Nerlov C, 2014. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med 211, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF, 2011. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 118, 6258–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck JE, Chen Y, Grann VR, Slavkovich V, Parvez F, Ahsan H, 2008. Arsenic exposure and anemia in Bangladesh: a population-based study. J Occup Environ Med 50, 80–87. [DOI] [PubMed] [Google Scholar]
- Hopenhayn C, Bush HM, Bingcang A, Hertz-Picciotto I, 2006. Association between arsenic exposure from drinking water and anemia during pregnancy. J Occup Environ Med 48, 635–643. [DOI] [PubMed] [Google Scholar]
- Huang YK, Pu YS, Chung CJ, Shiue HS, Yang MH, Chen CJ, Hsueh YM, 2008. Plasma folate level, urinary arsenic methylation profiles, and urothelial carcinoma susceptibility. Food Chem Toxicol 46, 929–938. [DOI] [PubMed] [Google Scholar]
- Hughes MF, 2002. Arsenic toxicity and potential mechanisms of action. Toxicol Lett 133, 1–16. [DOI] [PubMed] [Google Scholar]
- Kile ML, Faraj JM, Ronnenberg AG, Quamruzzaman Q, Rahman M, Mostofa G, Afroz S, Christiani DC, 2016. A cross sectional study of anemia and iron deficiency as risk factors for arsenic-induced skin lesions in Bangladeshi women. BMC Public Health 16, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinross KM, Clark AJ, Iazzolino RM, Humbert PO, 2006. E2f4 regulates fetal erythropoiesis through the promotion of cellular proliferation. Blood 108, 886–895. [DOI] [PubMed] [Google Scholar]
- Koulnis M, Pop R, Porpiglia E, Shearstone JR, Hidalgo D, Socolovsky M, 2011. Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flowcytometric assay. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulnis M, Porpiglia E, Porpiglia PA, Liu Y, Hallstrom K, Hidalgo D, Socolovsky M, 2012. Contrasting dynamic responses in vivo of the Bcl-xL and Bim erythropoietic survival pathways. Blood 119, 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koury MJ, 2014. Abnormal erythropoiesis and the pathophysiology of chronic anemia. Blood Rev 28, 49–66. [DOI] [PubMed] [Google Scholar]
- Lin S, Cullen WR, Thomas DJ, 1999. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem Res Toxicol 12, 924–930. [DOI] [PubMed] [Google Scholar]
- Lin YC, Chen WJ, Huang CY, Shiue HS, Su CT, Ao PL, Pu YS, Hsueh YM, 2018. Polymorphisms of Arsenic (+3 Oxidation State) Methyltransferase and Arsenic Methylation Capacity Affect the Risk of Bladder Cancer. Toxicol Sci 164, 328–338. [DOI] [PubMed] [Google Scholar]
- Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M, 2006. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood 108, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Li Y, Gao Y, Zhao L, Feng H, Wei W, Qiu C, He Q, Zhang Y, Fu S, Sun D, 2018. Association between arsenic metabolism gene polymorphisms and arsenic-induced skin lesions in individuals exposed to high-dose inorganic arsenic in northwest China. Sci Rep 8, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina S, Xu H, Wang SC, Lauer FT, Liu KJ, Burchiel SW, 2017. Low level arsenite exposures suppress the development of bone marrow erythroid progenitors and result in anemia in adult male mice. Toxicol Lett 273, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA, 2013. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect 121, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez F, Medina S, Santella RM, Islam T, Lauer FT, Alam N, Eunus M, Rahman M, Factor-Litvak P, Ahsan H, Graziano JH, Liu KJ, Burchiel SW, 2017. Arsenic exposures alter clinical indicators of anemia in a male population of smokers and nonsmokers in Bangladesh. Toxicol Appl Pharmacol 331, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Vasken Aposhian H, 2000. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol 163, 203–207. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Jagadish B, Mash EA, Aposhian HV, 2001. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem Res Toxicol 14, 651–656. [DOI] [PubMed] [Google Scholar]
- Pevny L, Lin CS, D'Agati V, Simon MC, Orkin SH, Costantini F, 1995. Development of hematopoietic cells lacking transcription factor GATA-1. Development 121, 163–172. [DOI] [PubMed] [Google Scholar]
- Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D'Agati V, Orkin SH, Costantini F, 1991. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260. [DOI] [PubMed] [Google Scholar]
- Pop R, Shearstone JR, Shen Q, Liu Y, Hallstrom K, Koulnis M, Gribnau J, Socolovsky M, 2010. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Bryder D, 2011. Flow cytometry-based identification of immature myeloerythroid development. Methods Mol Biol 699, 275–293. [DOI] [PubMed] [Google Scholar]
- Shen S, Li XF, Cullen WR, Weinfeld M, Le XC, 2013. Arsenic binding to proteins. Chem Rev 113, 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuga J, Zhang J, Samson LD, Lodish HF, Griffith LG, 2007. In vitro erythropoiesis from bone marrow-derived progenitors provides a physiological assay for toxic and mutagenic compounds. Proc Natl Acad Sci U S A 104, 8737–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M, Grillot D, Benito A, Richard C, Nunez G, Fernandez-Luna JL, 1996. Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood 88, 1576–1582. [PubMed] [Google Scholar]
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF, 1999. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell 98, 181–191. [DOI] [PubMed] [Google Scholar]
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF, 2001. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood 98, 3261–3273. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ, 2000. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol 74, 289–299. [DOI] [PubMed] [Google Scholar]
- Styblo M, Serves SV, Cullen WR, Thomas DJ, 1997. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem Res Toxicol 10, 27–33. [DOI] [PubMed] [Google Scholar]
- Styblo M, Yamauchi H, Thomas DJ, 1995. Comparative in vitro methylation of trivalent and pentavalent arsenicals. Toxicol Appl Pharmacol 135, 172–178. [DOI] [PubMed] [Google Scholar]
- Surdu S, Bloom MS, Neamtiu IA, Pop C, Anastasiu D, Fitzgerald EF, Gurzau ES, 2015. Consumption of arsenic-contaminated drinking water and anemia among pregnant and non-pregnant women in northwestern Romania. Environ Res 140, 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallack MR, Keys JR, Perkins AC, 2007. Erythroid Kruppel-like factor regulates the G1 cyclin dependent kinase inhibitor p18INK4c. J Mol Biol 369, 313–321. [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Styblo M, Lin S, 2001. The cellular metabolism and systemic toxicity of arsenic. Toxicol Appl Pharmacol 176, 127–144. [DOI] [PubMed] [Google Scholar]
- Tsiftsoglou AS, Vizirianakis IS, Strouboulis J, 2009. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 61, 800–830. [DOI] [PubMed] [Google Scholar]
- Tyler CR, Allan AM, 2014. The Effects of Arsenic Exposure on Neurological and Cognitive Dysfunction in Human and Rodent Studies: A Review. Curr Environ Health Rep 1, 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahter M, 2002. Mechanisms of arsenic biotransformation. Toxicology 181-182, 211–217. [DOI] [PubMed] [Google Scholar]
- Weiss MJ, Keller G, Orkin SH, 1994. Novel insights into erythroid development revealed through in vitro differentiation of GATA-1 embryonic stem cells. Genes Dev 8, 1184–1197. [DOI] [PubMed] [Google Scholar]
- Weiss MJ, Orkin SH, 1995. Transcription factor GATA-1 permits survival and maturation of erythroid precursors by preventing apoptosis. Proc Natl Acad Sci U S A 92, 9623–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ, 2004. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147. [DOI] [PubMed] [Google Scholar]
- WHO.Organization, W.H. 2015. The Global Prevalence of Anaemia in 2011, www.who.int/nutrition/publications/micronutrients/global_prevalence_anaemia_2011/n/.
- Wu H, Liu X, Jaenisch R, Lodish HF, 1995. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 83, 59–67. [DOI] [PubMed] [Google Scholar]
- Xu H, Lauer FT, Liu KJ, Hudson LG, Burchiel SW, 2016a. Environmentally relevant concentrations of arsenite and monomethylarsonous acid inhibit IL-7/STAT5 cytokine signaling pathways in mouse CD3+CD4-CD8- double negative thymus cells. Toxicol Lett 247, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, McClain S, Medina S, Lauer FT, Douillet C, Liu KJ, Hudson LG, Styblo M, Burchiel SW, 2016b. Differential sensitivities of bone marrow, spleen and thymus to genotoxicity induced by environmentally relevant concentrations of arsenite. Toxicol Lett 262, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Medina S, Lauer FT, Douillet C, Liu KJ, Styblo M, Burchiel SW, 2017. Genotoxicity induced by monomethylarsonous acid (MMA(+3)) in mouse thymic developing T cells. Toxicol Lett 279, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RC, Hsu KH, Chen CJ, Froines JR, 2000. Arsenic methylation capacity and skin cancer. Cancer Epidemiol Biomarkers Prev 9, 1259–1262. [PubMed] [Google Scholar]
- Zhou X, Medina S, Bolt AM, Zhang H, Wan G, Xu H, Lauer FT, Wang SC, Burchiel SW, Liu KJ, 2020. Inhibition of red blood cell development by arsenic-induced disruption of GATA-1. Sci Rep 10, 19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun X, Mobarak C, Gandolfi AJ, Burchiel SW, Hudson LG, Liu KJ, 2014. Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins. Chem Res Toxicol 27, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.