
Figure 1: Complement activation and its effects in periodontitis.
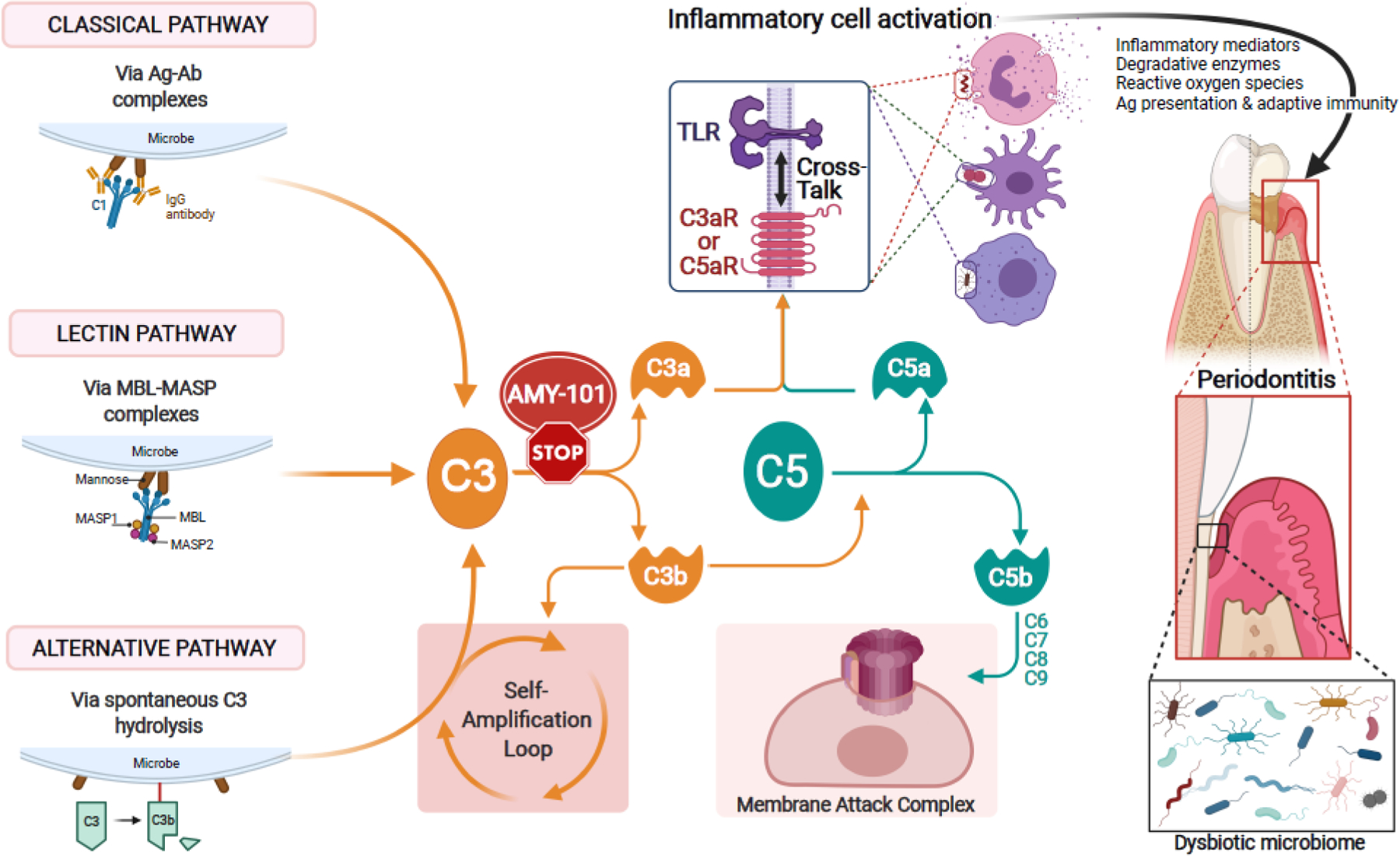
The complement cascade can be triggered by distinct initiation mechanisms: The classical pathway is initiated by antigen (Ag) – antibody (Ab) complex–mediated activation of the C1 complex. The lectin pathway is activated when complexes of mannose-binding lectin (MBL) and MBL-associated serine proteases (MASPs) bind to microbial surfaces. The alternative pathway can be triggered by a ‘tick-over’ mechanism involving spontaneous hydrolysis of C3, if a regulatory mechanism is absent (as is typically the case with microbial surfaces). The so-called ‘alternative pathway (AP)-amplification loop’ (in which more C3 is cleaved into more C3b which further fuels the loop) amplifies complement activation independently of the initiating mechanism, thereby contributing most of the downstream terminal pathway-mediated effector responses (i.e., C5a, membrane attack complex [MAC]). All three mechanisms of complement activation and amplification converge at C3, which is thus an attractive target of pharmacological interception, e.g., by the cyclic inhibitory peptide AMY-101. If C3 is not blocked, downstream effects include the generation of effectors (C3a and C5a) that promote inflammation and the generation the C5b-C9 MAC with potential antimicrobial but also tissue destructive capacity. Whereas the role of MAC in periodontitis is uncertain, C3a and C5a activate their cognate G-protein-coupled receptors (C3aR and C5aR1), which cross-talk with Toll-like receptors. This cross-talk synergistically activates inflammatory leukocytes, which can directly or indirectly mediate destructive periodontal tissue inflammation and bone loss in periodontitis [46, 56]. Complement-mediated inflammation also promotes the dysbiosis of the periodontal microbial community [32, 33]. C3 blockade can prevent these downstream effects and thereby offer broader protection against periodontitis [33, 37]. TLR, toll-like receptor.