

Abstract
We examined the way body-weight patterns through the first 4 decades of life relate to gene expression signatures of common forms of morbidity, including cardiovascular disease (CVD), type 2 diabetes (T2D), and inflammation. As part of wave V of the nationally representative National Longitudinal Study of Adolescent to Adult Health (1997–2018) in the United States, mRNA abundance data were collected from peripheral blood (n = 1,132). We used a Bayesian modeling strategy to examine the relative associations between body size at 5 life stages—birth, adolescence, early adulthood, young adulthood, and adulthood—and gene expression–based disease signatures. We compared life-course models that consider critical or sensitive periods, as well as accumulation over the entire period. Our results are consistent with a sensitive-period model when examining CVD and T2D gene expression signatures: Birth weight has a prominent role for the CVD and T2D signatures (explaining 33.1% and 22.1%, respectively, of the total association accounted for by body size), while the most recent adult obesity status (ages 33–39) is important for both of these gene expression signatures (24.3% and 35.1%, respectively). Body size in all life stages was associated with inflammation, consistent with the accumulation model.
Keywords: birth weight, cardiovascular disease, gene expression, inflammation, life course, obesity, type 2 diabetes
Abbreviations
- Add Health
National Longitudinal Study of Adolescent to Adult Health
- BMI
adult body mass index
- CVD
cardiovascular disease
- T2D
type 2 diabetes
Birth weight is a risk factor for cardiovascular disease (CVD), type 2 diabetes (T2D) (1), and generalized inflammation in later life (2). These associations likely reflect diverse mechanisms, including maternal and offspring genotype, maternal health, organ remodeling, epigenetic modifications, and cellular senescence (3–5). Although there is considerable interest in the long-term consequences of birth weight, adult body mass index (BMI) is also a well-established risk factor for such outcomes. Moreover, mounting evidence suggests that obesity and overweight status across periods of the early life course predict CVD, T2D, and generalized inflammation (6–8).
The role of body size at different points in the life course in the development of chronic diseases is not well understood (9–12). An increasing number of studies examine childhood and adolescent histories of body weight and their long-term implications for disease (7, 8). Some research suggests that a long duration of obesity is a risk factor for diabetes, especially if it occurs in young adulthood (13, 14), while other studies emphasize the role of weight gain for diabetes (15). Researchers have also devoted considerable attention to the long-term implications of birth weight for inflammation, CVD, and T2D (16–18). However, the relative contributions of all of these life-course aspects of body mass for common forms of morbidity, such as CVD and T2D, have not yet been studied.
The present paper examines body size at 5 life stages—birth, adolescence, early adulthood, young adulthood, and adulthood—to predict mRNA-based signatures of CVD, T2D, and inflammation. We used 3 different classes of life-course models to examine the association between early life-course patterns of body size and signatures: accumulation, and critical and sensitive periods (19). The accumulation model posits that body size in each life-course phase contributes equally to health outcomes later in life. The critical- and sensitive-period models posit increased importance of body size at specific points in life. The former posits only 1 significant age period in the life course, while the latter hypothesizes that some age periods have heightened salience.
The disease signatures—constructed via empirically derived a priori gene sets—reveal the mRNA abundance levels for genes that are significantly related to these common forms of morbidity. As such, the mRNA signatures provide an opportunity to observe the molecular underpinnings of disease in the decades before the diseases become prevalent in the population (and thus during a period when interventions might be feasible). The study of mRNA signatures aims to better understand the interaction between exposures and systemic, precursor biology associated with these later disease states (20, 21). According to the social signal transduction model, increases in body mass (and other exposures) alter gene expression which, in turn, alters the probability of diagnosed disease (22). Fallin et al. (23) call for more such work, which integrates different types of genetic measurements in order to reshape future goals of clinical and analytical epidemiology. Similarly, Jones et al. (24) call for a greater integration of life-course approaches into the understanding of biological mechanisms.
The relevance of the current study is highlighted by the pronounced secular trend of increasing BMI among the young in the United States (25) coupled with essentially steady rates of low birth weight (i.e., less than 5.5 pounds (2.5 kg)) (26). However, clinical manifestations of T2D and CVD often take many decades to appear. The average age of onset for T2D in the United States is approximately 45, and the risk of stroke, transient ischemic attacks, and diagnosed heart attacks is also notably increased in the mid-40s (27). Indeed, studies of these conditions are typically based on samples of older adults.
Early adulthood in the United States, extending into the fourth decade of life, is characterized as a period of life that is generally healthy but vulnerable due to high rates of obesity. Measuring health in such a population poses significant challenges but can also reveal important insights into the human biology underlying different health outcomes that emerge later in life. Further, the emphasis on the life-course aspect of obesity in early adulthood fills an important gap in biomedical research (28). Many studies analyze the association between early-life conditions and chronic diseases later in life, but they give limited attention to the time periods in between, especially young adulthood (29, 30).
METHODS
Study population
The data used in this study are from the National Longitudinal Study of Adolescent to Adult Health (Add Health), a representative study of US adolescents in grades 7–12 in 1994–1995 who were followed into adulthood over 5 waves of data collection (31). Data from all waves are used: waves I and II (age range 12–18 years; mean age 15.3), wave III (age range 19–24; mean age 21.7), wave IV (age range 25–31; mean age 28.1), and wave V, sample 1 (age range 33–39; mean age 36.4), which was collected in 2016–2017.
The wave V interviews for Add Health were conducted in 2016–2018 and included the collection of mRNA abundance data from peripheral blood samples. We used data from 1,132 people, the first mRNA data from wave V to have been released. The final sample size was 788 individuals after missing data on covariates were removed using listwise deletion (see Web Table 1 for comparison with the full sample and Web Figure 1 for flow diagram, available at https://doi.org/10.1093/aje/kwab049).
Gene expression signature scores
Gene expression data were normalized by applying a common reference gene technique using 11 housekeeping genes (32, 33). The gene candidates used to construct the gene expression signatures were derived from other studies, including genome-wide association studies, which identified replicated statistically significant genes: CVD genes from Nikpay et al. (34), T2D genes from Xue et al. (35), and inflammatory genes from Fredrickson et al. (36) and Levine et al. (37). The gene sets included 137 genes related to T2D, 71 genes related to CVD, and 19 inflammatory genes. After standard procedures were conducted to correct for genes with zero counts or insufficient variation, the sets included 30 genes related to CVD, 67 genes related to T2D, and 19 related to inflammation (see Web Appendix 1 for details and Web Tables 2 and 3 for gene lists). We created a gene expression score by averaging the mRNA abundance data for each outcome. As a robustness check we considered the direction of the association of each gene by dividing the gene set into up-regulated and down-regulated using internal information (see Web Tables 4 and 5 for details).
Moreover, the validity of these gene signatures was tested both internally and externally; the results show consistent patterns for various diseases proxies (see Web Figure 2 for external and Web Figure 3 for internal validation). We exploited gene expression data from Grayson et al. (38), which addresses both T2D and CVD clinical diagnosis, to ask whether there was an association between the clinical outcome per se and our disease sets. This (omnibus) test was implemented by simply inspecting whether at least 1 corrected P value in our disease set was significant within a standard mass univariate linear model framework supported by limma (see Web Tables 6 and 7 for full results).
Moreover, we implemented a rotation gene test as suggested by Wu et al. (39), showing how the genes are contributing to the significance of the signatures. This represents an extensive effort to validate the disease signatures with clinical outcomes, although we were constrained in performing an exact validation because of the absence of the same covariates in external data sets.
Measurements of body size over the life course
Body size over the life course was measured using 5 indicators covering different life stages. First, body size at birth was determined using birth weight information provided in parental reports from wave I, which was supplemented with self-reported information from wave V. Two indicators were constructed: low birth weight (microsomia: <5.5 pounds (2.5 kg)) and high birth weight (macrosomia: >8.8 pounds (4 kg)). Only low birth weight was used to model inflammation (2), whereas a combined measure of low and/or high birth weight was used for CVD and T2D signatures because metabolic diseases are sensitive to both micro- and macrosomia (3, 4). Second, body size over the other life-course periods was modeled by creating indicator variables for obesity status (BMI > 30). During waves II, III, IV, and V, field examiners collected height and weight measurements for each respondent. Self-reported height and weight were available for waves I and V (measured height and weight were also collected during wave V). Four age categories (in years: 12–18, 19–24, 25–32, and 33–39) were created to characterize the different life-course periods. Due to modeling requirements, only individuals with complete information across waves were included in the analysis. The reliability of the anthropometric measures collected for Add Health is high (40). Table 1 shows the increasing obesity trend over the early life course for this cohort, which reaches 40% for adults aged 33–39. Comparisons reveal that the mRNA subsample is similar to the wave V sample 1 (excluding the mRNA subsample) in terms of obesity at ages 33–39, as indicated by a t test.
Table 1.
Body Mass Index Categories Among Men and Women Across Age Groups, National Longitudinal Study of Adolescent to Adult Health, United States, 1997–2018
| Ages 12–18 Years | Ages 19–24 Years | Ages 25–32 Years | Ages 33–39 Years | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BMI Category a | Male | Female | Male | Female | Male | Female | Male | Female | ||||||||
| No. | Proportion | No. | Proportion | No. | Proportion | No. | Proportion | No. | Proportion. | No. | Proportion | No. | Proportion | No. | Proportion | |
| Underweight (<18.5) | 86 | 0.17 | 29 | 0.09 | 27 | 0.05 | 7 | 0.02 | 9 | 0.01 | 2 | 0.00 | 7 | 0.01 | 2 | 0.00 |
| Normal weight (18.5–24.9) | 317 | 0.66 | 206 | 0.66 | 231 | 0.48 | 123 | 0.39 | 176 | 0.36 | 91 | 0.29 | 150 | 0.31 | 62 | 0.20 |
| Overweight (25.0–29.9) | 46 | 0.09 | 50 | 0.16 | 112 | 0.23 | 107 | 0.34 | 118 | 0.24 | 103 | 0.33 | 116 | 0.24 | 122 | 0.39 |
| Obese (30.0–39.9) | 29 | 0.06 | 24 | 0.07 | 90 | 0.18 | 64 | 0.20 | 130 | 0.27 | 93 | 0.30 | 156 | 0.32 | 99 | 0.32 |
| Morbid obesity (≥40.0) | 1 | 0.00 | 0 | 0.00 | 19 | 0.03 | 8 | 0.02 | 46 | 0.09 | 20 | 0.06 | 50 | 0.10 | 24 | 0.07 |
Abbreviation: BMI, body mass index.
a Weight (kg)/height (m)2.
Other measurements
Based on a literature review we constructed a causal diagram to assess which variables might confound the relationship between mRNA signature and obesity over the life course (see Web Figure 4). As controls, we included birth year, age at waves I or II, biological sex, region of residence, and self-reported race/ethnicity, current smoking status, binge drinking in the past 12 months (>4 (for female participants) and >5 (for male participants) drinks in a row), educational attainment at ages 33–39, preterm birth status, an indicator for pubertal development, and maternal education. An extensive set of controls referring to the circumstances of the blood draw was included. Two mRNA technical controls were also included: sample-specific quality control measures for mRNA and indicators for assay batch. Finally, because cell type heterogeneity could be a potential confounder for our analysis (41), the cell composition was estimated with CIBERSORT32 (42), and cell type composition was included as a covariate in the model. Additional analyses were conducted to explore the distribution of different cell types using a compositional approach (43).
Statistical analysis
We modeled associations between body size in 5 life stages  (from birth to adulthood
(from birth to adulthood 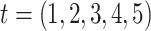 ) and gene expression signatures
) and gene expression signatures 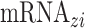 with
with 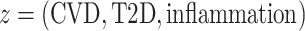 . The goal was to estimate the relative contributions of body size in different life-course periods to gene expression signatures in adulthood. The model was proposed by Madathil et al. (44) and it was used to estimate
. The goal was to estimate the relative contributions of body size in different life-course periods to gene expression signatures in adulthood. The model was proposed by Madathil et al. (44) and it was used to estimate 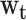 , a compositional vector that sums to 1 and represents the relative importance of various life-course periods. This kernel permits us to extend the general linear framework in order to isolate relative (
, a compositional vector that sums to 1 and represents the relative importance of various life-course periods. This kernel permits us to extend the general linear framework in order to isolate relative (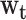 ) and absolute (
) and absolute (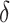 ) effects:
) effects:
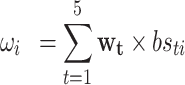 |
(1) |
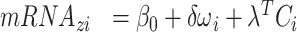 |
(2) |
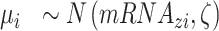 |
(3) |
where 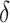 is the lifetime effect for body size and
is the lifetime effect for body size and 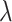 is the column vector of coefficients for the
is the column vector of coefficients for the 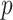 covariates
covariates 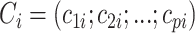 . Our priors are
. Our priors are 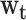 ~ Dirichlet (1);
~ Dirichlet (1); 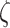 ~ logNormal (1);
~ logNormal (1); 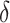 ~ Normal
~ Normal  ; and
; and 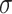 ~ logNormal (1). The parameters
~ logNormal (1). The parameters 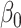 ,
, 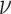 , and
, and 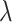 have uninformative priors following a Cauchy distribution. The model assumes the same direction of associations in all periods and no confounding by variables that change over time. This modeling allows for correlations between body sizes over time. Simulations show that the statistical model can detect the correct life-course hypothesis even with substantial correlations across time in body size (results available on request). Moreover, the reported association between mRNA gene expression signatures and body size at a certain time period t was adjusted for body size at each of the other time periods.
have uninformative priors following a Cauchy distribution. The model assumes the same direction of associations in all periods and no confounding by variables that change over time. This modeling allows for correlations between body sizes over time. Simulations show that the statistical model can detect the correct life-course hypothesis even with substantial correlations across time in body size (results available on request). Moreover, the reported association between mRNA gene expression signatures and body size at a certain time period t was adjusted for body size at each of the other time periods.
Within this framework, we compared the relative weights 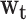 , which were predicted by the critical-period, sensitive-period, and accumulation models, with a posteriori weights derived from the data. The accumulation model posits that the weights are the same in each life-course period (i.e., 0.2 for each of 5 measurement occasions), and the critical-period model holds that only 1 of the life-course periods is important (only one of the weights is 1 and the others are zero). The sensitive-period models propose that all periods matter to some extent, and therefore the weight in each life-course period is on a continuum between 0 and 1, but together they must sum to 1.
, which were predicted by the critical-period, sensitive-period, and accumulation models, with a posteriori weights derived from the data. The accumulation model posits that the weights are the same in each life-course period (i.e., 0.2 for each of 5 measurement occasions), and the critical-period model holds that only 1 of the life-course periods is important (only one of the weights is 1 and the others are zero). The sensitive-period models propose that all periods matter to some extent, and therefore the weight in each life-course period is on a continuum between 0 and 1, but together they must sum to 1.
We tested 3 sets of weights referring to different sensitive-period hypotheses. In the first, birth is the most prominent sensitive period (0.5, 0.125, 0.125, 0.125, 0.125). In the second, birth and adulthood matter most (0.35, 0.1, 0.1, 0.1, 0.35), and in the third, current status is the most important (0.125, 0.125, 0.125, 0.125, 0.50). Comparisons were then made based on the distribution of the metric Aitchison’s distance between the posterior weight distributions and the theoretically derived weights. Bayesian inference was implemented in Stan (45). This framework allowed us to formally examine when, in the life course, body size matters for mRNA signatures associated with common forms of adult morbidity.
Additional tests were conducted to understand whether there was an overrepresentation of certain cell types for individuals with obesity at ages 33–39 using a compositional approach (42). Several robustness checks were also carried out. First, individuals were categorized based on whether they were overweight or obese (BMI > 25). Second, we modeled BMI rather than relying on obesity dichotomies. Third, we performed sensitivity analyses by including only low or high birthweight (instead of both low and high birthweight); the results are qualitatively similar, showing that the different life-course periods have similar degrees of importance whichever measurement strategy is used. Fourth, the models were also estimated stratifying the sample by sex. Fifth, we divided the up-regulated and down-regulated genes in the CVD and T2D signatures using the information concerning disease proxies in Add Health, and we created 2 new scores by averaging the up-regulated genes and the down-regulated ones. Finally, we also replicated the analysis by removing individuals who reported having been diagnosed with T2D (61 out of 788) and those who had had a heart attack or had undergone heart surgery for clogged coronary arteries (8 out of 788).
RESULTS
Table 2 presents means and 95% credible intervals of posterior distributions for the parameters resulting from the Bayesian estimation. The main parameters of interest are the lifetime effect of body size (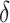 ) and the set of 5 weights for the relative importance of specific life-course periods estimated for different models (
) and the set of 5 weights for the relative importance of specific life-course periods estimated for different models ( ). The lifetime effect reveals that body size across the early life course is related to the 3 gene expression signatures associated with CVD, T2D, and inflammation.
). The lifetime effect reveals that body size across the early life course is related to the 3 gene expression signatures associated with CVD, T2D, and inflammation.
Table 2.
Means and 95% Credible Intervals of Posterior Distributions of Weights and Lifetime Effects by Outcome (n = 788), National Longitudinal Study of Adolescent to Adult Health, United States, 1997–2018a
| Coefficient | CVD Expression | T2D Expression | Inflammation Expression | |||
|---|---|---|---|---|---|---|
| Mean | 95% CrI | Mean | 95% CrI | Mean | 95% CrI | |
| Lifetime effect, body weight (δ) | 0.088 | 0.02, 0.15 | 0.090 | 0.04, 0.14 | 0.115 | 0.04, 0.19 |
| Weights (wt) | ||||||
| Birth (low birth weight/high birth weight)b | 0.331 | 0.044, 0.639 | 0.221 | 0.014, 0.478 | 0.243 | 0.014, 0.540 |
| Obesity, ages 12–18 years | 0.214 | 0.007, 0.539 | 0.133 | 0.003, 0.390 | 0.217 | 0.011, 0.541 |
| Obesity, ages 19–24 years | 0.110 | 0.003, 0.366 | 0.098 | 0.002, 0.321 | 0.188 | 0.009, 0.508 |
| Obesity, ages 25–32 years | 0.102 | 0.002, 0.360 | 0.197 | 0.009, 0.522 | 0.196 | 0.009, 0.518 |
| Obesity, ages 33–39 years | 0.243 | 0.016, 0.572 | 0.351 | 0.049, 0.680 | 0.156 | 0.005, 0.452 |
| No. of observations | 788 | 788 | 788 | |||
Abbreviations: CrI, credible interval; CVD, cardiovascular disease; T2D, type 2 diabetes.
a Sample-specific quality control measures for mRNA and indicators for assay batch are also included.
b Only low birth weight in the case of inflammation.
The relative importance of the different weights (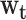 ) shows 3 interesting patterns. First, for genes expression related to CVD, birth weight played a prominent role, accounting for 33.1% of the total association between CVD and lifetime body weight, followed by obesity in the most recent adulthood period (ages 33–39), which accounted for 24.3%. Second, for the expression of T2D-related genes, obesity in the most recent adulthood period (ages 33–39) had the greatest relative importance at 35.1%, followed by 22.1% for macro- and microsomia at birth. Third, for the inflammation signature, low birth weight had a more prominent role (24.3%) than obesity at other points in the life course (especially at ages 25–31 and 33–39), with diminishing relative contributions with age.
) shows 3 interesting patterns. First, for genes expression related to CVD, birth weight played a prominent role, accounting for 33.1% of the total association between CVD and lifetime body weight, followed by obesity in the most recent adulthood period (ages 33–39), which accounted for 24.3%. Second, for the expression of T2D-related genes, obesity in the most recent adulthood period (ages 33–39) had the greatest relative importance at 35.1%, followed by 22.1% for macro- and microsomia at birth. Third, for the inflammation signature, low birth weight had a more prominent role (24.3%) than obesity at other points in the life course (especially at ages 25–31 and 33–39), with diminishing relative contributions with age.
Given the estimated weights showing the relative importance of different periods, we can evaluate which life-course model best describes the association between early life-course patterns of body weight and the disease signatures. We made this evaluation by examining the posterior distribution of the Aitchison’s distances between the estimated weight and each of the hypothesized ones. Table 3 shows the mean distances such that the life-course hypothesis associated with the shortest distance (indicating best fit) is the one that best describes the data. For CVD- and T2D-related genes, the sensitive-period model positing that both birth weight (either low or high birth weight) and adulthood obesity (ages 33–39) had the highest relative importance was best supported by the data (for CVD, Aitchison’s distance 2.17, 95% credible interval: 0.7, 4.6; for T2D, Aitchison’s distance 2.1, 95% credible interval: 0.7, 4.5), although other time points mattered as well. Finally, in the case of inflammation, the accumulation hypothesis was the best performing model (Aitchison’s distance 2.0, 95% credible interval: 0.6, 4.4).
Table 3.
Measures of Posterior Fit Comparing Observed and Theoretical Weights, National Longitudinal Study of Adolescent to Adult Health, United States, 1997–2018a
| Life-Course Model | CVD | T2D | Inflammation | |||
|---|---|---|---|---|---|---|
| Mean | 95% CrI | Mean | 95% CrI | Mean | 95% CrI | |
| Accumulation, all ages | 2.40 | 0.8, 5.0 | 2.29 | 0.7, 4.8 | 2.02 | 0.6, 4.4 |
| Critical period | ||||||
| 1, birth only | 3.96 | 2.6, 6.0 | 4.46 | 3.1, 6.7 | 4.32 | 2.9, 6.7 |
| 2, ages 12–18 years only | 4.61 | 3.0, 7.2 | 5.10 | 3.4, 7.8 | 4.48 | 2.9, 6.9 |
| 3, ages 19–24 years only | 5.33 | 3.5, 8.0 | 5.40 | 3.7, 8.0 | 4.65 | 3.0, 7.0 |
| 4, ages 25–32 years only | 5.41 | 3.6, 8.0 | 4.65 | 3.0, 7.1 | 4.59 | 3.0, 7.0 |
| 5, ages 33–39 years only | 4.40 | 2.9, 6.6 | 3.86 | 2.5, 5.8 | 4.87 | 3.2, 7.5 |
| Sensitive period | ||||||
| 1, mainly birth | 2.26 | 0.7, 4.8 | 2.47 | 0.9, 4.9 | 2.21 | 0.7, 4.6 |
| 2, birth + ages 33–39 years | 2.17 | 0.7, 4.6 | 2.10 | 0.6, 4.5 | 2.45 | 0.9, 4.9 |
| 3, mainly ages 33–39 years | 2.51 | 0.9, 5.0 | 2.14 | 0.7, 4.6 | 2.54 | 1.0, 5.0 |
Abbreviations: CrI, credible interval; CVD, cardiovascular disease; T2D, type 2 diabetes.
a Means and 95% credible intervals of posterior distributions of Aitchison’s distances.
The additional tests conducted to understand whether there was an overrepresentation of certain cell types for individuals with obesity at ages 33–39 showed that people who are obese in adulthood (ages 33–39) had a larger proportion of naive B cells and plasma cells than the nonobese at this age, controlling for previous obesity (Web Figure 5 and Web Table 8). Previous research has shown the pivotal role of B cells for diabetes development and cardiovascular health (46–48). The results from the overweight or obese classification are relatively similar to those reported in Table 2 (Web Table 9). The sex-specific results showed patterns consistent with those reported in Table 2, although with less precision given the smaller sex-specific sample sizes. Similarly, results remained mostly unchanged by removing individuals who reported having been diagnosed with T2D and those who had had a heart attack or had undergone heart surgery for clogged coronary arteries (Web Table 10). Additional robustness checks were carried out to understand the sensitivity of the results to different specifications of birth-weight disadvantage (Web Tables 11–14). Despite observing similar coefficients across different specifications in the weight vector, estimates for the weights are compositional and thus sum to 1. Therefore, different specifications of body size as well as of age groups (Web Table 15) and priors (Web Table 16) can lead to different distributions of the weight vector estimates, although they follow similar qualitative patterns.
DISCUSSION
The prevalence of obesity and overweight status among children and young adults has increased considerably over the past several decades (49). Obesity and overweight status have profound consequences for life expectancy and morbidity rates (50–52). Among the diseases related to elevated BMI, previous research has highlighted the increased risk for T2D (53), CVD (12, 14, 54, 55), cancer (56), deficits in physical functioning (57), and systemic inflammation (58). Moreover, the long-term consequences of birth weight on cardiovascular and metabolic health outcomes have also attracted considerable interest (2, 59–61). However, clinically significant indications of these diseases often do not begin to manifest until the late 40s and 50s. The present research examines whether birth weight and obesity through the early life course (from birth through young adulthood) are associated with the expression of genes (as indicated by mRNA abundance) related to CVD, T2D, and inflammation already in the 30s.
Several life-course hypotheses were examined (19, 62–66): an accumulation hypothesis, a critical-period hypothesis, and several sensitive-period hypotheses. This work finds support for a sensitive-period model in the case of the CVD and T2D gene expression signatures. The most relevant life-course periods for gene expression signatures related to CVD were found to be birth and adulthood (ages 33–39); obesity status in adulthood alone is not decisive. While CVD and T2D gene expression are associated with greater relative importance of low/high birth weight and adulthood obesity, low birth weight and obesity status at all ages contribute to the expression of inflammation-related genes, supporting an accumulation model. These results contribute to the literature by suggesting the importance of birth weight (both micro- and macrosomia) for CVD and T2D (56) and the prominent role of body size over the entire early adulthood life course, especially for inflammation (67).
This work is not free from limitations. First, the design does not allow us to make causal statements about the role of different life-course periods. Second, the gene expression scores are markers of pre-disease, but they are probabilistic markers of disease and the degree of their specificity and sensitivity is presently unknown. Moreover, the gene sets are derived from previous studies, which might not necessarily have identified causal variants. Third, the design does not allow us to disentangle associations due to recency and ages 33–39, which are confounded. Finally, recent studies suggest that visceral adipose tissue and proportionality at birth, which unfortunately were not measured in Add Health, play important roles in the development of CVD and T2D (5, 68). Nevertheless, the present study is the first, to our knowledge, to examine associations between patterns of body weight across the early life course and mRNA risk signatures for major forms of morbidity in a population-representative study.
Public health research faces the increasing challenge of integrating proliferating genetic data with data from more traditional channels (21). Understanding when body size matters most in pre-disease pathways can inform the design of effective policy interventions to reduce the impact of obesity on public health outcomes. This work contributes to emerging efforts in the literature on integrating life-course longitudinal studies and gene expression data (69) using a priori selected gene sets (37, 70). Our study suggests that efforts to reduce the burden of disease should start from very early in life with the promotion of healthy birth weights. Simultaneously, public health measures aimed at reducing obesity in adolescence and adulthood could represent important actions to prevent the development of T2D and CVD and reduce inflammatory burden.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Jacobs Center for Productive Youth Development, University of Zürich, Zürich, Switzerland (Cecilia Potente, Justin Chumbley, Wenjia Xu, Michael J. Shanahan); Carolina Population Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, United States (Kathleen Mullan Harris, Brandt Levitt); Department of Sociology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, United States (Kathleen Mullan Harris); David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California, United States (Steven W. Cole); Norman Cousins Center for Psychoneuroimmunology, Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, Los Angeles, California, United States (Steven W. Cole); Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, California, United States (Steven W. Cole); Center for Medicine, Health, and Society, Vanderbilt University, Nashville, Tennessee, United States (Lauren Gaydosh); and Department of Sociology, University of Zürich, Zürich, Switzerland (Michael J. Shanahan).
M.J.S. and K.M.H. acknowledge support by the National Institutes of Health (grants R01-HD087061, P30-AG017265, R01-AG043404, and R01-AG033590); C.P., M.J.S., J.C., and W.X. acknowledge support by the Jacobs Center for Productive Youth Development, University of Zürich. This research uses data from Add Health, a program project directed by K.M.H. and designed by J. Richard Udry, Peter S. Bearman, and K.M.H. at the University of North Carolina at Chapel Hill, and funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (grant P01-HD31921), with cooperative funding from 23 other federal agencies and foundations.
A preliminary version of this work was presented at 2019 Annual Meeting of the Population Association of America, April 10–13, 2019, Austin, Texas. Data availability: Add Health data are available at https://www.cpc.unc.edu/projects/addhealth/documentation/. R code used in these analyses is available upon request from the corresponding authors.
Conflict of interest: none declared.
REFERENCES
- 1.Feng C, Osgood ND, Dyck RF. Low birth weight, cumulative obesity dose, and the risk of incident type 2 diabetes. J Diabetes Res. 2018;8435762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDade TW, Metzger MW, Chyu L, et al. Long-term effects of birth weight and breastfeeding duration on inflammation in early adulthood. Proc Roy Soc B. 2014;281(1784):20133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin-Gronert MS, Ozanne SE. Mechanisms underlying the developmental origins of disease. Rev Endocr Metab Disord. 2012;13(2):85–92. [DOI] [PubMed] [Google Scholar]
- 4.Knop MR, Geng TT, Gorny AW, et al. Birth weight and risk of type 2 diabetes mellitus, cardiovascular disease, and hypertension in adults: a meta-analysis of 7 646 267 participants from 135 studies. J Am Heart Assoc. 2018;7(23):e008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warrington NM, Beaumont RN, Horikoshi M, et al. Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet. 2019;51(5):804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlsson T, Rask-Andersen M, Pan G, et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med. 2019;25(9):1390–1395. [DOI] [PubMed] [Google Scholar]
- 7.Tirosh A, Shai I, Afek A, et al. Adolescent BMI trajectory and risk of diabetes versus coronary disease. N Engl J Med. 2011;364(14):1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohlsson C, Bygdell M, Sondén A, et al. Association between excessive BMI increase during puberty and risk of cardiovascular mortality in adult men: a population-based cohort study. Lancet Diabetes Endocrinol. 2016;4(12):1017–1024. [DOI] [PubMed] [Google Scholar]
- 9.McCarron P, Smith GD, Okasha M, et al. Blood pressure in young adulthood and mortality from cardiovascular disease. Lancet. 2000;355(9213):1430–1431. [DOI] [PubMed] [Google Scholar]
- 10.Rademacher ER, Jacobs DRJ, Moran A, et al. Relation of blood pressure and body mass index during childhood to cardiovascular risk factor levels in young adults. J Hypertens. 2009;27(9):1766–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakurai Y, Teruya K, Shimada N, et al. Association between duration of obesity and risk of non-insulin-dependent diabetes mellitus. The Sotetsu Study. Am J Epidemiol. 1999;149(3):256–260. [DOI] [PubMed] [Google Scholar]
- 12.Shihab HM, Meoni LA, Chu AY, et al. Body mass index and risk of incident hypertension over the life course: the Johns Hopkins Precursors Study. Circulation. 2012;126(25):2983–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JM, Gebremariam A, Vijan S, et al. Excess body mass index-years, a measure of degree and duration of excess weight, and risk for incident diabetes. Arch Pediatr Adolesc Med. 2012;166(1):42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reis JP, Loria CM, Lewis CE, et al. Association between duration of overall and abdominal obesity beginning in young adulthood and coronary artery calcification in middle age. JAMA. 2013;310(3):280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Y, Manson JE, Yuan C, et al. Associations of weight gain from early to middle adulthood with major health outcomes later in life. JAMA. 2017;318(3):255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDade TW, Ryan C, Jones MJ, et al. Social and physical environments early in development predict DNA methylation of inflammatory genes in young adulthood. Proc Natl Acad Sci U S A. 2017;114(29):7611–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawlor DA, Leon DA. Association of body mass index and obesity measured in early childhood with risk of coronary heart disease and stroke in middle age. Circulation. 2005;111(15):1891–1896. [DOI] [PubMed] [Google Scholar]
- 18.Lawlor DA, Martin RM, Gunnell D, et al. Association of body mass index measured in childhood, adolescence, and young adulthood with risk of ischemic heart disease and stroke: findings from 3 historical cohort studies. Am J Clin Nutr. 2006;83(4):767–773. [DOI] [PubMed] [Google Scholar]
- 19.Kuh D, Ben-Shlomo Y, Lynch J, et al. Life course epidemiology. J Epidemiol Community Health. 2003;57(10):778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole SW. Social regulation of human gene expression: mechanisms and implications for public health. Am J Public Health. 2013;103(suppl 1):S84–S92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole SW, Shanahan MJ, Gaydosh L, et al. Population-based RNA profiling in Add Health finds social disparities in inflammatory and antiviral gene regulation to emerge by young adulthood. Proc Natl Acad Sci U S A. 2020;117(9):4601–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11(9):625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fallin MD, Duggal P, Beaty TH. Genetic epidemiology and public health: the evolution from theory to technology. Am J Epidemiol. 2016;183(5):387–393. [DOI] [PubMed] [Google Scholar]
- 24.Jones NL, Gilman SE, Cheng TL, et al. Life course approaches to the causes of health disparities. Am J Public Health. 2019;109(suppl 1):S48–S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hales CM, Carroll MD, Fryar CD, et al. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief. 2017;1(288):1–8. [PubMed] [Google Scholar]
- 26.Brosco JP, Sanders LM, Guez G, et al. Historical trends in low birth weight. Arch Pediatr Adolesc Med. 2010;164(1):99–100. [DOI] [PubMed] [Google Scholar]
- 27.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics—2015 update. Circulation. 2015;131(4):e29–e322. [DOI] [PubMed] [Google Scholar]
- 28.Harris KM, Schorpp KM. Integrating biomarkers in social stratification and Health Research. Annu Rev Sociol. 2018;44(1):361–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crimmins EM, Finch CE. Infection, inflammation, height, and longevity. Proc Natl Acad Sci U S A. 2006;103(2):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith CJ, Ryckman KK. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab Syndr Obes. 2015;8:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris KM, Halpern CT, Whitsel EA, et al. Cohort profile: the National Longitudinal Study of Adolescent to Adult Health (Add Health). Int J Epidemiol. 2019;48(5):1415–1415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29(10):569–574. [DOI] [PubMed] [Google Scholar]
- 33.Evans C, Hardin J, Stoebel DM. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief Bioinform. 2018;19(5):776–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue A, Wu Y, Zhu Z, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9(1):2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fredrickson BL, Grewen KM, Coffey KA, et al. A functional genomic perspective on human well-being. Proc Natl Acad Sci U S A. 2013;110(33):13684–13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levine ME, Crimmins EM, Weir DR, et al. Contemporaneous social environment and the architecture of late-life gene expression profiles. Am J Epidemiol. 2017;186(5):503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grayson BL, Wang L, Aune TM. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun. 2011;12(5):341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D, Lim E, Vaillant F, et al. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26(17):2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hussey JM, Nguyen QC, Whitsel EA, et al. The reliability of in-home measures of height and weight in large cohort studies: evidence from Add Health. Demogr Res. 2015;32(1):1081–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belsky DW, Snyder-Mackler N. Invited commentary: integrating genomics and social epidemiology-analysis of late-life low socioeconomic status and the conserved transcriptional response to adversity. Am J Epidemiol. 2017;186(5):510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erb I, Notredame C. How should we measure proportionality on relative gene expression data? Theory Biosci. 2016;135(1–2):21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madathil S, Joseph L, Hardy R, et al. A Bayesian approach to investigate life course hypotheses involving continuous exposures. Int J Epidemiol. 2018;47(5):1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gelman A, Lee D, Guo J. Stan: a probabilistic programming language for Bayesian inference and optimization. J Educ Behav Stat. 2015;40(5):530–543. [Google Scholar]
- 46.Zhai X, Qian G, Wang Y, et al. Elevated B cell activation is associated with type 2 diabetes development in obese subjects. Cell Physiol Biochem. 2016;38(3):1257–1266. [DOI] [PubMed] [Google Scholar]
- 47.Sage AP, Tsiantoulas D, Binder CJ, et al. The role of B cells in atherosclerosis. Nat Rev Cardiol. 2019;16(3):180–196. [DOI] [PubMed] [Google Scholar]
- 48.Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. 2017;8:1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skelton JA, Cook SR, Auinger P, et al. Prevalence and trends of severe obesity among US children and adolescents. Acad Pediatr. 2009;9(5):322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peeters A, Barendregt JJ, Willekens F, et al. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Ann Intern Med. 2003;138(1):24–32. [DOI] [PubMed] [Google Scholar]
- 51.GBD 2015 Obesity Collaborators . Health effects of overweight and obesity in 195 countries over 25 years. New Engl J Med. 2017;377(1):13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhaskaran K, Dos-Santos-Silva I, Leon DA, et al. Association of BMI with overall and cause-specific mortality: a population-based cohort study of 36 million adults in the UK. Lancet Diabetes Endocrinol. 2018;6(12):944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boone-Heinonen J, Sacks RM, Takemoto EE, et al. Prenatal development and adolescent obesity: two distinct pathways to diabetes in adulthood. Child Obes. 2018;14(3):173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nordestgaard BG, Palmer TM, Benn M, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9(5):e1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Owen CG, Whincup PH, Orfei L, et al. Is body mass index before middle age related to coronary heart disease risk in later life? Evidence from observational studies. Int J Obes (Lond). 2009;33(8):866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Renehan AG, Tyson M, Egger M, et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371(9612):569–578. [DOI] [PubMed] [Google Scholar]
- 57.Rogers NT, Power C, Pinto Pereira SM. Birthweight, lifetime obesity and physical functioning in mid-adulthood: a nationwide birth cohort study. Int J Epidemiol. 2020;49(2):657–665. [DOI] [PubMed] [Google Scholar]
- 58.Ellulu MS, Patimah I, Khaza'ai H, et al. Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci. 2017;13(4):851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barker DJP. Fetal origins of cardiovascular disease. Ann Med. 1999;31(sup1):3–6. [DOI] [PubMed] [Google Scholar]
- 60.Gluckman PD, Hanson MA, Buklijas T, et al. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol. 2009;5(7):401–408. [DOI] [PubMed] [Google Scholar]
- 61.Goosby BJ, Cheadle JE, McDade T. Birth weight, early life course BMI, and body size change: chains of risk to adult inflammation? Soc Sci Med. 2016;148:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ben-Shlomo Y, Cooper R, Kuh D. The last two decades of life course epidemiology, and its relevance for research on ageing. Int J Epidemiol. 2016;45(4):973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mishra G, Nitsch D, Black S, et al. A structured approach to modelling the effects of binary exposure variables over the life course. Int J Epidemiol. 2009;38(2):528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuh D, Ben-Shlomo Y. A life course approach to obesity. In: Kuh D, Ben-Shlomo Y, Ezra S, eds. A Life Course Approach to Chronic Disease Epidemiology. Oxford, UK: Oxford University Press; 2004:189–217. [Google Scholar]
- 65.Singh-Manoux A, Ferrie JE, Chandola T, et al. Socioeconomic trajectories across the life course and health outcomes in midlife: evidence for the accumulation hypothesis? Int J Epidemiol. 2004;33(5):1072–1079. [DOI] [PubMed] [Google Scholar]
- 66.Dunn EC, Soare TW, Raffeld MR, et al. What life course theoretical models best explain the relationship between exposure to childhood adversity and psychopathology symptoms: recency, accumulation, or sensitive periods? Psychol Med. 2018;48(15):2562–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singer K, Lumeng CN. The initiation of metabolic inflammation in childhood obesity. J Clin Invest. 2017;127(1):65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forsén T, Eriksson JG, Tuomilehto J, et al. Growth in utero and during childhood among women who develop coronary heart disease: longitudinal study. BMJ. 1999;319(7222):1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belsky DW. Life-course longitudinal studies are needed to advance integration of genomics and social epidemiology. Am J Epidemiol. 2018;187(6):1337–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang JY, Gavin AR, Richardson TS, et al. Accounting for life-course exposures in epigenetic biomarker association studies: early life socioeconomic position, candidate gene DNA methylation, and adult cardiometabolic risk. Am J Epidemiol. 2016;184(7):520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.