
Table 2.
Mechanism of action and structure of Polycomb modulators
| Target subunit | Ligand | Mechanism of action | Structure | Reference | Associated Clinical Trials (NCT number) |
|---|---|---|---|---|---|
| PRC2 | |||||
| EZH2 | 3-Deazane-planocin A | Inhibits the histone methyltransferase activity of EZH2 while inducing degradation of the PRC2 core subunits EZH2, EED, and SUZ12. In immunocompromised mice, this compound reduced the time of formation of tumors originating from prostate cancer cells |

|
(240) |
|
| EZH2 | GSK926 | SAM-e competitive inhibitor discovered from a high-throughput screening of the GSK compound collection. Reduces H3K27me3 levels in a breast cancer cell line and inhibits cell proliferation in breast and prostate cancer cell-based models |

|
(241) |
|
| EZH2 | GSK126 | In a similar fashion as GSK926, GSK126 was discovered from a high-throughput screening of the GSK compound collection. Highly selective for EZH2 over other methyltransferases. Inhibits cell proliferation in B-cell lymphoma cell-based and murine models that contain an EZH2-activating mutation |

|
(159) | NCT02082977 |
| EZH2 | EPZ005678 | Selectively reduces H3K27 methylation by EZH2 in vitro and in lymphoma cell-based models. Treatment of lymphoma cells bearing a mutant EZH2, leads to antiproliferative effects, indicating that these cancers are critically dependent on mutant EZH2 |

|
(158) |
|
| EZH2 | EPZ-6438 (tazemetostat) | Discovered along with EPZ005678 but shows better potency and oral bioavailability in animals. Treatment of mice bearing a lymphoma xenograft with mutant EZH2 reduces cell growth in a concentration dependent manner. FDA-approved for follicular lymphoma and epithelioid sarcoma with SNF5 deletions |
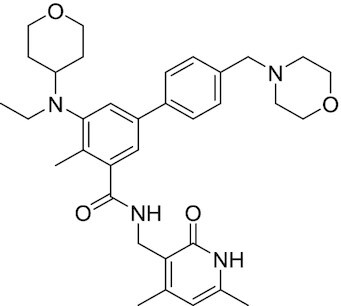
|
(243) |
NCT03456726 NCT02601950 NCT01897571 NCT03213665 NCT02860286 NCT02601937 NCT04557956 NCT04762160 NCT03010982 NCT03874455 NCT02875548 NCT04179864 NCT03854474 NCT03028103 NCT04917042 |
| EZH2 | EPZ011989 | Inhibits EZH2 in a mouse xenograft model of DLBCL, resulting in tumor growth inhibition while showing oral bioavailability |
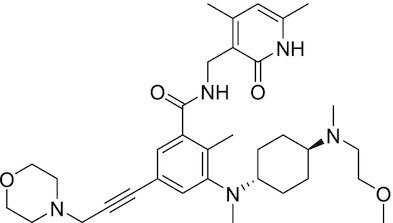
|
(247) |
|
| EZH2 | EI1 | Inhibits the methyl-transferase activity of EZH2/PRC2 leading to reduction of H3K27 methylation over other H3 methylation marks. EI1 shows antiproliferative effects and down-regulates the proliferation gene signature in DLBCL |
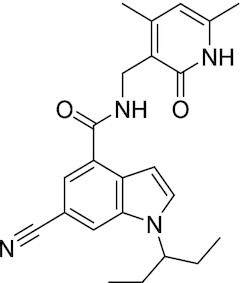
|
(248) |
|
| EZH2 | UNC1999 | Inhibits the methyl-transferase activity of EZH2 and EZH1 by acting as competitor of SAM-e. UNC1999 is orally bioavailable and shows no adverse effects in Swiss albino mice |

|
(249) |
|
| EZH2 | CPI-360 | Competes with SAM-e for the EZH2 SET domain, leading to reduction of H3K27 trimethylation levels without affecting the protein levels of EZH2, SUZ12, and EED. CPI-360 has antiproliferative effects in different lymphoma cell-based models as well as in a human B-cell non-Hodgkin lymphoma murine model |

|
(251) |
|
| EZH2 | CPI-1205 | Binds to the EZH2 catalytic domain. CPI-1205 proved to be efficacious, well-tolerated and highly bioavailable in a lymphoma xenograft model. Currently under clinical trials |

|
(291) |
NCT02395601 NCT03480646 NCT03525795 |
| EZH2 | PF06821497 | EZH2 catalytic inhibitor effective in mouse xenograft model of DLBCL |

|
(253) | NCT03460977 |
| EZH1/2 | Valemetostat (DS-3201b) | EZH1/EZH2 dual inhibitor with activity in DLBCL, as well as AML, TAL and urogenital cancers |

|
(255) |
NCT04703192 NCT04842877 NCT04102150 NCT04388852 |
| EZH2 | MS1943 | First-in-class EZH2 degrader, selective for EZH2 over other methyltransferases. Induces EZH2 degradation and cytotoxicity in triple-negative breast cancer cell-based models |

|
(199) |
|
| EZH2 | Ebastine | Initially discovered as an antihistamine drug, repurposed as an EZH2 inhibitor by decreasing EZH2 expression and reducing the levels of H3K27me3 in breast cancer and prostate cancer cells. Also active in a triple-negative breast cancer murine model |

|
(229) |
|
| EZH2-EED | Astemizole | Disrupts EZH2-EED protein-protein interaction, which results in inhibition of the methyltransferase activity of PRC2. Astemizole inhibits proliferation of DLBCL cells |

|
(258) |
|
| EZH2-EED | SAH-EZH2 | Peptidomimetic of stabilized alpha-helix of EZH2 which disrupt the EZH2-EED interaction leading to reduced H3K27me3 and EZH2 protein levels. SAH-EZH2 is capable of inducing growth arrest in leukemia cells and shows antiproliferative effects in B-cell lymphoma cell lines. | FSSNRQKILERTEILNQEWKQRRIQPV | (259) |
|
| EZH2-EED | DC-PRC2in-01 | Inhibits EZH2-EED interaction leading to reduced H3K27me3, as well as degradation of PRC2 core subunits. DC-PRC2in-01 inhibits PRC2-driven lymphomas cell growth and demonstrates cell cycle arrest at G0/G1 phase |

|
(260) |
|
| EED | A-395 | Inhibits EED H3K27me3 recognition by binding to the H3K27me3 binding pocket. Inhibits growth in DLBCL cell lines that have acquired resistance to EZH2 inhibitors. Also active in a xenograft murine model |

|
(292) |
|
| EED | EED226 | Binds to the EED binding pocket that recognizes H3K27me3. Reduces H3K27 methylation in a human B-cell non-Hodgkin lymphoma cell line and inhibits tumor proliferation in a human B-cell non-Hodgkin lymphoma murine model |

|
(263) |
|
| EED | MAK-683 | Currently under Phase I/II study to be evaluated as an anti-tumor agent in DLBCL, nasopharyngeal carcinoma (NPC) or other advanced solid tumors for whom no further effective standard treatment is available | Unknown | (265) | NCT02900651 |
| EED | UNC5115 | Discovered with UNC5114. Binds to the H3K27me3 binding pocket and inhibits the catalytic activity of PRC2 |

|
(266) |
|
| EED | Compound 19 | Acts as a competitor for the H3K27me3 binding pocket in EED, leading to reduction in the methyltransferase activity of PRC2. Inhibits growth in a DLBCL cell line. |

|
(267) |
|
| EED | UNC6852 | Bivalent chemical degrader that binds to EED and leads to degradation of PRC2. Derived from EED226 and a VHL ligand. Decreases H3K27me3 levels in DLBCL cell lines |

|
(268) |
|
| EED | PROTAC 2 | Degrades EED along with EZH2 and SUZ12. PROTAC 2 is a more potent degrader than its analogue PROTAC 1. Both molecules inhibit growth in a DLBCL cell line as well as a rhabdoid cancer cell line |

|
(269) |
|
| EED | UNC5636 | Peptidomimetic compound that selectively activates EED bearing a I363M mutation. This promotes PRC2 catalytic activity shown by the incorporation of a methyl group to lysine 27 of H3 peptide |

|
(270) |
|
| PRC1 | |||||
| RING | PRT4165 | Inhibits H2A ubiquitination of topoisomerase Top2α at double-strand break sites in cells |

|
(275) |
|
| RING | RB-3 | Binds RING1B and alters protein conformation, preventing association with histones and subsequent H2A119Ub |

|
(276) |
|
| PCGF4 (BMI-1) | PTC-209 | Inhibits colorectal cancer-initiating cells by reducing the protein levels of PCGF4 (BMI-1) |

|
(238) |
|
| PCGF4 (BMI-1) | PTC-596 | Reduces the levels of functional BMI-1 by inducing its hyper-phosphorylation. Currently under Phase 1 clinical trials |

|
(277) |
NCT03206645 NCT03761095 NCT03605550 NCT02404480 |
| PCGF4 (BMI-1) | QW24 | Induces BMI-1 protein degradation through the autophagy-lysosome pathway, leading to inhibition of colorectal CICs’ self-renewal |
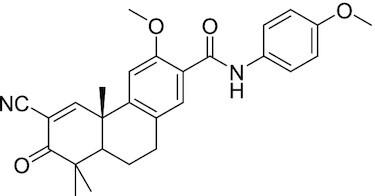
|
(278) |
|
| CBX4/7 | UNC3866 | Binds to the ChD of the CBX paralogs, preventing them from binding methyllysine. UNC3866 inhibits proliferation of PC3 prostate cancer cells |

|
(282) |
|
| CBX7 | MS37452 | Displaces CBX7 from the INK4A/ARF locus in prostate cancer cells, hence de-repressing the transcription of p16/CDKN2A |

|
(283) |
|
| CBX7 | MS351 | Discovered via structure-guided drug design, MS351 inhibits CBX7 binding to H3K27me3 when it is bound to RNA. It also derepresses CBX7 target genes in both mouse embryonic stem cells and PC3 prostate cancer cells |
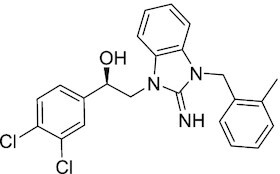
|
(284) |
|
| CBX7 | Compound 33F | Developed using rational design to modify a L3MBTL1 methyllysine binding inhibitor |

|
(285) |
|
| CBX4/7 | UNC4976 | Allosteric modulator of CBX7, abrogating its function as reader of H3K27me3 marks and increasing its non-specific binding to DNA |

|
(286) |
|
| CBX6 | Ligand 5 | Binds to the beta groove of CBX6, which includes the lysine trimethylation binding pocket along with a (−2) pocket and a hydrophobic cleft extending from the binding site |

|
(287) |
|
| CBX6/8 | Ligand 22 | Selectively binds to both CBX6 and CBX8 over other CBX ChDs |

|
(288) |
|
| CBX8 | SW2_110A | Binds to the ChD of CBX8 and prevents its association with chromatin, leading to inhibition of proliferation and deactivation of MLL-AF9 target genes in THP1 leukemia cells |

|
(175) |
|
| CBX8 | UNC7040 | Allosteric modulator of CBX8, abrogating its function as reader of H3K27me3 marks and increasing its non-specific binding to DNA, leading to inhibition of proliferation in lymphoma cells |

|
(290) |
|
| CBX2 | SW2_152F | Selective CBX2 chromodomain inhibitor. Prevents and reverts neuroendocrine differentiation in prostate cancer cells |
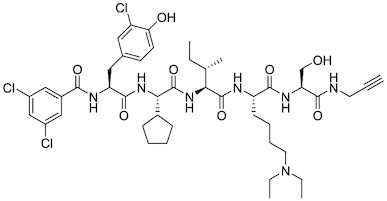
|
(226) |
|