

Abstract
Biomolecular simulations, which were once batch queue or compute limited, have now become data analysis and management limited. In this paper we introduce a new management system for large biomolecular simulation and computational chemistry datasets. The system can be easily deployed on distributed servers to create a mini-grid at the researcher’s site. The system not only offers a simple data deposition mechanism but also a way to register data into the system without moving the data from their original location. Any registered dataset can be searched and downloaded using a set of defined metadata for Molecular Dynamics and Quantum Mechanics, and visualized through a dynamic web interface.
Keywords: Biomolecular simulation, database, molecular dynamics, cheminformatics
Graphical Abstract
INTRODUCTION
Biomolecular simulations aim to study the structure, dynamics, interactions, and energetics of complex biomolecular systems. Understanding biological phenomena with these methods may facilitate the design of better drugs, therapies, catalysts and nanotechnology.1,2,3 With the recent advances in hardware, it is now not only possible to use more complex and accurate models, but also to reach time scales that are biologically significant. When simulating biomolecular dynamics on the microsecond time scale for example, one can easily generate molecular dynamics trajectories of the time series of atomic positions that represent terabytes (TB) of data on disk. More recently, special-purpose hardware such as the Anton machine has allowed researchers to reach millisecond time scales,4 increasing the size of the resulting data even further. While the computing power has dramatically increased in the last decade, our ability to manage, store, analyze, and move large datasets is still limited. Central repositories for the community or even at the lab level are desirable to facilitate data management, analysis, and sharing. This will require both new methods to catalog existing datasets by keeping them in place and improved mechanisms for facilitating and cataloguing data storage and movement.
Biomolecular simulations and computational chemistry are dominated by two classes of methods: Molecular dynamics (MD) and quantum mechanics (QM). Many variations (based on parameter choice or approximations) of the methods exist, along with hybrid approaches that combine different methods. A wide variety of MD and QM codes are available to the scientific community. AMBER,5 NAMD,6 CHARMM,7 GROMACS,8 and LAMMPS,9 are some of the most popular MD simulation codes in use today to simulate proteins, nucleic acids, or even larger molecules. Gaussian,10 NWChem,11 GAMESS,12 Q-Chem,13 Jaguar,14 and VASP15 on the other hand, are popular QM packages, typically used to study small molecules such as drug compounds. The heterogeneity of the data resulting from the simulations (e.g. QM calculation vs. MD atom trajectories), and the format of input and output files makes data management non-trivial. Moreover, each simulation software package has its own way to represent simulation parameters (e.g. simulated time, method), molecule topologies, and resulting data (e.g. trajectories of the times series of atomic positions). Additionally, each lab has multiple researchers (including students, post-docs, staff) using local and national resources, different software packages and methods, different file naming conventions, and different analysis workflows. As a result it can become quite complicated for investigators to manage this distributed multi-user environment and retrieve summaries of simulations that were run in the past.
The heterogeneity of biomolecular simulation data and the distributed nature of the resources used by researchers become even more obvious as we move towards collaboration between labs, and across institutions. Nevertheless, sharing data outside the owner’s institution has a scientific purpose. As theoretical models (e.g. basis sets, force-fields) and implementations evolve developers need to validate their code by comparing results to existing implementations. Creating collaborative networks for developers of a particular software package would increase the number of testing and validation datasets available to them. For biomedical researchers, as more datasets become available to the community, the easier it is to expose correlations between experiments and provide insight into biological structure and function. A successful example is the ABC (Ascona B-DNA Consortium) initiative, led by multiple laboratories distributed all over the world. A large series of MD simulations of B-DNA were run by the many groups in a divide-and-conquer manner to expose sequence-specific nucleic acid structure and dynamics.16,17,18,19 A significant challenge has been to aggregate the data. Such initiatives could be facilitated if labs had tools to manage and share their data within a collaborative network or with the community at large.
Sharing raw simulation data with the community would also facilitate replication of results and increase the trustworthiness of related publications. For a single software package, there might be hundreds of different parameters a user can set, and related publications typically will not include all of them. Replication of a simulation run will then require guesses if the original input files are not made publicly available. Finally, there may be unanticipated uses of MD data that will prove community-level databases to be desirable (e.g. the development of coarse-grained force fields parameterization or novel analyses of the existing data).
Because of the amount of data researchers have to deal with, it is not always practical to centralize the data for collaboration. Distributed systems offer a good solution for scientific research in general. Distributed data sources can be aggregated as a single resource despite being physically distant, and local control over the data at each node can be conserved. This is very important as researchers tend to be reluctant to expose all their data or give up ownership. Distributed systems, such as the Grid,20 allow researchers to keep control over their own data (storage, backup, security) while offering the tools to expose them to the community with authentication and authorization mechanisms.
Although data management systems at the community level are important, new mechanisms are needed to facilitate or even automate the integration of local data owned by individual researchers into collaborative or public repositories. While local data is usually unorganized (file system versus database) and dynamic by nature, public repositories tend to be more static and more structured to enable domain-specific queries by researchers. Mapping these two approaches seamlessly is not a trivial task. Three levels of granularity for data management should be considered. First, at the lowest level, tools should provide a means for individual researchers to effectively catalogue, browse, and search their data, and expose features across datasets. In the case of MD simulation data, such features might include, beyond the raw simulation data and input files, summaries of the analysis such as root-mean-squared deviation (RMSD) plots versus time, molecular graphics of average structures, and/or sequence/topology information. The tools used to catalogue and collect this data should not be onerous or complicated. They also need to run in closed environments where the data owner might not have root privileges (e.g. national computer resources). Finally, data presentation should be customizable so that the user can specify which analysis results should be considered for display to summarize a particular experiment. At the next level, data management tools should allow users to share information (and customizations) within their group or lab. Ultimately, these tools should allow users to share their data with the community either by granting access to their existing data in a secured fashion or by copying the data and its description (i.e. the metadata) to a public repository.
An important aspect of biomolecular simulation data management is the ability to catalogue the data not only at the level of an individual simulation - typically physically represented by a single set of files or a single directory of data on a file system - but also across larger experiments or projects distributed among multiple file systems and directories of data. In the context of this work we consider an experiment or project as a set of dependent QM or MD runs. For example MD experiments usually require a minimization and an equilibration pre-processing phase. Here the minimization-equilibration-production runs would be considered as a single experiment. Experiments can be grouped together to form experiment sets, for example independent runs of a similar system with different force fields or simulation protocols (i.e. related but independent simulations, results and files). By providing organization not only at the level of individual simulations but across related experiment sets, the user is provided with a greater ability to manage and search physical data (files and directories) and logical sets.
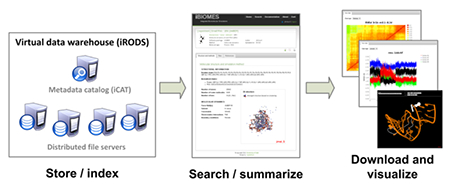
In this paper, we introduce iBIOMES (integrated BIOMolEcular Simulations), a distributed system for biomolecular simulation data management. Input and output files can be easily registered into the system and indexed using a set of metadata, automatically generated by format-specific parsers. Servers containing existing datasets can be easily integrated into the system to avoid large data movements and still benefit from the indexing capabilities of iBIOMES. A prototype is deployed at the University of Utah and is being developed to expose a subset of the MD and QM datasets generated by our lab over the years. Data is managed via a Java API and exposed via a web portal (http://ibiomes.chpc.utah.edu).
Several projects have tried to tackle the problem of molecular simulation data storing and/or sharing. We can distinguish two types of infrastructure: one that is purely based on relational databases, and one that keeps references to the raw input and output files and only stores simulation metadata in a relational database. The BioSimGrid project21 and the Dynameomics project22 belong to this first category, where trajectory information is stored directly into database tables, using one entry for each atom and for each time frame. Scalability of pure relational databases using this approach becomes problematic as we reach larger molecular systems and biologically-relevant time scales. For example, in our lab we have over 200 TB of raw MD simulation data including multiple microsecond scale simulations containing millions of frames of trajectory data; replicating the raw data into a database is impractical, wasteful of disk resources, and would be extremely slow to process. Another issue for these databases is the lack of analysis tools as most current analysis tools perform their calculations on the raw files, and not on database tables. The eMinerals project23,24 and the MoDEL (Molecular Dynamics Extended Library)25,26 databases adopted a different approach where the raw output files (or a compressed version) are made available and searchable through a database that stores information about the runs (e.g. PDB ID, molecule name). The advantage of keeping the raw files is that it becomes easier to replicate the results if necessary and existing tools can be used to perform the analysis of trajectory files.
For the iBIOMES project, we designed and implemented a distributed solution to data storage and sharing across research labs using this second approach. Simplicity was one of the key concerns for the development of this system. Users should be able to deposit, search, and retrieve data into and from the system easily through simple commands, similar to those offered by the Bookshelf system.27 The iBIOMES system provides such a command-line interface along with a web interface which offers extra visualization components. Another key concern was the ability to deploy the system locally without interfering with the lab workflow. Data can be “deposited” into the system – i.e. copied from a remote resource to a resource that is part of the system — or simply “registered” in place if the host server is integrated into the system. This becomes a crucial necessity as labs tend to have multiple servers storing terabytes of data and moving this data to be tracked by the system is not practical. The underlying data handling system, based on the iRODS (Integrated Rule Oriented Data System) framework,28 creates a virtual data warehouse at the researcher’s site, where data can be distributed among multiple servers and searched through metadata query. Metadata include system information (e.g. file location, file name, permissions, registration date) and iBIOMES-defined metadata (e.g. simulation description, title, force field used) that are used to index MD simulations or QM calculations. iRODS provides a command-line interface to manage all the servers and the files that are registered into the system. iBIOMES offers several other commands that are used to publish simulation files into the system and automatically generate metadata. A web portal and a REST (REpresentational State Transfer29) interface are also available to facilitate queries of MD and QM data for the end-user and external systems. In the next sections, we will give more details about the iRODS data-handling system, the metadata being used, and the different user interfaces that were specifically developed for iBIOMES.
THE iRODS DATA-HANDLING SYSTEM
The Integrated Rule Oriented Data System (iRODS)28 is a file management system that provides the tools to register, move, and lookup files that are distributed over the network and stored in different types of disk (e.g. HPC servers, files servers, archive tapes). iBIOMES uses iRODS as its underlying data handling system to manage distributed resources. Files that are registered into an iRODS zone are accessed using a virtual path that hides the physical location of the files (and servers), which makes it simple for users to logically organize their own data in a distributed environment. Information about the resources and the files registered into an iRODS zone are stored into the iCAT (iRODS CATalog) database. This database keeps track of the system information (e.g. file location, file name, owner) and user-defined metadata that allow any triplet “attribute, value, unit” (AVU). A simplified example of a user metadata table is given in Table 1. User-defined metadata can be used to search and retrieve distributed data that are registered in iRODS.
Table 1.
Simplified view of the iRODS user-metadata table. The first column is a reference to the file for which the metadata triplet applies. The Unit value is not mandatory.
| File ID | Attribute | Value | Unit |
|---|---|---|---|
| 1 | molecule type | Protein | |
| 1 | simulated time | 0.5 | ms |
| 1 | software | AMBER | |
| 2 | molecule type | RNA | |
| 2 | temperature | 300 | K |
A command line interface is available to manage this virtual warehouse. The “i-commands” provide the necessary functionalities one would need in a Unix-like environment to move data between servers, manage file permissions, users and groups, etc. Commands are also available to check data integrity, i.e. whether a registered file physically exists and if its content has not been altered outside iRODS. The ifsck command can be used to compare the size or checksum of the physical file with its corresponding entry in the system, while the iscan command can parse the file system to check if a physical file or directory is already registered into iRODS. iRODS also provides a powerful rule engine to manage policies and respond to specified conditions (e.g. registration of a new file) by applying a defined rule (e.g. synchronize the file with another server). Command-line and web interfaces are provided to lookup files based on user-defined metadata or system metadata. iRODS is supported by the Data Intensive Cyber Environment (DICE), which is also responsible in part for the development of the Storage Resource Broker (SRB).30 Although SRB is still supported, iRODS became the DICE-recommended framework to manage distributed data. Several national and international scientific projects have already successfully adopted iRODS for their cyberinfrastructure needs. The Wellcome Trust Sanger Institute and the Broad Institute currently use iRODS to manage sequencing data.31 The iPlant Collaborative project32 uses iRODS to manage data gathered from all plant sciences, including genotypic and phenotypic data. iRODS has also been used to manage astronomy data, typically images in the gigabyte range (National Optical Astronomy Observatory (NOAO), International Virtual Observatory Alliance (IVOA)). National computational Grids have also started to use iRODS for data management in their widely distributed environments. XSEDE (Extreme Science and Engineering Discovery Environment, https://www.xsede.org/), a large cyberinfrastructure project in the US, now offers data replication services based on iRODS at a number of its sites (e.g. National Center for Supercomputing Applications, Pittsburgh Supercomputing Center, Texas Advanced Computing Center). The Open Science Grid (OSG) is following the trend and is currently integrating iRODS into their cyberinfrastructure (www.opensciencegrid.org). This adoption by major computational centers is very important. First it creates a strong community of users and developers. Then it facilitates the federation of remote sites together, and therefore the deployment of systems such as iBIOMES to fulfill the needs of scientists in a particular area. While iRODS provides generic data and metadata storage and query capabilities, iBIOMES offers a domain-specific metadata catalog and customized user interfaces for biomolecular simulation data.
IBIOMES ARCHITECTURE
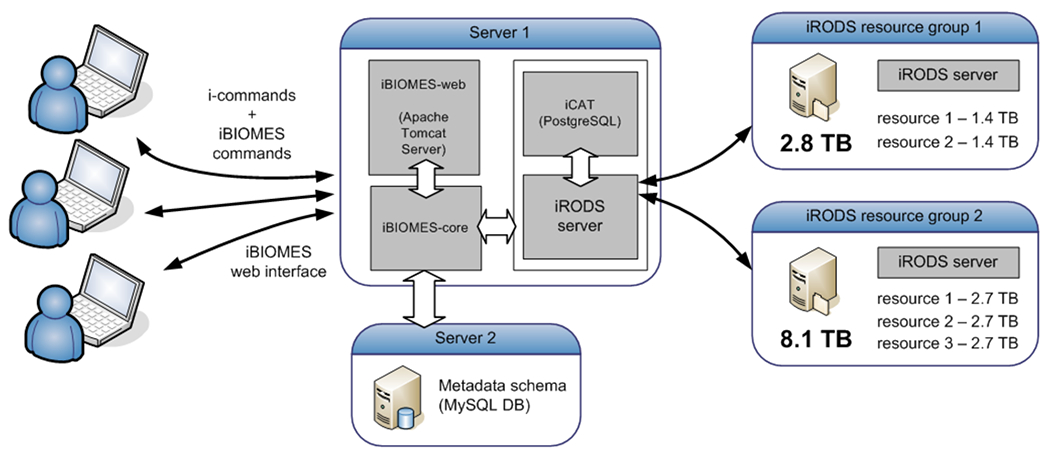
The general architecture of iBIOMES is presented in Figure 1. At the lowest level, iRODS stores the file/collection metadata in a PostgreSQL database (http://www.postgresql.org), and provides interfaces to manage the distributed resources integrated into the system. A MySQL database (http://www.mysql.com) was added to store MD and QM related metadata definitions and dictionaries such as lists of force-fields, basis sets, software, and definitions of experiment sets. Each experiment set can be assigned a name, description, and a set of metadata. While each experiment is assumed to be a physical directory somewhere in the system, sets are logical groups of experiments where each experiment can be part of multiple sets. A Java API (iBIOMES-core) was created to programmatically access iRODS resources and to manage metadata that are specific to biomolecular simulations. The API also helps to generate metadata by parsing the files that are being registered into the system in order to avoid manual annotation by the data owner. Access to iRODS functionalities is facilitated through the Jargon Java API provided by iRODS. Finally, a RESTful interface and a web portal provide access to the registered data in a more user-friendly fashion.
Figure 1.
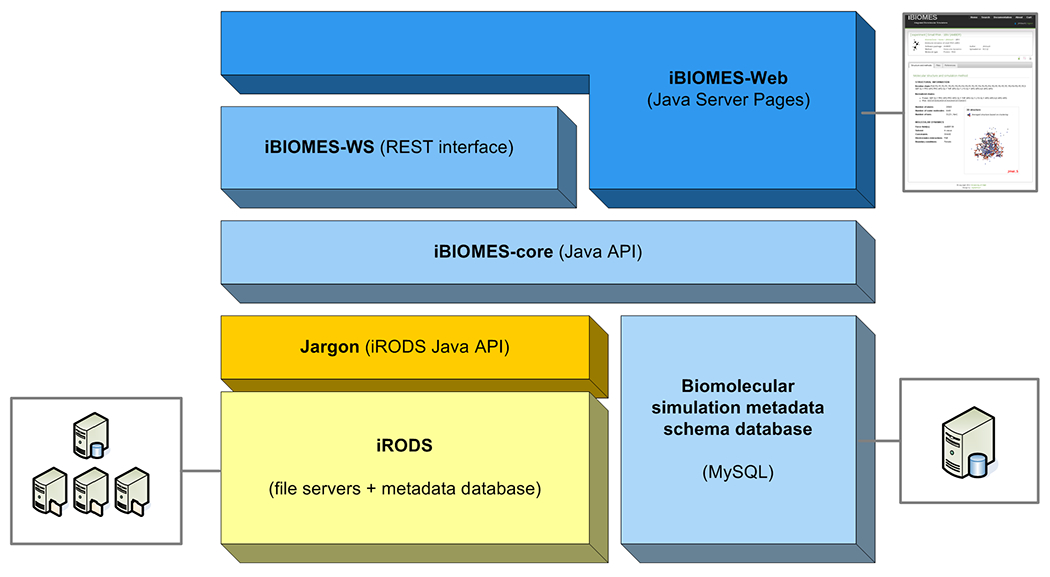
General architecture of iBIOMES. At the lowest level, iRODS stores the file metadata while a separate MySQL database enforces standard metadata use and allows definitions of experiment sets. A REST interface and a web client provide query and update capability to the metadata catalog through the iRODS API (Jargon) and an iBIOMES-specific API (iBIOMES-core).
METADATA
When working with biomolecular simulation data, several pieces of information are needed to summarize and index the experiments. Our current list of metadata covers the following categories: authorship (e.g. owner, related publications), methods (e.g. MD or QM, basis set, force field, parameters), molecular system (e.g. topology, type of molecule), platform (hardware and software information), and files (e.g. format). Our goal is to develop a list of core metadata that would be software-independent, and sufficient to retrieve raw data files that contain the necessary details to replicate an experiment. The metadata schema database contains the current list of metadata attributes and their definitions. A subset of the metadata attributes defined in iBIOMES is given in Table 2. This database also contains several dictionaries such as lists of force fields, basis sets, or software packages that users can use to facilitate queries or annotations of experiments. This list is extensible and allows custom user-defined metadata.
Table 2.
A subset of the metadata attributes defined in iBIOMES.
| Category | Attribute | Example values |
|---|---|---|
| Molecular system | Water count | Integer |
| Atom count | Integer | |
| Ion count | Integer | |
| Molecule type | Protein, RNA, DNA, chemical compound | |
| Residue sequence | ATTCGAAT, ALA PRO HIS LEU, APHL | |
| Reference structure | PDB:1BIV, PubChem:2733526 | |
|
| ||
| Method (general) | General method | Molecular dynamics, Quantum Mechanics, Coarse-grain Dynamics, QM/MM |
| Boundary conditions | Periodic, non-periodic | |
| Solvent | Implicit, explicit, in vacuum | |
|
| ||
| Molecular Dynamics | Force field | AMBER FF 99, GROMOS 43A1, ReaxFF |
| Barostat | Andersen, Berendsen, Parrinello-Rahman | |
| Thermostat | Berendsen, Nose, Nose-Poincare | |
| Molecular mechanics integrator | Verlet, Leapfrog | |
| Electrostatics modeling | Cutoff, Classic ewald, PME, reaction field | |
|
| ||
| Quantum Mechanics | General QM method | Hartree-Fock, Moeller-Plesset, DFT, Configuration interaction |
| Level of theory | SCF, MP2, MP4, CCSD(T) | |
| Basis set | STO-3G, 6-31++G*, cc-pCDVZ | |
| Spin multiplicity | 0, 2 | |
| Total charge | −1, 0, 1, 2 | |
The distinction between experiment and experiment set is important when registering data into iBIOMES. Metadata is automatically generated for the files through the API’s parsers then pushed up to the experiment level. For example, in a directory containing AMBER simulation data, the topology-related metadata is parsed from AMBER topology files, or PDB files if not available. The new topology metadata set is then added to the root directory, which is considered to be the representation of the experiment. Currently, no metadata is generated for experiment sets, but the owner can easily pick one of the experiments or a file to push metadata to the experiment set level. For example if the topology information is the same for all experiments within the set, this information can be easily pulled and applied to the set level via the web interface.
Currently, automatic metadata generation is supported for PDB files, MOL/SDF files, Mol2 files, AMBER topology, input, and output files, GROMACS Include Topology (.itp), System Topology (.top), and parameter input (.mdp) files, Protein Structure Files (.psf), NWChem, Gaussian, and GAMESS input files. Each parser implementation is based on the conceptual model summarized in Figure 2. File parser classes inherit from AbstractTopologyFile, AbstractParameterFile, or AbstractParameterAndTopologyFile, whether the target file format defines topology information, calculation parameter, or both. For example the Gaussian input file parser inherits from AbstractParameterAndTopology since it needs to parse the QM calculation parameters (e.g. basis set, level of theory) and the compound topology, while the PDB parser only looks at topology information and inherits from AbstractTopologyFile.
Figure 2.
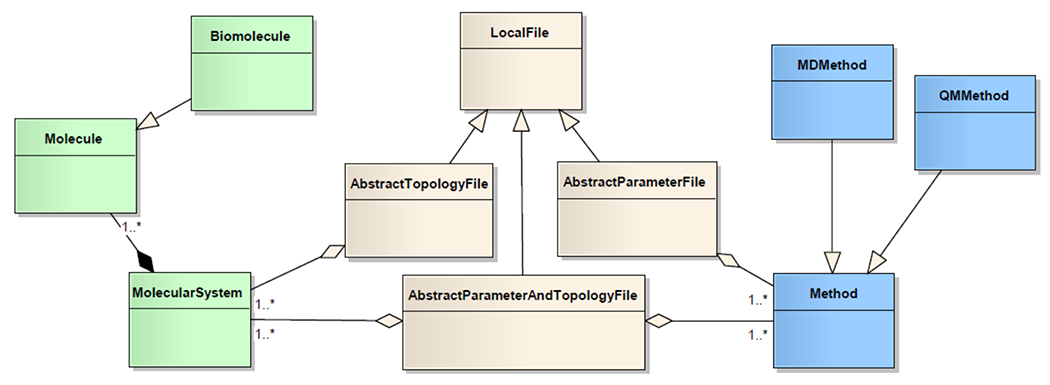
Simplified class diagram representing the file parser implementations.
In order to implement a new parser one needs to create a new Java class that inherits from one of the abstract classes and write a parsing function that will build the Method and/or MolecularSystem (i.e. a set of molecules) objects. Mapping between this data model and the iBIOMES metadata is done through the getMetadata () method available for each of the classes inheriting from Method and Molecule. This method is automatically called when registering the files into iBIOMES.
While in most cases rules for parsing files can be applied solely based on the file name extension (e.g. .pdb), there are cases where the format of a file cannot be determined based on its extension. To overcome this issue and enable automatic metadata assignment and extensibility, a set of rules can be defined in an XML descriptor file. Rules can define metadata for files or directories with names matching a specified pattern. Examples of such rules are given in Figure 3. In this example the first rule defines possible file extensions for AMBER topology files (.prmtop, .topo, .top, or .parm). The second rule targets files that are the result of an MD trajectory clustering algorithm. The clustering tool generates averaged structures in PDB format but omits the .pdb file extension. By applying this rule these files are recognized as PDB files when registered into the system and viewable as 3D structures. The last rule targets a CSV (comma-separated value) file that represents a time series, generated by an analysis script. As the same script and name conventions are used in our lab, this rule helps define the labels (e.g. Time, Density), titles (e.g. Evolution of density over time), and units (e.g. ps, g/cm3) for the data contained in the file. Once registered, this file can be automatically displayed through the web interface as a 2D plot with the correct legends and axis titles.
Figure 3.

Example of XML rule set used to customize the publication process. The first rule associates file extensions to a particular file format (AMBER topology). The second and third rules associate a particular set of metadata to analysis output files that follow a standard nomenclature in our lab.
This rule set can be customized to fit the needs of a particular lab or user. Experience showed that file name convention for a particular software package run (e.g. AMBER) and the following analysis vary only slightly for the same user. Therefore the XML file will be reusable. Once a simulation and its associated files are registered into iBIOMES, the owner or the authorized users can still edit the metadata through the web interface (or any iRODS interface).
INTERFACES
Web interfaces
A REST interface was developed to offer web services for access to the metadata catalog and dictionaries. The metadata catalog is open access as it only contains general definitions of biomolecular simulation related metadata. The related services are mainly used to auto-complete user entries in the web interface (e.g. software name, force field). The current web portal builds upon this REST interface and allows authenticated and authorized users to manage and search data registered in iBIOMES (Figure 4). Users can create queries based on the standard metadata catalog to retrieve simulations of interest. The queries can either target files, experiments (collections of files), or experiment sets. A simple web interface is available to query data files and experiments based on common attributes such as methods, molecule type (e.g. DNA, RNA, protein) or residue chain (nucleotide or amino-acid sequence). Residue chains are normalized and used as file or experiment metadata, along with the software-specific residue chains. The normalized residue chains are sequences of 1-letter nucleotide or amino acid codes. For example one could search for a particular protein / RNA system using the following AVUs:
Figure 4.
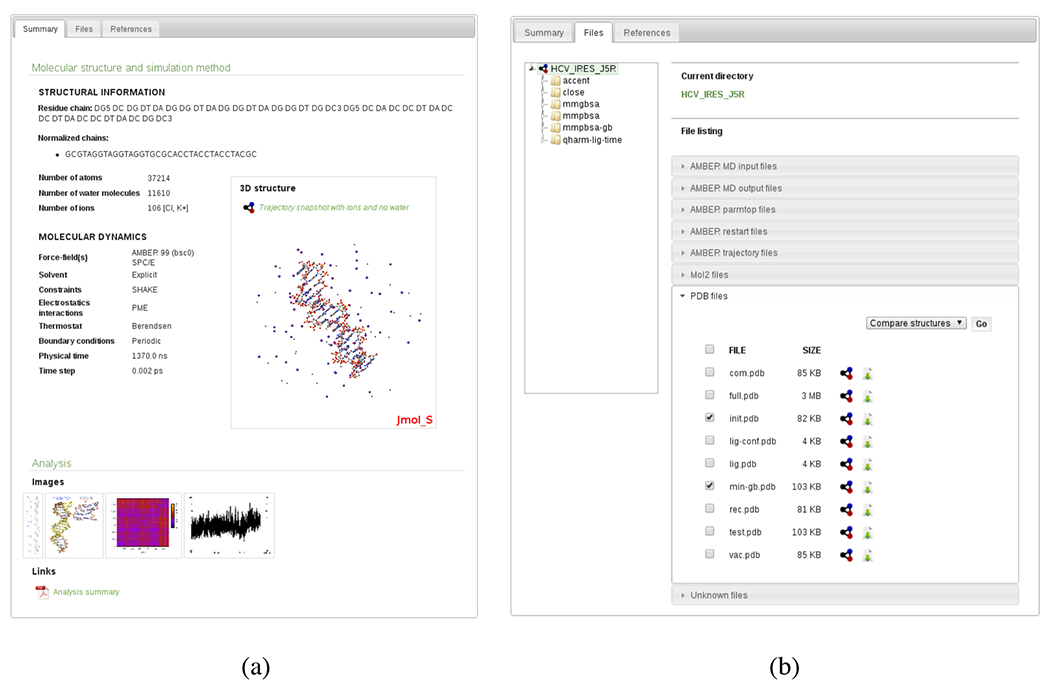
iBIOMES web interface. (a) Summary page for an MD simulation of DNA including analysis data and a representative 3D structure. (b) File listing for a particular experiment.
RESIDUE_CHAIN_NORM = “%GGCUCGUGUAGCUCAUUAGCUCCGAGCC%”
RESIDUE_CHAIN_NORM = “%SGPRPRGTRGKGRRIRR%”
Or using AMBER-specific residue chains:
RESIDUE_CHAIN = “%RG5 RG RC RU RC RG RU RG RU RA RG RC RU RC RA RU RU RA RG RC RU RC RC RG RA RG RC RC3%”
RESIDUE_CHAIN = “%SER GLY PRO ARG PRO ARG GLY THR ARG GLY LYS GLY ARG ARG ILE ARG ARG%”
Although the first approach enable searches through experiments generated by different software packages, the second approach is still useful as certain residue codes are meaningful only in the context of a particular software package or within a community.
Experiments can also be retrieved by simply entering keywords, in which case the metadata attribute is bypassed and the query only uses the value component of the AVU triplets to find matches. Advanced queries can be built as well. The user can pick and choose metadata attributes from the iBIOMES metadata catalog or manually enter user-specific attributes, then assign values to each attribute. Figure 5 shows how one could build a query through the web interface using the catalog of standard iBIOMES metadata.
Figure 5.
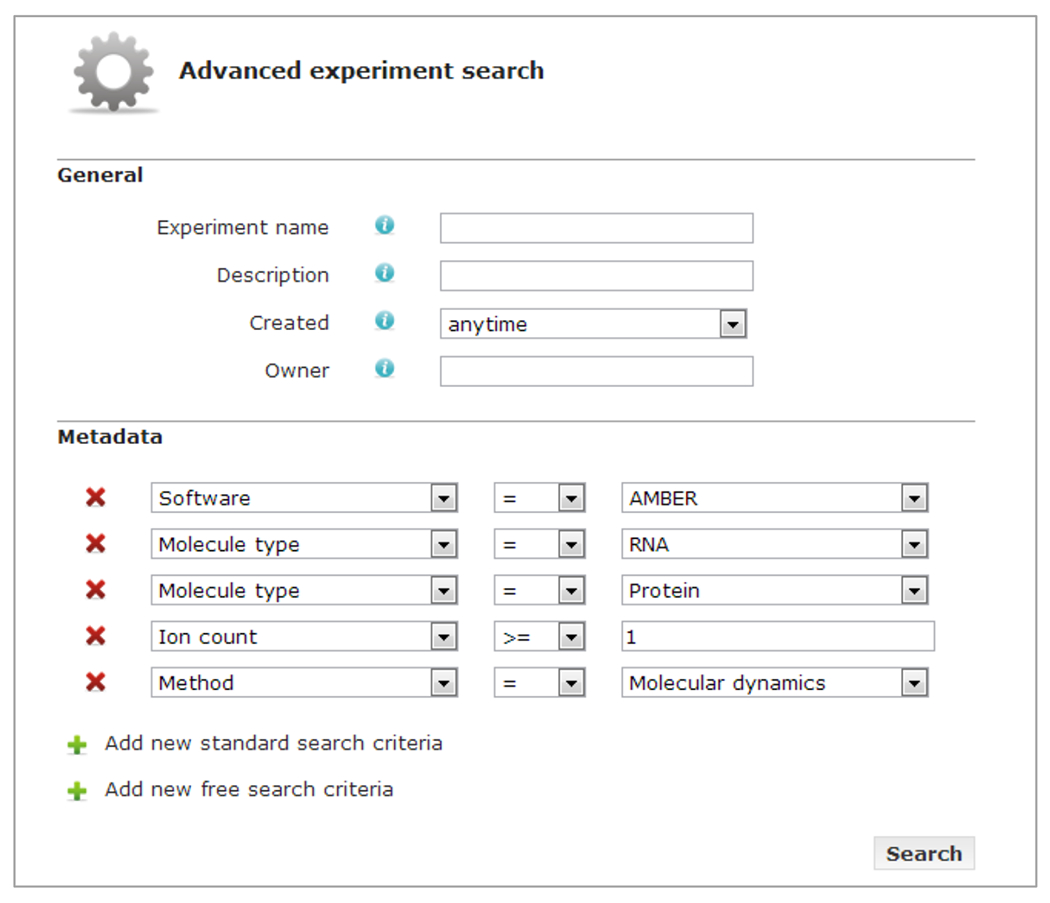
Advanced experiment search through the web interface. Users can pick metadata attributes and values from the standard catalog or create free-text criteria. This particular example shows how one would search MD simulations of protein/RNA complexes run with AMBER.
Matching experiments and files can be downloaded and data content can be summarized directly through different applets if the user has the right permissions. For example Jmol33 is used for 3D rendering of molecules described in PDB, Mol2, MOL/SDF or Gaussian log files (Figure 6c). Users can pick Jmol-supported files and load them into the applet to compare structures or create multi-frame animations. 2D data such as time series in comma-separated or tab-delimited value format can be dynamically plotted through a service based on the JFreeChart (http://www.jfree.org/jfreechart/) library (Figures 6a–b). Supported graphs include multi-line plots (e.g. comparison of RMSd of multiple runs), scatter plots, and heatmaps (2D-RMSd matrix). A “shopping cart” based on DICE’s iDrop applet (https://code.renci.org/gf/project/irodsidrop) also allows users to pick and choose files or collections of files they want to download in a bulk fashion (Figure 6d).
Figure 6.
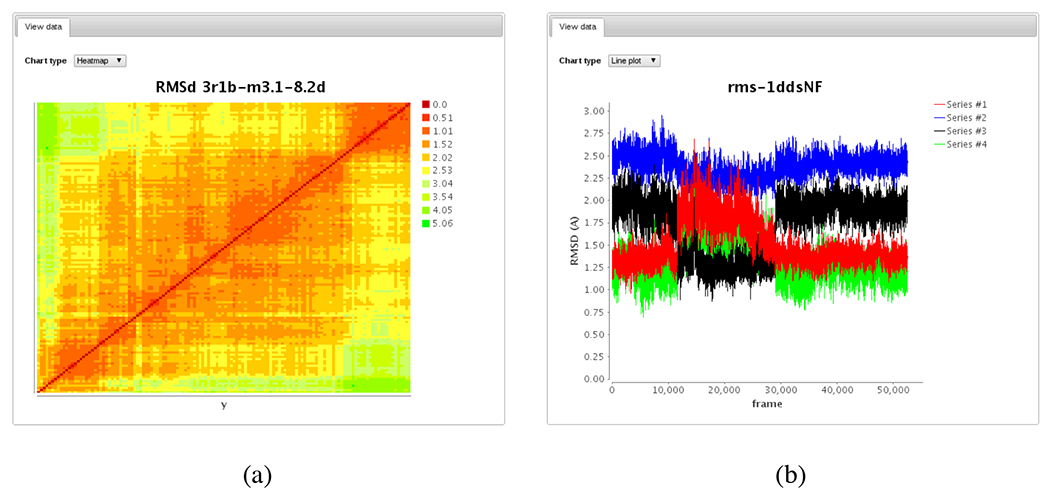
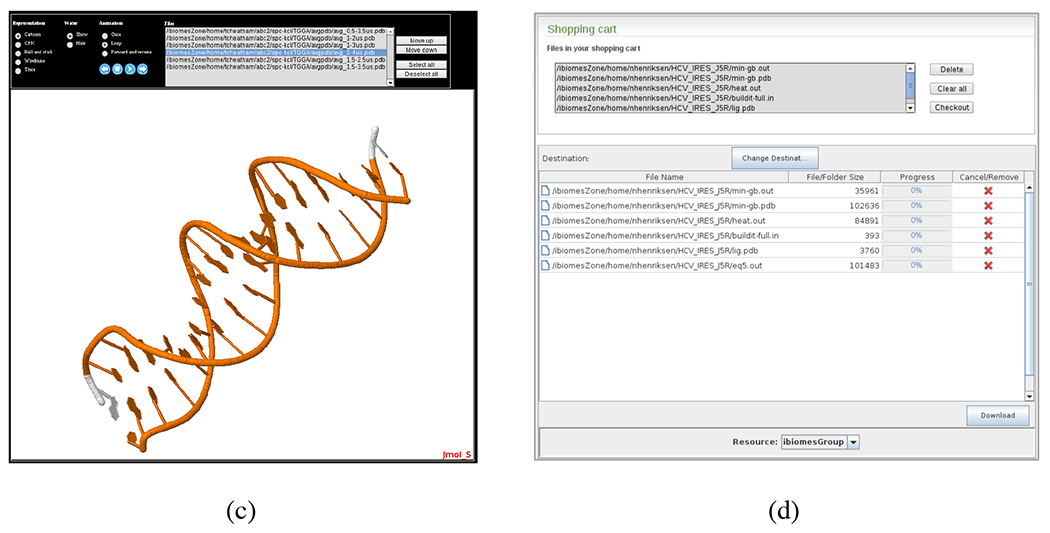
Graphical tools used in the iBIOMES web interface for data visualization and bulk downloads. A plotting service based on the JFreeChart library enables comparison of multiple RMSd (root mean square deviation) plots (a) and rendering of RMSd 2D matrices as heatmaps (b). Jmol is used to render and manipulate 3D structures (c). The iDrop Lite applet is used for bulk downloads of files through the shopping cart service (d).
Experiments sets can be created through the web interface as well. Set owners can define the list of referenced experiments and metadata for a particular set directly from the corresponding experiment set summary page. Experiment sets can be made public or private.
More options are available to experiment data owners or users with write permissions. For example they can manage permissions at the collection or file level and update the associated metadata. iBIOMES-defined metadata can be easily edited using the available dictionaries. User-defined metadata that are not defined in the iBIOMES catalog can be added as well, and used to build queries. While metadata is automatically generated during data publication into the system, the set of metadata might be incomplete or not totally accurate. The web interface allows the user to update topology-specific metadata or method-specific metadata by specifying which files should be used as templates. In the case of the topology for AMBER data, this could be a topology file or a PDB file; for the methods, this could be an MD input or output file. Finally, the main page for a particular experiment can be customized by specifying which 3D structures should be displayed, and which files should be presented to summarize the results. Related publications and published structures (e.g. from the Protein Data Bank,34 PubChem,35 or the Cambridge Structural Database36) can be added as well for reference.
The web portal was built with Java Server Pages (JSP) and Spring MVC (http://www.springsource.org/). This code, along with the main Java API (iBIOMES-core) was integrated into Maven (http://maven.apache.org/) to manage external dependencies and automate builds.
Data registration
One of the goals of iBIOMES is to make the data publication process as easy as possible. Two scenarios are supported: registration of data into the system without moving the files, and registration after data transfer from a local or remote resource (e.g. desktop, remote computational resource) to an iBIOMES node. Both registration options are available through Unix-like commands that can be run from the machine where the data resides. For in-place registration, the host needs to be integrated to the target iBIOMES zone. Usage of these commands is given in Figure 7.
Figure 7.


iBIOMES commands for in-place registration and standard publication with data transfer.
DEPLOYMENT AT THE UNIVERSITY OF UTAH
iBIOMES installation requirements
iBIOMES requires a Java Runtime Environment (1.7) to be installed on the host machine. iBIOMES-core is packaged into a single JAR (Java ARchive) file including all the dependencies (e.g. iRODS Java API). As iBIOMES is dependent on iRODS, iRODS should be installed first on the servers that need to be integrated to the system, then the iBIOMES-core library and scripts can be copied on these machines. To host the web application, a web server such as Apache Tomcat (http://tomcat.apache.org) is required to deploy the iBIOMES-web and iBIOMES-ws codes, which are packaged as two WAR (Web application ARchive) files.
iRODS configuration
The current iBIOMES setup for our lab is presented in Figure 8. Although all the components of iBIOMES could be installed on a single physical server, we decided to deploy the system in a distributed environment to assess a more likely scenario where data needs to be scattered among multiple disks. The primary iRODS server along with the iCAT database were installed on a Linux server (CentOS 5.8). Two file servers (Red Hat Enterprise Linux Server 6.3) were integrated into the same iRODS zone (“ibiomesZone”) to provide over 10 TB of disk space overall. Each file server runs an iRODS server instance, and each disk on the servers is exposed as an iRODS resource. Resources can be grouped together to apply data storage policies managed by iRODS. For example one could define a policy to enforce data replication on all resources of the same group, or to order resources in the group to define which resource should be used for storage first. For our case, the 5 resources (5 disks in 2 separate servers) were grouped together and managed through a load balancing policy defined in iRODS. A rule periodically triggers the activation of a resource monitoring system and calculates the load factor on each machine. The iRODS administrator can customize the way the load factor is calculated by assigning a weight to the disk space resource, the CPU load, the memory load, etc. The administration of iRODS servers (start/stop, resource definition, rule control) is made simple through the i-commands and other scripts that can be run only by an iRODS administrator.
Figure 8.
Configuration of the iBIOMES infrastructure at the University of Utah (Cheatham lab). Storage resources are distributed over 2 servers and currently offer a 10 TB capacity.
iBIOMES deployment
An Apache Tomcat 7 server was installed on the first server to host the web portal and the REST services. The iBIOMES metadata schema database (MySQL) was installed on a second Linux server (CentOS 5.8). This was done through a set of SQL scripts that create the database schema and populate the biomolecular simulation metadata catalog and the dictionaries. The iBIOMES client tools (scripts and JAR file) can be copied to remote resources (e.g. HPC facility) by users to enable data transfer and registration into the system directly from resources outside the defined iRODS zone.
Data summary
Our lab currently owns over 200 TB of both MD simulation and QM calculation datasets. For this prototype we decided to expose a subset of this data that would still be representative of the type of simulation that is done in our lab. Our current projects involve mainly nucleic acid force field developments and P450 QM studies. This is reflected in the datasets currently published in our iBIOMES instance, which for now contains MD simulations of RNA for force-field assessment (AMBER FF 10), and QM calculations that were performed in Gaussian 03 to generate AMBER-compatible heme parameters for various states of the P450 cycle.37 Because of licensing restrictions, our Gaussian datasets could not be released for public access yet. On the other hand a series of MD simulations of RNA was released, along with a subset of the data derived from the ABC consortium’s study on B-DNA.17 The ABC set currently includes a series of experiments with final stripped trajectories (~20-60 GB each) and basic analysis data (e.g. RMSd, radial plots).
A guest account was created to enable read access for anybody interested in these public datasets. Guests can search experiments, read summaries, and graphically visualize data from this subset. Currently the shopping cart service for bulk downloads is not available for guest logins. Guests can still download files individually. The iBIOMES prototype can be accessed via the guest login option at: http://ibiomes.chpc.utah.edu.
DISCUSSION
In this paper we presented a new distributed system developed to manage large biomolecular simulation datasets. The underlying data handling system based on the iRODS framework creates a virtual data warehouse at the researcher’s site, where data can be distributed among multiple servers. Both iRODS and iBIOMES are easy to deploy through a set of scripts. Existing archive servers can be integrated into iBIOMES without a need for a physical reorganization of the files, saving the cost of moving terabytes of data. The current implementation of iBIOMES uses the native iRODS password mechanism to authenticate users. iRODS also supports the Grid Security Infrastructure (GSI) which will facilitate the integration of iBIOMES into scientific Grids. Support for LDAP has been recently added as well. The burden of creating and maintaining iRODS-specific accounts can then be avoided by system administrators, who in turn can deploy iRODS in closed environments with existing security mechanisms and user accounts.
The publication process is facilitated by parsers that automatically generate metadata during file registration, and can be customized for the need of a particular user or lab through XML descriptors. Although our efforts have mainly focused on supporting AMBER and Gaussian datasets, we are currently working on improving our parsers for other popular MD and QM software packages, including GROMACS, CHARMM, Gaussian, GAMESS, and NWChem. Experiments registered into iBIOMES can be easily retrieved through simple keyword searches or queries built upon data elements defined in a metadata catalog for MD simulations and QM calculations. We are currently gathering feedback from the community to define a list of core metadata that would be sufficient to search and retrieve simulation datasets. A data model will be designed to define relationships between the concepts represented by these metadata, and facilitate future semantic integration with external systems, such as scientific grids. In order to enable researchers outside the field of computational chemistry to query data in a meaningful way, it will be necessary to facilitate the annotation of experiments using biological metadata (e.g. molecule name, organism). Currently this type of metadata would have to be entered manually via the web interface after data publication. This process could be facilitated in the future through a web service that would query common databases such as the Protein Data Bank to automatically generate these data elements based on the PDB ID.
Metadata is represented by AVU triplets that can be either tied to the iBIOMES metadata catalog, or customized to represent concepts that are specific to a user or a lab. This provides a very flexible data annotation model compared to a standard relational database schema, where model modifications require an intervention from the database administrators. One limitation of the AVU model is the lack of relations between AVUs. For example, one cannot assign properties to two different molecules (e.g. name, type, residue chain) represented in the same experiment, as attribute names will be the same for both molecules, and cannot be distinguished, as shown in the following example:
MOLECULE_TYPE = “RNA”
RESIDUE_CHAIN = “GGCUCGUGUAGCUCA…”
MOLECULE_TYPE = “Protein”
RESIDUE_CHAIN = “SER GLY PRO ARG PRO ARG…”
In the current implementation of iBIOMES relations between AVUS cannot be determined. While this is not required for indexing purpose, this becomes necessary to provide a clear conceptual view of the data to the users. To create a more structured metadata schema the iCAT database can be extended with custom tables and enable queries on these tables via the standard iRODS interfaces. Such capability could help us keep track of metadata in a more structured way, especially for multi-molecule systems and experiments based on multiple runs using different methods.
The current prototype deployed for our lab demonstrated the ability of iRODS and iBIOMES to manage large biomolecular simulation datasets in a distributed environment. The iBIOMES web portal provides a rich and dynamic user interface to search, download, and visualize data registered into the system. Advanced features are available for data owners to manage permissions, annotate experiments, and customize data display in the web interface. Direct data analysis via iBIOMES is currently not supported. The analysis output has to be explicitly registered into the system and described via metadata to enable visualization through Jmol or the plotting service. This can be achieved automatically by customizing the XML rule set descriptor before data publication or directly via the web interface after data deposit. Thanks to these features users can easily extend the web interface to include new pictures, spreadsheets, or links to any type of data file. The current focus of iBIOMES is not to enable deep analysis of the derived data but instead to provide the means to display, catalogue and share information about biomolecular simulations. As we move forward the system will be enhanced to add simple analysis support (e.g. RMSd calculations, data extraction from time series datasets). Our long-term goal is to provide a complete framework where data can be tracked locally, analyzed via automated processes, and registered seamlessly into a global system such as iBIOMES. For now we hope to learn more from the current iBIOMES system, and define more clearly the needs of the users, such as:
Which data elements are required or missing for indexing and search purpose?
How would users interact with iBIOMES to execute complex analysis workflows?
What can be improved to facilitate education, networking or collaboration between users?
CONCLUSION
iBIOMES is a new distributed system for biomolecular simulation data management. The data registration process is simple and supported by metadata generators, customizable by the user if needed. Registration does not require physical transfer of the data, which makes it a great solution for researchers who want to expose existing datasets. Finally data summarization and management are facilitated through a rich web interface that offers different visualization components for 3D structures and analysis data (e.g. time series). Guest access to our web portal is currently available at http://ibiomes.chpc.utah.edu.
With the adoption of iRODS across the world, and across scientific domains, we believe that iBIOMES has a strong potential to create collaborative networks within the field of biomolecular simulation, for users, developers, and new comers to the field.
ACKNOWLEDGMENT
Thanks to Mike Conway and DICE for their support with iRODS, Jargon, and iDrop. Thanks to the CHPC staff at the University of Utah for computer time allocations and hardware and software support, and to Anita Orendt and Wim Cardoen who provided valuable input for the definition of the metadata. Thanks to the National Science Foundation’s XSEDE program (MCA-015027) for computer time.
Funding Sources
This work has been supported by a grant from the National Institutes of Health R01-GM081441. JCT received partial support from the National Library of Medicine Training grant #LM007124 and JCF was partially supported by National Center for Advancing Translational Sciences, National Institutes of Health, through Grant s UL1-RR025764 and C06-RR11234.
ABBREVIATIONS
- MD
Molecular Dynamics
- QM
Quantum Mechanics
- AVU
Attribute-Value-Unit
- HPC
High-Performance Computing
REFERENCES
- 1.Dror RO; Dirks RM; Grossman JP; Xu H; Shaw DE, Biomolecular Simulation: a Computational Microscope for Molecular Biology. Annu. Rev. Biophys 2012, 41, 429–452. [DOI] [PubMed] [Google Scholar]
- 2.Alonso H; Bliznyuk AA; Gready JE, Combining Docking and Molecular Dynamic Simulations in Drug Design. Med. Res. Rev 2006, 26 (5), 531–568. [DOI] [PubMed] [Google Scholar]
- 3.Klein ML; Shinoda W, Large-Scale Molecular Dynamics Simulations of Self-Assembling Systems. Science 2008, 321 (5890), 798–800. [DOI] [PubMed] [Google Scholar]
- 4.Shaw DE; Deneroff MM; Dror RO; Kuskin JS; Larson RH; Salmon JK; Young C; Batson B; Bowers KJ; Chao JC In Anton, a Special-Purpose Machine for Molecular Dynamics Simulation, ACM SIGARCH Computer Architecture News, ACM: 2007; pp 1–12. [Google Scholar]
- 5.Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26 (16), 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K, Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks BR; Bruccoleri RE; Olafson BD; Swaminathan S; Karplus M, CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 2004, 4 (2), 187–217. [Google Scholar]
- 8.Hess B; Kutzner C; van der Spoel D; Lindahl E, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput 2008, 4 (3), 435–447. [DOI] [PubMed] [Google Scholar]
- 9.Plimpton S, Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys 1995, 117 (1), 1–19. [Google Scholar]
- 10.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JJA; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision C. 01; Gaussian, Inc: Wallingford, CT, 2009. [Google Scholar]
- 11.Valiev M; Bylaska EJ; Govind N; Kowalski K; Straatsma TP; Van Dam HJJ; Wang D; Nieplocha J; Apra E; Windus TL, NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun 2010, 181 (9), 1477–1489. [Google Scholar]
- 12.Schmidt MW; Baldridge KK; Boatz JA; Elbert ST; Gordon MS; Jensen JH; Koseki S; Matsunaga N; Nguyen KA; Su S, General Atomic and Molecular Electronic Structure System. J. Comput. Chem 2004, 14 (11), 1347–1363. [Google Scholar]
- 13.Kong J; White CA; Krylov AI; Sherrill D; Adamson RD; Furlani TR; Lee MS; Lee AM; Gwaltney SR; Adams TR, Q-Chem 2.0: A High-Performance Ab Initio Electronic Structure Program Package. J. Comput. Chem 2000, 21 (16), 1532–1548. [Google Scholar]
- 14.Jaguar, Version 7.5; Schrödinger, L.L.C.: New York, NY, 2008. [Google Scholar]
- 15.Vienna Ab Initio Simulation Package (VASP), Version 5.3.3; 2012. [Google Scholar]
- 16.Beveridge DL; Cheatham TE III; Mezei M, The ABCs of Molecular Dynamics Simulations on B-DNA, Circa 2012. J. Biosci. (Bangalore, India) 2012, 37 (3), 379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavery R; Zakrzewska K; Beveridge D; Bishop TC; Case DA; Cheatham T III; Dixit S; Jayaram B; Lankas F; Laughton C; Maddocks JH; Michon A; Osman R; Orozco M; Perez A; Singh T; Spackova N; Sponer J, A Systematic Molecular Dynamics Study of Nearest-Neighbor Effects on Base Pair and Base Pair Step Conformations and Fluctuations in B-DNA. Nucleic Acids Res. 2010, 38 (1), 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beveridge DL; Barreiro G; Byun KS; Case DA; Cheatham TE; Dixit SB; Giudice E; Lankas F; Lavery R; Maddocks JH; Osman R; Seibert E; Sklenar H; Stoll G; Thayer KM; Varnai P; Young MA, Molecular Dynamics Simulations of the 136 Unique Tetranucleotide Sequences of DNA Oligonucleotides. I. Research Design and Results on d(CpG) Steps. Biophys. J 2004, 87 (6), 3799–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixit SB; Beveridge DL; Case DA; Cheatham TE; Giudice E; Lankas F; Lavery R; Maddocks JH; Osman R; Sklenar H; Thayer KM; Varnai P, Molecular Dynamics Simulations of the 136 Unique Tetranucleotide Sequences of DNA Oligonucleotides. II: Sequence Context Effects on the Dynamical Structures of the 10 Unique Dinucleotide Steps. Biophys. J 2005, 89 (6), 3721–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Grid 2: Blueprint for a New Computing Infrastructure. second ed.; Morgan Kaufmann: San Francisco, CA, 2003. [Google Scholar]
- 21.Ng MH; Johnston S; Wu B; Murdock SE; Tai K; Fangohr H; Cox SJ; Essex JW; Sansom MSP; Jeffreys P, BioSimGrid: Grid-Enabled Biomolecular Simulation Data Storage and Analysis. Future Generation Computer Systems 2006, 22 (6), 657–664. [Google Scholar]
- 22.Simms AM; Toofanny RD; Kehl C; Benson NC; Daggett V, Dynameomics: Design of a Computational Lab Workflow and Scientific Data Repository for Protein Simulations. Protein Eng., Des. Sel 2008, 21 (6), 369–377. [DOI] [PubMed] [Google Scholar]
- 23.Alfredsson M In eMinerals: Science Outcomes Enabled by New Grid Tools, Proc. UK eScience All Hands Meeting, 2005; pp 788–795. [Google Scholar]
- 24.Calleja M; Bruin R; Tucker MG; Dove MT; Tyer R; Blanshard L; Van Dam KK; Allan RJ; Chapman C; Emmerich W, Collaborative Grid Infrastructure for Molecular Simulations: The eMinerals Minigrid as a Prototype Integrated Compute and Data Grid. Molecular Simulation 2005, 31 (5), 303–313. [Google Scholar]
- 25.Meyer T; D’Abramo M; Hospital A; Rueda M; Ferrer-Costa C; Perez A; Carrillo O; Camps J; Fenollosa C; Repchevsky D; Lluis Gelpi J; Orozco M, MoDEL (Molecular Dynamics Extended Library): A Database of Atomistic Molecular Dynamics Trajectories. Structure 2010, 18 (11), 1399–1409. [DOI] [PubMed] [Google Scholar]
- 26.Hospital A; Andrio P; Fenollosa C; Cicin-Sain D; Orozco M; Lluis Gelpi J, MDWeb and MDMoby: An Integrated Web-Based Platform for Molecular Dynamics Simulations. Bioinformatics 2012, 28 (9), 1278–1279. [DOI] [PubMed] [Google Scholar]
- 27.Vohra S; Hall BA; Holdbrook DA; Khalid S; Biggin PC, Bookshelf: A Simple Curation System for the Storage of Biomolecular Simulation Data. Database: the Journal of Biological Databases and Curation 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajasekar A; Moore R; Hou C; Lee CA; Marciano R; de Torcy A; Wan M; Schroeder W; Chen SY; Gilbert L, iRODS Primer: Integrated Rule-Oriented Data System. Synthesis Lectures on Information Concepts, Retrieval, and Services 2010, 2 (1), 1–143. [Google Scholar]
- 29.Fielding RT, Chapter 5: Representational State Transfer (REST). Architectural Styles and the Design of Network-based Software Architectures, Dissertation2000. [Google Scholar]
- 30.Baru C; Moore R; Rajasekar A; Wan M In The SDSC Storage Resource Broker, Proceedings of the 1998 Conference of the Centre for Advanced Studies on Collaborative research, IBM Press: 1998; p 5. [Google Scholar]
- 31.Chiang G-T; Clapham P; Qi G; Sale K; Coates G, Implementing a Genomic Data Management System Using iRODS in the Wellcome Trust Sanger Institute. BMC Bioinf. 2011, 12, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goff SA; Vaughn M; McKay S; Lyons E; Stapleton AE; Gessler D; Matasci N; Wang L; Hanlon M; Lenards A; Muir A; Merchant N; Lowry S; Mock S; Helmke M; Kubach A; Narro M; Hopkins N; Micklos D; Hilgert U; Gonzales M; Jordan C; Skidmore E; Dooley R; Cazes J; McLay R; Lu Z; Pasternak S; Koesterke L; Piel WH; Grene R; Noutsos C; Gendler K; Feng X; Tang C; Lent M; Kim S-J; Kvilekval K; Manjunath BS; Tannen V; Stamatakis A; Sanderson M; Welch SM; Cranston KA; Soltis P; Soltis D; O’Meara B; Ane C; Brutnell T; Kleibenstein DJ; White JW; Leebens-Mack J; Donoghue MJ; Spalding EP; Vision TJ; Myers CR; Lowenthal D; Enquist BJ; Boyle B; Akoglu A; Andrews G; Ram S; Ware D; Stein L; Stanzione D, The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front. Plant. Sci 2011, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herráez A, Biomolecules in the Computer: Jmol to the Rescue. Biochem. Mol. Biol. Educ 2006, 34 (4), 255–261. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein FC; Koetzle TF; Williams GJB; Meyer EF; Brice MD; Rodgers JR; Kennard O; Shimanouchi T; Tasumi M, The Protein Data Bank. Eur. J. Biochem 2008, 80 (2), 319–324. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y; Xiao J; Suzek TO; Zhang J; Wang J; Bryant SH, PubChem: A Public Information System for Analyzing Bioactivities of Small Molecules. Nucleic Acids Res. 2009, 37 (suppl 2), W623–W633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allen FH; Taylor R, Research Applications of the Cambridge Structural Database (CSD). Chem. Soc. Rev 2004, 33 (8), 463–475. [DOI] [PubMed] [Google Scholar]
- 37.Shahrokh K; Orendt A; Yost GS; Cheatham TE 3rd, Quantum Mechanically Derived AMBER-Compatible Heme Parameters for Various States of the Cytochrome P450 Catalytic Cycle. J. Comput. Chem 2012, 33 (2), 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]