

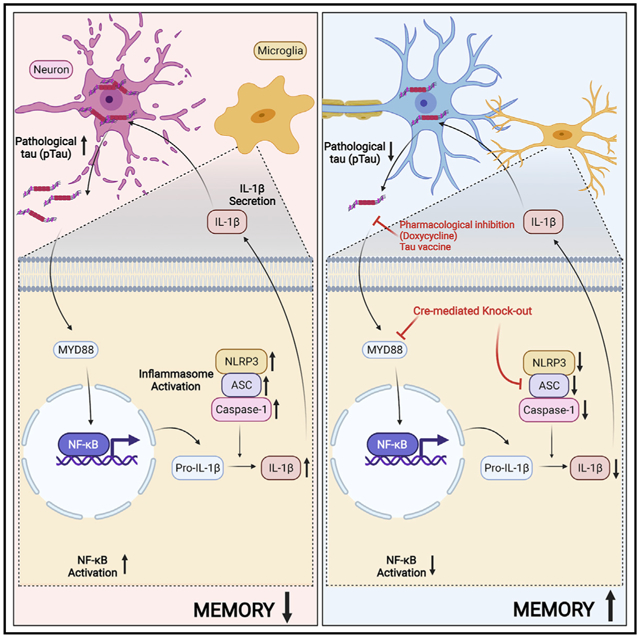
SUMMARY
Pathological hyperphosphorylation and aggregation of tau (pTau) and neuroinflammation, driven by interleukin-1β (IL-1β), are the major hallmarks of tauopathies. Here, we show that pTau primes and activates IL-1β. First, RNA-sequence analysis suggests paired-helical filaments (PHFs) from human tauopathy brain primes nuclear factor κB (NF-κB), chemokine, and IL-1β signaling clusters in human primary microglia. Treating microglia with pTau-containing neuronal media, exosomes, or PHFs causes IL-1β activation, which is NLRP3, ASC, and caspase-1 dependent. Suppression of pTau or ASC reduces tau pathology and inflammasome activation in rTg4510 and hTau mice, respectively. Although the deletion of MyD88 prevents both IL-1β expression and activation in the hTau mouse model of tauopathy, ASC deficiency in myeloid cells reduces pTau-induced IL-1β activation and improves cognitive function in hTau mice. Finally, pTau burden co-exists with elevated IL-1β and ASC in autopsy brains of human tauopathies. Together, our results suggest pTau activates IL-1β via MyD88- and NLRP3-ASC-dependent pathways in myeloid cells, including microglia.
Graphical Abstract
In brief
Jiang et al. show pathological tau primes and activates interleukin-1β in microglia via MyD88-, NLRP3-, and ASC-dependent pathways. Suppressing tau, MyD88, or ASC reduces tau pathology and inflammasome activation and improves cognitive function in the hTau mice. Tau burden co-exists with elevated IL-1β and ASC in human tauopathy brains.
INTRODUCTION
Accumulation of hyperphosphorylated microtubule-associated protein tau (Mapt) as paired-helical filaments (PHFs) or straight filaments (SFs) and eventually into neurofibrillary tangles (NFTs) is a prominent pathological hallmark of many tauopathies including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), tangle-predominant senile dementia (TPSD), Pick’s disease (PiD), primary age-related tauopathy (PART), and frontotemporal lobar dementia-tau (FTLD-Tau) (Lee et al., 2001). Several recent studies have shown that tau pathology tightly correlates with cognitive decline in various tauopathies (Nelson et al., 2012; Schöll et al., 2016; Smith et al., 2016). Interestingly, pathologically hyperphosphorylated and/or aggregated forms tau (pTau) and reactive microglia spatially co-exist in the same “at risk” brain regions in tauopathies (Gerhard et al., 2004, 2006; Ishizawa and Dickson, 2001). However, it is unclear what drives inflammatory responses in amyloid-independent, non-AD tauopathies, specifically during prodromal stages. Multiple lines of evidence supports that there is ample opportunity for microglia to “sense” pTau as a “danger signal” in the extracellular milieu and set-off innate immune cascade. This includes, the presence of pTau in the cerebrospinal fluid (CSF) of patients with AD (Bateman et al., 2012), “prion”-like cell-to-cell propagation of misfolded tau (Kfoury et al., 2012), exosome-based release of hyperphosphorylated tau to the extracellular space (Saman et al., 2012), and uptake of pathological tau by neurons (Wu et al., 2013) or microglia (Asai et al., 2015). Presence of pTau within microglia are reported in brains of frontotemporal dementia and parkinsonism linked to chromosome 17 with mutations in the tau gene (FTDP-17T) at advanced stages of disease (Bellucci et al., 2011), and activation of inflammasome is also evident in FTLD-tau (Ising et al., 2019). Finally, expression of misfolded tau can trigger neuroinflammation (Zilka et al., 2009, 2012) and suppression pTau in rTg4510 mouse model of FTDP-17T (Wes et al., 2014) reduced inflammatory responses. We have previously demonstrated that genetic deficiency of tau in lipopolysaccharide (LPS)-treated Cx3cr1−/− mouse model of neuroinflammation (Maphis et al., 2015a) reduces neuroinflammation and neurotoxicity, suggesting that pTau may likely induce inflammatory responses in the brain. However, cell-autonomous effects of pTau-driven immune responses in tauopathies are still unknown.
RESULTS
RNA-sequence analysis suggests that human PHFs primes nuclear factor κB, chemokine, and interleukin-1β signaling clusters in human primary microglia
First, we performed an unbiased bulk RNA-sequencing (RNA-seq) analysis in human primary microglia treated with human PHFs (hPHFs) (Figure 1A; Table S1). A total of 708 mRNAs, 1,263 long non-coding RNAs (lncRNAs), and 17 circular RNAs (circRNAs) were differentially altered in hPHF-treated microglia (Figure S1). These two samples showed separate unsupervised clustering in the principal component analysis (Mendeley dataset, see data and code availability). We validated the RNA-seq results by qRT-PCR analysis for CD40, interleukin-1β (IL-1β), and nuclear factor κB (NF-κB), which all showed significantly increased expression in both RNA-seq and qRT-PCR analysis, whereas the NLRP1 showed significantly reduced expression (Figures 1D, S1A, and S1D). Next, we plotted the five top and bottom significantly up- and downregulated genes (mRNA), lncRNA, and circRNA and observed that mRNA transcripts of certain chemokine signaling (e.g., CCL15, CCL8, CCL4L2, etc.), lncRNAs for NADPH and inositol triphosphosphate receptors (e.g., lnc-NQO2-5 and lnc-ITPR2-3) and circRNA (e.g., chr15_73052748_73067438, “circADPGK”) were among the most significantly upregulated RNA species in hPHF-treated primary microglia compared to controls (Figures S1A–S1C). Similarly, mRNA transcripts of mitochondrial humanins (e.g., MTRNR2), lncRNA for transcription factors (e.g., TFEC-13), and circRNA (e.g., chr18_54423814_54426184, “circWDR7”) were among the most significantly downregulated RNA species with hPHF treatment (Figures S1A–S1C). Surprisingly, numerous genes in the NF-κB (e.g., NFKBIA, NFKBIE, NFKBIZ, NFKB1, and NFKB2) and IL1B signaling were among the most significantly upregulated mRNAs (Figure 1B). An Ingenuity Pathway Analysis (IPA) of mRNA data revealed that a significant number of gene transcripts were associated with granulocyte adhesion/migration, diapedesis, pattern-recognition receptors (PRRs), NF-κB, and tumor necrosis factor (TNF) canonical pathways (Figure S1E) (Mendeley dataset, see data and code availability). Similarly, we found that 49 gene transcripts overlapped with the top three functional and disease pathways (Figure S1F) (Mendeley dataset, see data and code availability). We also plotted significantly altered upstream regulators of altered genes from our RNA-seq analysis and observed that various regulators including TNF, PYCARD, and IL1RAP were altered after hPHF treatment (Mendeley dataset, see data and code availability). As a secondary analysis for the lncRNA datasets, we compared differentially expressed mRNAs with intronic, overlapping, or antisense to protein-coding gene’s lncRNAs. Significantly altered lncRNAs were chosen that display a Pearson correlation coefficient of more than +0.5 or less than −0.5 with the mRNAs for the gene from which they are expressed (that also were differentially expressed). A total of 122 lncRNAs met those criteria. We then used IPA to display the network of such lncRNAs (Figure 1C, gene name is shown instead of lncRNA ID, e.g., lncIL1A was displayed as IL1A in IPA networks in Figure 1C) and observed that lncRNA for IL1 and IL6 were among those significantly altered by hPHF treatment. Similarly, we compared differentially expressed circRNAs (17 in total) that were generated from exons of protein coding genes to the mRNA levels of their linear counterparts, and we calculated Pearson correlation coefficients. Due to the low number of altered circRNAs, no pathway analysis was possible. Instead, we plotted a heatmap of circRNAs with their parent genes and observed that certain circRNA both positively and negatively correlated with the expression of their target genes (Figure S1G). Finally, because of significant increase in IL1B expression, we assessed if markers in inflammasome network is altered. Instead of investigating individual inflammasomes, we chose to assess PYCARD (the gene for apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain or “ASC”), which is a common adaptor protein required for the assembly of many inflammasomes (Lamkanfi and Dixit, 2009). ASC-interacting network was assessed using STRING Database because of ASC being a common adopter for several Nod-like receptor (NLR) containing inflammasomes. Interestingly, many mRNAs (e.g., PSTIP1, TP53, MMS19, CDK9, etc.) and lncRNAs (including the lnc-PYCARD) in the ASC protein network were differentially altered in hPHF-treated human microglia compared to VEH-treated controls (Figures 1E–1G). Taken together, the RNA-seq analysis suggested that hPHF can induce significant upregulation of various RNA species in the PYCARD, NF-κB, chemokine, TNF-α, and IL-1β pathways.
Figure 1. hPHFs primes NF-κB, chemokine, and IL-1β signaling in human primary microglia.
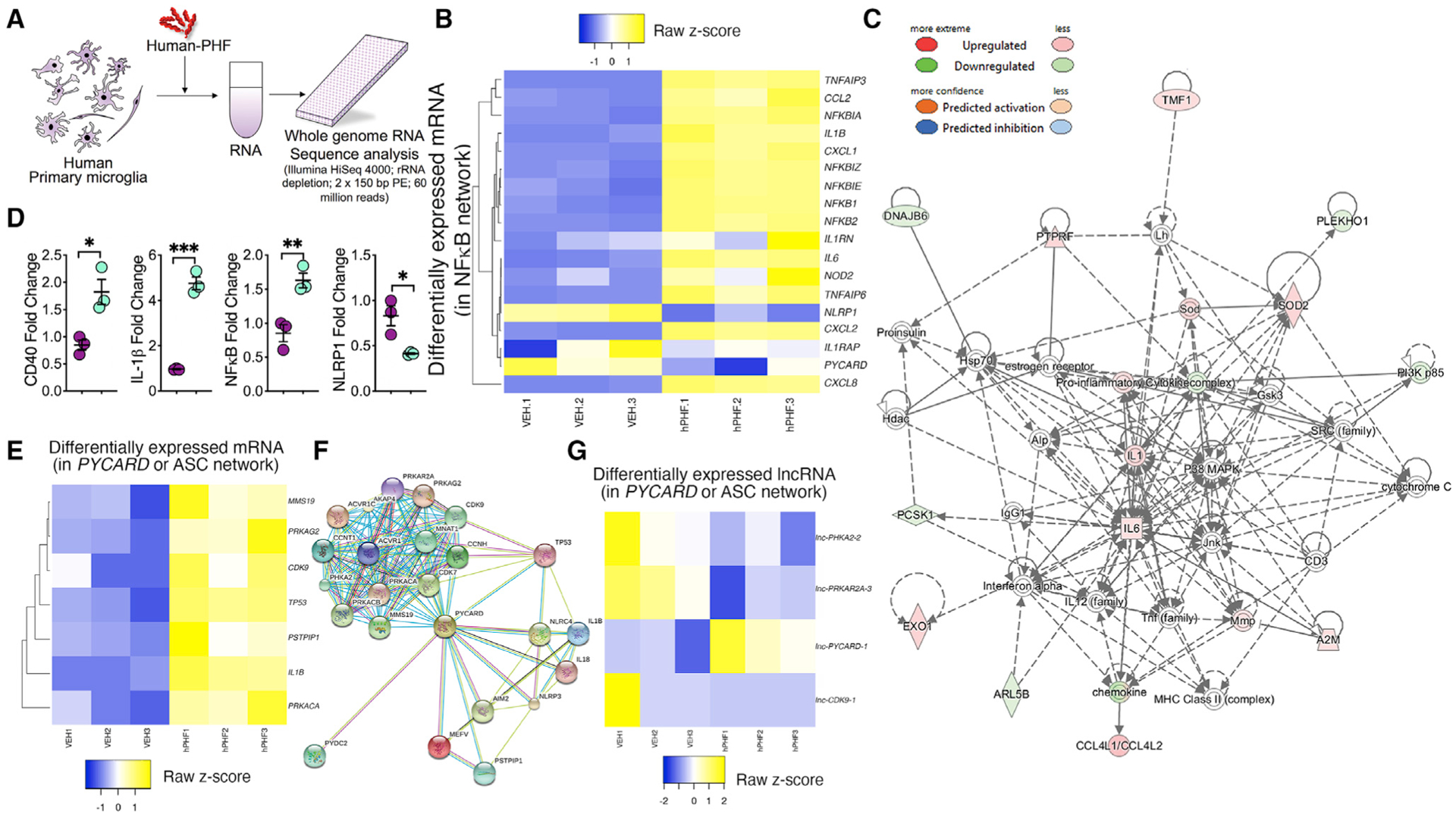
(A) Schematic of human primary microglia treated with hPHF (Sarkosyl insoluble paired helical filaments purified from human FTLD-tau brains) or VEH and harvested for RNA-seq analysis.
(B) Heatmap and unsupervised clustering of mRNA from human primary microglia treated with 2 μg/mL hPHF for 18 h. Genes in the VEH- and hPHF-treated conditions cluster closely together in three replicates of VEH or hPHF. Differentially down- and upregulated genes (mRNA) are pseudo-colored in blue and yellow, respectively. Note that genes in IL1B, chemokines, and NFKB1 pathways are differentially altered in hPHF-treated microglia compared to VEH-treated controls.
(C) IPA analysis shows the network map of 122 target genes from where the long non-coding RNA (lncRNA) is originated, and the mRNAs of these genes are differentially expressed (with Pearson correlation coefficient of greater than +0.5 or less than −0.5). Gene names are shown instead of lncRNA ID (e.g., lncIL1A was displayed as IL1A).
(D) RNA-seq (mRNA) validation by qRT-PCR for microglia treated with VEH or hPHF show increased fold change for CD40, IL1B, and NFKB1 and reduced fold change for NLRP1.
(E–G) Heatmap analysis of the differentially altered mRNA (E) and lncRNA (G) levels of the PYCARD (ASC) interacting genes (F) with VEH or PHF treatment. Data displayed as mean ± SEM, unpaired Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.005, n = 3 (D).
See also Figure S1, Table S1, and Mendeley dataset (see data and code availability).
Microglial cells take up pathological tau secreted from neuronal cells and lead to expression and activation of inflammasome-related genes
To assess whether altered inflammasome activation is triggered by tau phosphorylated at threonine 231 and serine 235 (AT180 antibody site, the earliest biomarker of tau pathology) (Ashton et al., 2021), we treated BV2 microglial cells with conditioned media (CM) derived from Neuro2a (N2a) cells expressing non mutant wild-type (WT) human tau with three microtubule-binding repeats (0N3R-WT) or 0N3R human tau with dual phosphorylation-mimicking mutations (T231D and S235D, mimics phosphorylation in AT180 site) (Figure 2A). AT180 site phosphorylation was previously shown to affect microtubule-binding and other functions of tau (Bhaskar et al., 2005; Eidenmüller et al., 2000). After 24 h, BV2 cells showed significant uptake and internalization of 0N3R-T231D/S235D tau compared to 0N3R-WT tau (Figures 2B–2D). Interestingly, 0N3R-T231D/S235D tau-internalized BV2 cells showed significant increase in the ASC, NLRP3, and cleaved-IL-1β levels and a 1.5-fold or higher increase in the levels of pro-IL-1β (Figures 2C and 2D). A modest increase in the mRNA levels of ASC and IL-1β, but not that of nucleotide oligomerization domain (NOD)-like receptor (NLR) family pyrin domain containing 3 (NLRP3), were also observed in N2a CM-treated BV2 cells (Figure 2E), suggesting that human tau containing N2a CM can prime BV2 cells to express certain inflammasome-related mRNAs and proteins. To assess if elevated levels of IL-1β, NLRP3, and ASC mRNA can form multi-protein inflammasome complex in response to pTau within BV2 cells, we utilized an ASC-mCerulean macrophage reporter cell-line (Heneka et al., 2013) and murine primary microglia, and treated them with N2a CM containing human tau (Figure 2F). As expected, the percentage of macrophages showing inflammasome complex (ASC-specks) was significantly higher following 0N3R-WT and 0N3R-T231D/S235D tau expressing N2a CM treatment compared to controls (Figures 2G, top panel, and 2H). ASC-specks were also discernable in mouse primary microglia treated with either 0N3R-WT or 0N3R-T231D/S235D tau containing N2a CM and analyzed by Cellomics high-content microscopy (Figures 2G, bottom panel, and 2I), suggesting that N2a CM with mutant human tau could induce inflammasome assembly in both macrophages and primary microglial. However, it is unclear whether the pTau is present as a soluble species or associated with exosomes, as it has been reported by multiple studies (Asai et al., 2015; Saman et al., 2012). To assess the effects of exosome-associated pTau in inducing inflammasome assembly, we purified exosomes from 0N3R-T231D/S235D tau-transfected N2a CM and confirmed the presence of tau via immune-electron microscopy (Figure S2A). We only tested in 0N3R-T231D/S235D tau because it showed the highest effects in Figures 2D and 2E. As expected, Tau12+ human tau was enriched in exosomes secreted by N2a cells (Figures S2A–S2F). Notably, BV2 cells treated with purified exosomes containing 0N3R-T231D/S235D tau showed significant increases in ASC, NLRP3, and pro-IL-1β protein levels (Figures S3A and S3B), which also increased speckling of ASC in macrophages (Figure S3C). Pharmacologically blocking the exosome secretion with GW4869 significantly reduced CD81+ exosomes and modestly reduced 0N3R-T231D/S235D tau in exosomes (Figures S3E and S3F). However, NLRP3 levels were unaltered in BV2 cells (Figures S3G and S3H), suggesting that non-exosome-associated tau (soluble tau) might be the major source of priming and activation of inflammasomes in myeloid cells. Because this interpretation was done with a small group size, current observation needs to be confirmed in an independent study.
Figure 2. Uptake of N2a-derived pTau by microglia leads to expression and activation of inflammasome-related genes.
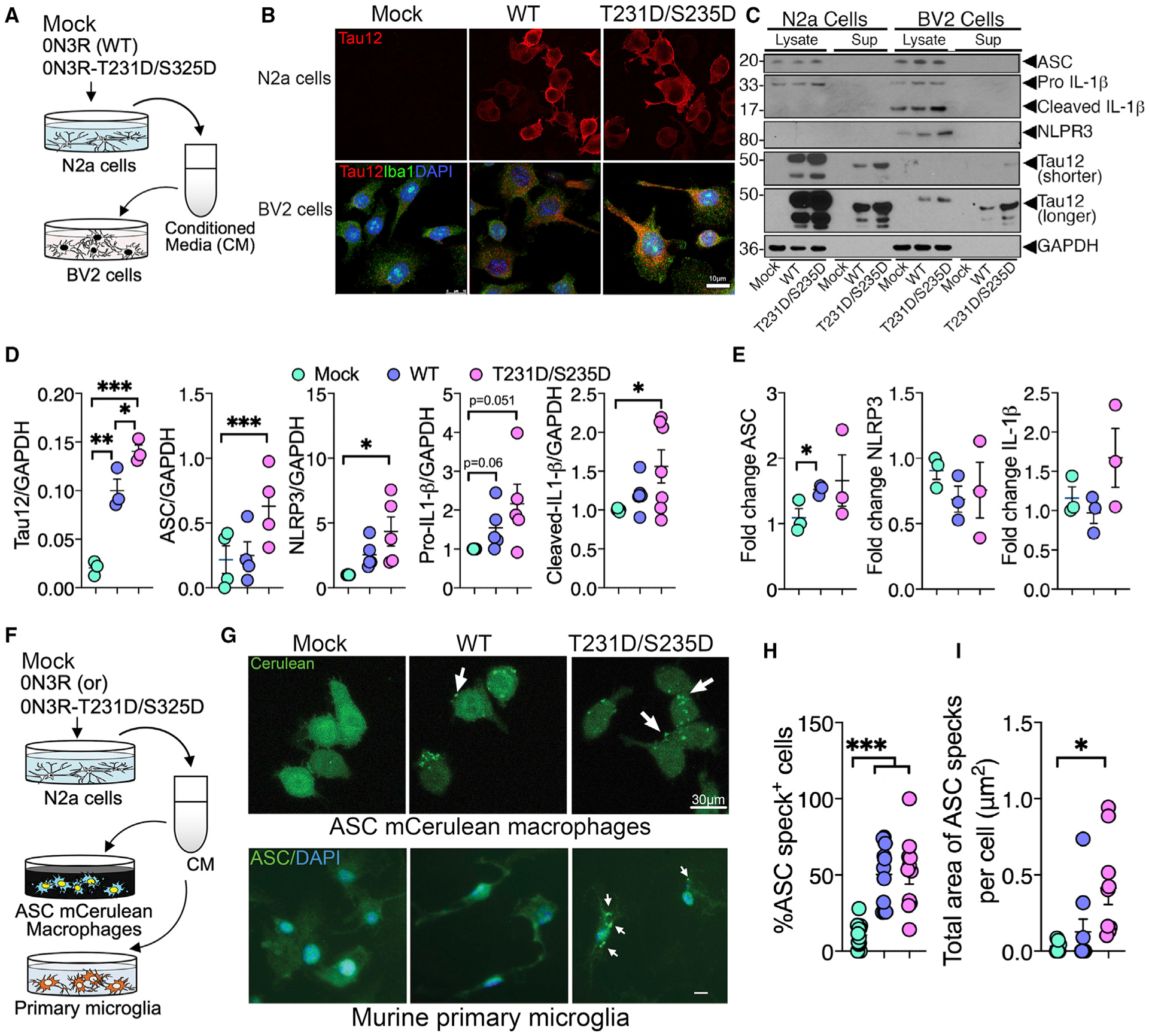
(A and B) LPS-primed (1 μg/mL LPS for 6 h) BV2 cells show internalization of human tau (Tau12, red; Iba1, green; DAPI, blue) when incubated with conditioned media (CM) from N2a cells expressing phosphorylation-mimicking tau (0N3R-T231D/S235D) for 24 h compared to CM from vector-transfected (“Mock”) or wild-type tau (0N3R-WT) transfected N2a cells (Tau12, red). Scale, 10 μm.
(C and D) Western blot and quantification of BV2 cell lysates shows significant uptake of human tau (Tau12–GAPDH ratio) and increased ASC, NLRP3, and cleaved(c)-IL-1β in BV2 cells treated with CM from WT (0N3R)- and (0N3R-T231D/S235D)-expressing N2a cells.
(E) qRT-PCR analysis of unprimed BV2 cells shows significantly elevated ASC (Pycard) mRNA when treated with CM from WT (0N3R)- and/or (0N3R-T231D/S235D)-expressing N2a cells. Both shorter and longer exposed blots for Tau12 are included.
(F–I) Immortalized mouse macrophages expressing ASC-mCerulean (via epifluorescence) and murine primary microglia (via Cellomics) show significant increase in the number of intracellular (white arrows) ASC-specks when treated with CM from N2a cells expressing 0N3R-T231D/S235D tau compared to mock control. Scale, 30 μm (top) or 10 μm (bottom).
Data displayed as mean ± SEM, one-way ANOVA with Tukey multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.005, n = 3–7 (C–E); n = 6–12 (G–I).
See also Figures S2 and S3 and Data S1.
Purified PHFs from human FTLD-Tau or rTg4510 mice brains induce inflammasome activation in an ASC-dependent manner
Because it is possible that many factors present in CM or exosomes could act as potential DAMPs to induce inflammasome priming and activation and the possibility that certain anti-inflammatory cytokines in CM may also potentially inhibit inflammasomes (e.g., IL-1β response in Figure 2E), we next assessed direct effects of purified hPHFs from the brains of rTg4510 mice or a patient with FTLD-tau (Figure 3A) could activate inflammasomes. Purified PHFs from human FTLD-Tau brain appeared as classical twisted filaments and were positive for human tau-specific antibody Tau12 (Figure 3B). The LPS-primed ASC-mCerulean macrophages responded to both hPHFs and mouse PHFs (mPHFs) and induced ASC specks within 30 min of treatment (Figures 3C–3E; Video S1). This response was comparable to ATP or nigericin treatment, which are potent activators of inflammasomes and were used as positive controls (Figure 3D). To quantitate the hPHF/mPHF-responsive ASC-specks, we utilized Amnis ImageStream flow cytometry and microscopy technique. ImageStream quantification revealed a significant increase in the percentage of LPS-unprimed macrophages showing ASC-specks (inflammasome assembly in the absence of LPS) with mPHF (and an increased trend with hPHF) treatment compared to the control group (Figure 3F). We confirmed the assembly of ASC-specks by hPHF in murine primary microglial cells (Figure S3H). We next validated functional inflammasome assembly in CD11b+ BV2 cells with hPHF treatment via FAM-FLICA caspase 1 assay. We observed a 2-fold increase in the percentage of FLICA-caspase-1+ macrophages with hPHF treatment (Figure 3G). As expected, like ATP and nigericin (30 min treatment), 18 h treatment with hPHF increased active IL-1β secretion in LPS-unprimed ASC-mCerulean macrophages (Figure 3H). Strikingly, PHF treatment also resulted in a significant upregulation of IL-1β mRNA expression (Figure 3I) in LPS-unprimed ASC-mCerulean macrophage cells, suggesting that priming of macrophages with LPS is not necessary to induce IL-1β expression by PHFs. Finally, small interfering RNA (siRNA)-mediated knockdown of ASC, caspase 1 (in both cell lysate and supernatant), or NLRP3 (in cell lysate), significantly inhibited active IL-1β following hPHF-treatment in ASC-mCerulean macrophages (Figures 3J and 3K). These results suggest that PHFs can induce inflammasome assembly, activation of caspase 1, and upregulate ASC and caspase 1 levels.
Figure 3. FTLD-Tau or rTg4510 mouse brain-derived PHFs induce inflammasome activation in an ASC-dependent manner.
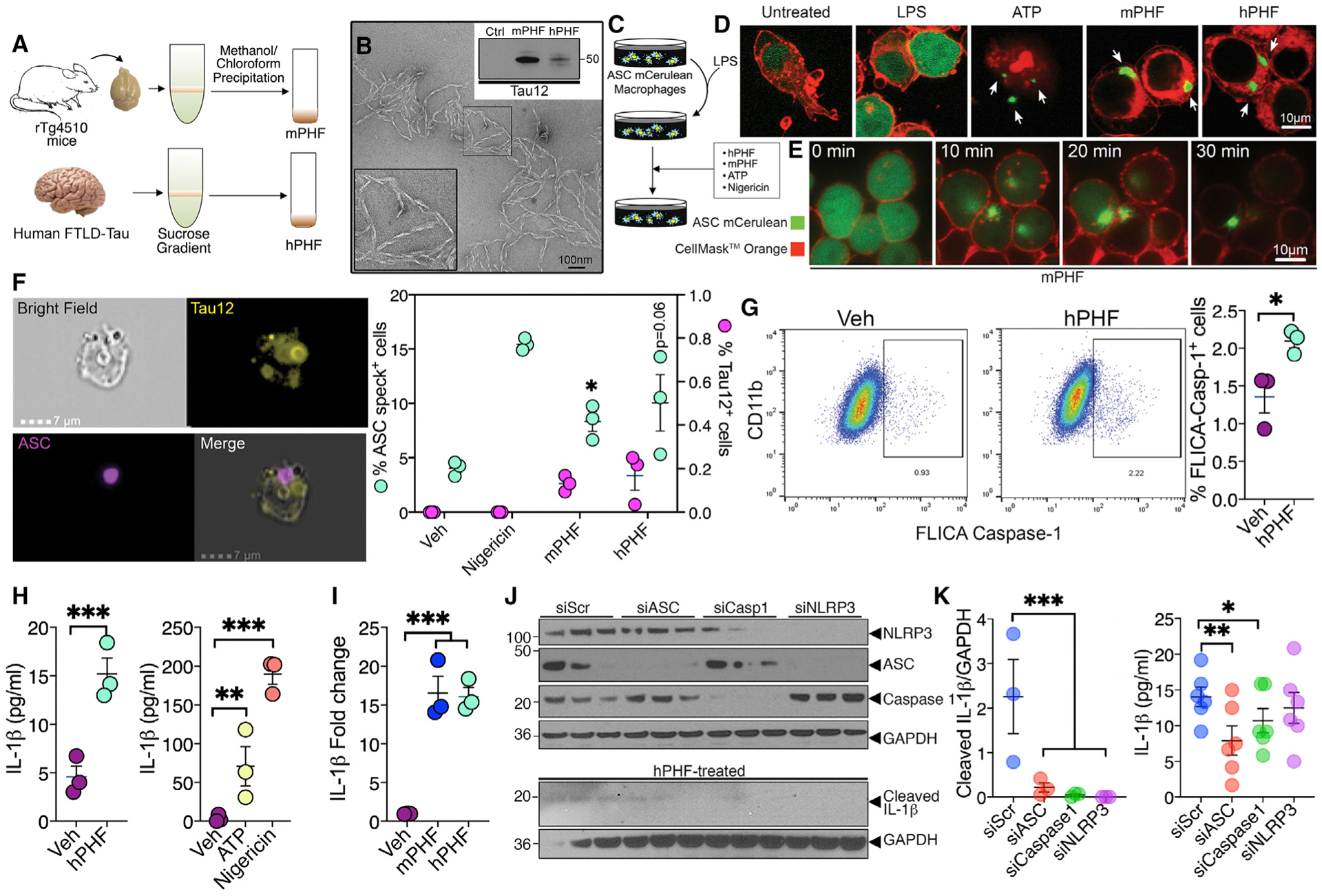
(A) Schematic of the Sarkosyl insoluble assay followed by sucrose gradient centrifugation and methanol/chloroform precipitation for the purification of mouse PHFs (mPHFs) and human PHFs (hPHFs) or the vehicle (VEH).
(B) Transmission electron microscopic image shows helical conformation of purified hPHF. Both mPHFs and hPHFs were reactive to human tau (Tau12) antibody (western blot inset). Scale, 100 nm.
(C–E) LPS-primed ASC-mCerulean macrophages show speckling of ASC (arrows) similar to ATP (positive control) when treated with 2 μg/mL h/m-PHF within 30 min. Scale, 10 μm.
(F) Amnis ImageStream flow-cytometry and microscopic analysis shows significant increase in the percentage of cells with ASC-specks following PHF treatment in unprimed ASC-mCerulean macrophages. Scale, 7 μm.
(G) Flow-cytometric based FAM-FLICA caspase 1 assay shows >2-fold increase in the percentage of CD11b+/FLICA-Caspase-1+ BV2 cells with hPHF treatment.
(H) ELISA analysis shows 3-fold increase in the secreted (active) IL-1β in the cell supernatant of hPHF-treated (2 μg/mL for 18 h), unprimed ASC-mCerulean macrophages compared to vehicle treatment. ATP- and nigericin-induced maturation of IL-1β shown as positive controls.
(I) Unprimed and mPHF-/hPHF-treated ASC-mCerulean macrophages show significant increase in IL-1β mRNA.
(J and K) Unprimed, hPHF-treated ASC-mCerulean macrophages show significantly reduced levels of cleaved IL-1β in the supernatant with siRNA against ASC (siASC), caspase 1 (siCaspase 1), or NLRP3 (siNLRP3) compared to siScramble (siScr) by western blot (J and left panel in K) and ELISA analyses (right panel in K).
Data displayed as mean ± SEM, unpaired Student’s t test (F, right), one-way ANOVA with Tukey multiple comparison test (H, right, I, and K–L), *p < 0.05, ***p < 0.0005 with n = 3 (F–I), n = 3–6 (K).
Suppression of human tau reduces levels of inflammasome markers in rTg4510 mice
Based on the role of hyperphosphorylated (in Figure 2) and aggregated tau (Figures 1 and 3) priming microglial cells and driving inflammasome gene expression, next we assessed the effects of suppression of pTau on IL-1β, ASC, and NLRP3 in rTg4510 mouse model of FTDP-17T expressing human tau with P301L mutation (Santacruz et al., 2005). The rTg4510 mice displayed age-dependent upregulation of NLRP1a/1b, ASC, IL-1α, IL-1β, and IL-18 mRNA levels from 4.5–9 months of age (Figure S4). When the human P301L tau expression was suppressed from 3 months until 6 months of age using doxycycline (DOX) (Figure 4A), it not only reduced mRNA for human tau (MAPT) and protein levels of total and pTau (Figures S5A–S5D), but also decreased the mRNA levels of ASC (Pycard), NLRP3, and IL-1β (Figure 4B). The protein levels of ASC, NLRP3, and IL-1β (Figures 4C and 4D) also showed modest to significant reduction following suppression of P301L tau with DOX. Coincidentally, the Iba1+ microglia appeared more ramified (resting) with thinner processes in the hippocampus of DOX-treated rTg4510 mice (Figures S5E and S5F). Further, the total Iba1+ area was significantly reduced following DOX treatment from 3–6 months (Figure S5F). On the other hand, human tau suppression by DOX from 5 until 9 months of age (Figure 4E) did not alter mRNA of NLRP3 or IL-1β but modestly reduced the mRNAs of ASC (Figures 4E and 4F). Strikingly, the suppression of human tau with DOX reduced protein levels of AT80+ (pThr231/pS235) and total human tau (Tau12), ASC, NLRP3n, and IL-1β in the 5- to 9-month-old group (Figures 4E–4H). Notably, suppression of human tau with DOX reduced tau mRNA levels (Figures 4F and S5G–S5I). Iba1+ microglia appeared ramified and less reactive, with ablated immunoreactivity in 5- to 9-month-old DOX groups (Figures S5E and S5F). Finally, based on a recent report (Xu et al., 2019) that DOX may directly inhibit inflammasome pathway, we used vaccine-based reduction of pTau as an alternative method to assess its effects on ASC and NLRP3 expression. By exploiting our previously reported tau-targeted vaccine strategy (Maphis et al., 2019), which uses Qβ bacteriophage virus-like particle (VLP) conjugated to 16-mer human tau peptide encompassing phospho-Thr181 (175TPPAPKpTPPSSGEGGC190 or Qβ-pT181) as a vaccine candidate, we confirmed that reduction of high-molecular weight AT180+ (pS231/pS235+) tau also reduced NLRP3, ASC, and total human tau in 4.5-month-old rT4510 mice vaccinated with Qβ-pT181 compared to Qβ controls (Figures S6A and S6B). Taken together, these results suggest that human FTLD-Tau (with P301L mutations) can induce significant upregulation of inflammasome genes. Conversely, suppression of human tau in rTg4510 mice can significantly reduce the expression of ASC, as well as NLRP3 and IL-1β even after the first appearance of pTau in these mice at ~2.5 months of age (Santacruz et al., 2005).
Figure 4. Reduction of P301L tau and ASC reduces inflammasome markers and tau pathology in rTg4510 and hTau/ASC−/− mice, respectively.
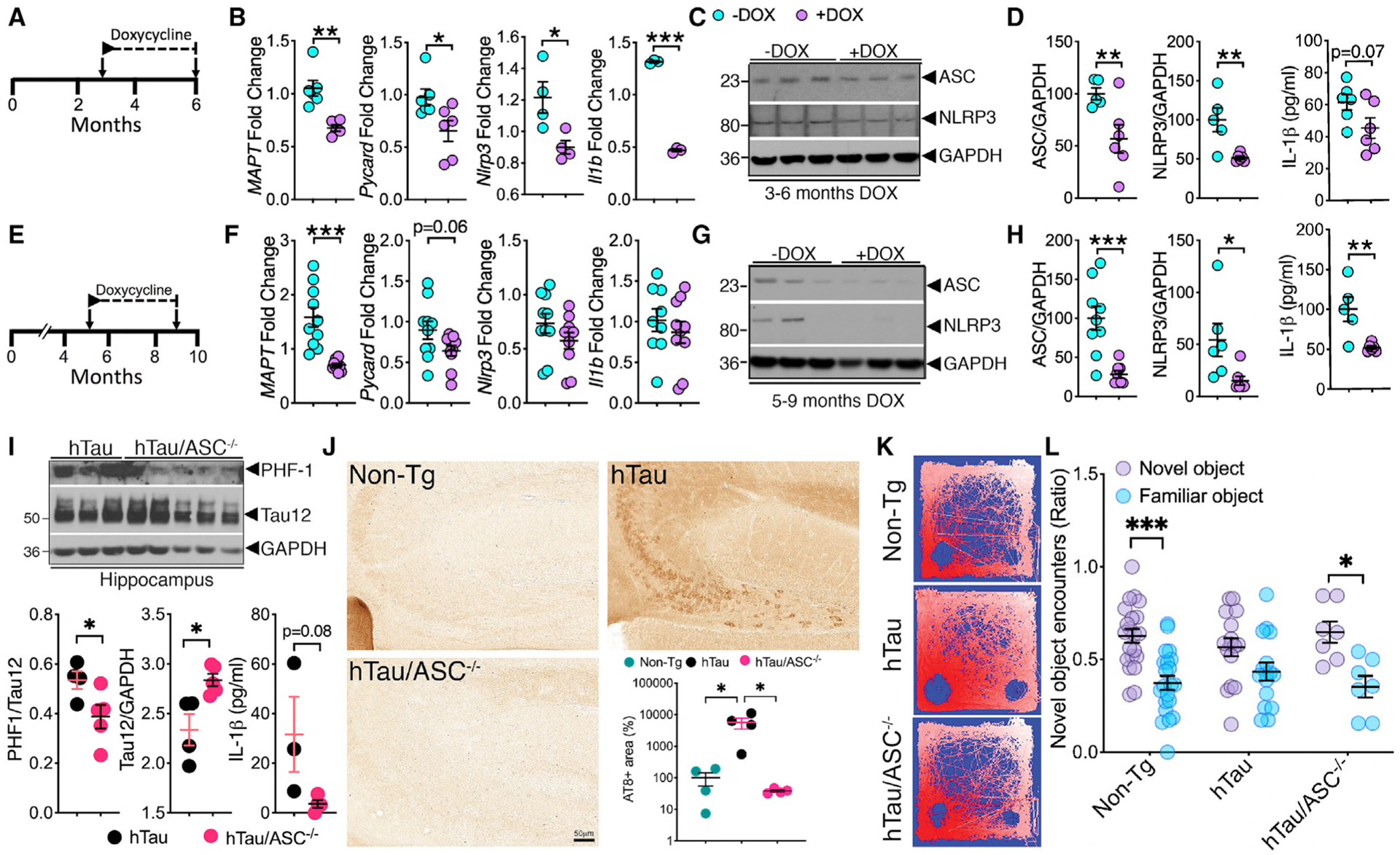
(A–D) qRT-PCR analysis show DOX treatment from 3–6 months in rTg4510 mice significantly reduced the mRNA expression of ASC, NLRP3, and IL-1β; protein levels of ASC, NLRP3, and modest reduction in IL-1β.
(E–H) DOX treatment from 5–9 months in rTg4510 mice showed modest reduction in mRNA for only ASC and significant reduction in protein levels of ASC, NLRP3, and IL-1β in the hippocampi.
(I) Western blot analysis and quantification for 6-month-old hTau/ASC−/− mice show significantly reduced PHF1+ (phosphorylated at ser396/ser404) in the hippocampi compared to age-matched hTau mice. ELISA analysis shows 10-fold reduction in the IL-1β levels in the hippocampi of 6-month-old hTau/ASC−/− mice.
(J) Significant reduction in the percentage of AT8+ area in the CA3/dentate gyrus of 6-month-old hTau/ASC−/− mice compared to age-matched hTau mice. Scale, 50 μm.
(K and L) Novel object recognition test shows hyperactivity and equal object preference for 6-month-old hTau mice (see representative heatmap of exploration in K) and significant impairment in distinguishing novel object from familiar object (ratio of encounters with novel object within 5 min) on the test day. This is rescued in 6-month-old hTau/ASC−/− mice.
Data displayed as mean ± SEM, unpaired Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.005, n = 4–6 (B and D); n = 5–9 (F and H); n = 3–5 (I), or one-way ANOVA followed by Tukey multiple comparison test, *p < 0.05, n = 4 (J); two-way ANOVA followed by Sidak’s multiple comparison test, *p < 0.05, ***p < 0.005, n = 21 for non-Tg; n = 16 for hTau; n = 7 for hTau/ASC−/− (K and L).
See also Figures S4, S5, S6, and S7 and Data S1.
Microglia-restricted deficiency of ASC reduces IL-1β maturation and tau pathology and improves cognitive function in hTau mouse model of tauopathy
ASC is a key adaptor protein necessary for the assembly of multi-protein inflammasome complex (Lamkanfi and Dixit, 2009), and our results suggest that pTau alters the expression and functional activation of ASC. Therefore, we next tested the effects of genetic deficiency of ASC in LPS-induced inflammation and hTau genomic mouse model of tauopathy. We have previously demonstrated that systemic LPS administration leads to tau hyperphosphorylation and neurodegeneration (Bhaskar et al., 2010; Maphis et al., 2015a). IL-1R1−/− and Mapt−/− mice were resistant to inflammation-induced hyperphosphorylation and/or neurodegeneration, respectively (Bhaskar et al., 2010; Maphis et al., 2015a). Here, we observed that ASC−/− mice showed significantly reduced pTau (phosphorylated at T231-AT180 site) following LPS treatment (Figures S7A and S7B). Notably, 6-month-old hTau/ASC−/− mice, which were deficient for ASC as expected (Figures S7C and S7D), displayed significantly reduced pTau (phosphorylated tau at S396/S404-PHF1 site, Figure 4I; and phosphorylated tau at S202/T205-AT8 site, Figure 4J) in the hippocampus compared to age-matched hTau (ASC+/+) mice. This decrease in pTau occurred despite significantly increased total tau (Tau12) levels in hTau/ASC−/− mice (Figure 4I). Most importantly, ASC deficiency in hTau mice showed >5-fold reduced IL-1β (Figure 4I). Overall, reduced tau pathology and IL-1β were accompanied with less hyperactivity and improved delay-dependent memory as measured in the novel object recognition task (NOR) in hTau/ASC−/− mice (Figures 4K, 4L, and S7E).
Next, to determine if pTau-driven NLRP3/ASC- inflammasome activation is cell-autonomous to myeloid cells including microglia (Figure 5A), we generated hTauCX3CR1Cre+ASCf/f mice. These mice express Cre recombinase under the CX3CR1 promoter and thus genetically delete floxed allele of ASC in all myeloid cells, including microglia in the brain. First, we confirmed complete deficiency of ASC in microglia purified from hTauCX3CR1Cre+ASCf/f mice by western blot and qRT-PCR analysis (Figures 5B–5D). However, no significant alterations in IL-1β mRNA expression were noted (Figure 5D). Strikingly, western blot analysis of the hippocampal lysates from 8-month-old hTauCX3CR1Cre+ASCf/f mice showed significant decrease in ASC level compared to age-matched hTau mice (Figures 5E and 5F), suggesting that the majority of ASC is expressed by microglia in the hippocampus of hTau mice. Previous reports have suggested ASC upregulation can occur in neurons (Meng et al., 2014), astrocytes (Couturier et al., 2016), and other cells in the CNS under inflammatory conditions (Chakraborty et al., 2010), which does not seem to be the case in the hippocampi of hTauCX3CR1Cre+ASCf/f mice. Levels of cleaved IL-1β and human tau phosphorylated at AT8, AT180, and PHF-1 sites were all significantly reduced in hTauCX3CR1Cre+ASCf/f mice compared to age-matched hTau control mice (Figures 5E and 5F). Interestingly, in the Morris water maze (MWM) test, the 8-month-old hTau mice displayed impairment in spatial learning, which was significantly improved in hTauCX3CR1Cre+ASCf/f mice and these were comparable to age-matched non-transgenic controls (Figure 5G). In probe trials of the MWM test (tested after removing the platform), hTauCX3CR1Cre+ASCf/f mice were able to spend significantly more time in the target quadrant versus other quadrants (as non-transgenic controls), suggesting a better spatial navigation memory compared to hTau mice, which were cognitively impaired (Figures 5G and 5H). Notably, there were no significant differences in the swim speed or the total distance traveled by non-transgenic, hTau, or hTauCX3CR1Cre+ASCf/f mice (Figure S7E–S7G). These results suggest that myeloid cell-/microglia-specific deficiency of ASC is sufficient to prevent pTau-induced inflammasome activation, neuroinflammation, and exacerbation of tau pathology in the hTau mouse model of FTLD-Tau.
Figure 5. Myeloid cell-restricted deletion of ASC reduces IL-1β maturation and tau pathology and improves cognitive function in hTau mouse model of tauopathy.
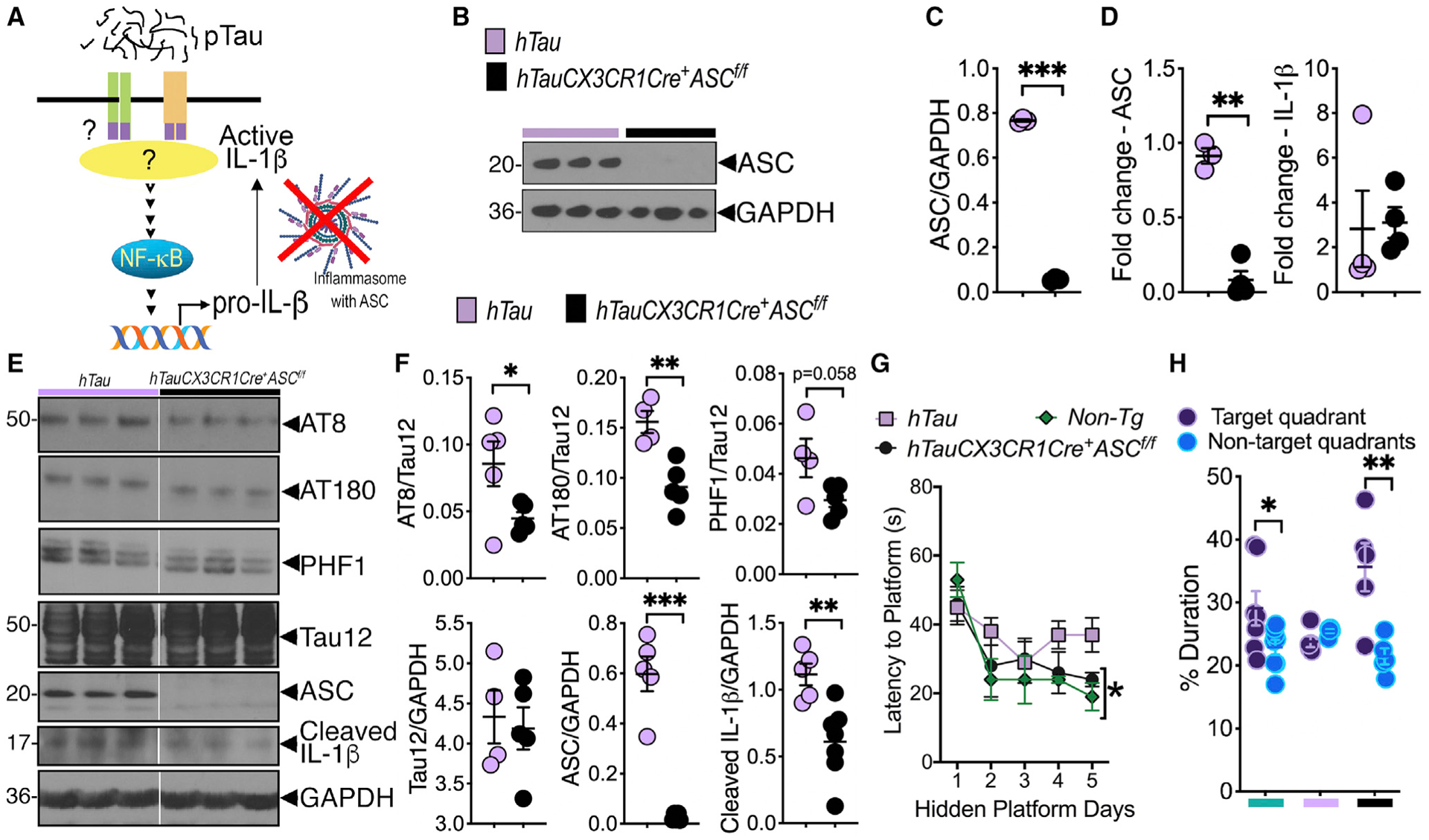
(A) Working model shows potential mechanism of pTau-induced ASC-inflammasome activation/IL-1β maturation, which can be blocked by myeloid/microglial cell-specific deletion of ASC.
(B and C) Western blot analysis and quantification shows complete deletion of ASC in purified microglia from hTauCX3CR1Cre+ASCf/f mice.
(D) Microglia-restricted deletion of ASC shows significant reduction of ASC, but not IL-1β, mRNA expression in the brains of hTauCX3CR1Cre+ASCf/f mice compared to hTau controls.
(E and F) Western blot analysis and quantification of 8-month-old hTauCX3CR1Cre+ASCf/f mice shows reduced tau pathology (on AT8, AT180, and PHF1 sites), cleaved (activated) IL-1β, and ASC in the hippocampus compared to age-matched hTau controls.
(G) Morris water maze (MWM) analysis shows improved spatial learning to reach hidden platform by day 5 in 8-month-old hTauCX3CR1Cre+ASCf/f mice that was comparable to non-transgenic mice. This learning measure was impaired in age-matched hTau mice.
(H) Probe trial analysis of MWM test shows hTauCX3CR1Cre+ASCf/f mice spent significantly more time in the target quadrant compared to average duration in all other non-target quadrants.
Data displayed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.005, unpaired Student’s t test, n = 3–5 (B–D), n = 4–7 (E and F), or two-way ANOVA followed by Tukey’s multiple comparison test, n = 5–10 (G), multiple t test, n = 5–10 (H).
See also Figures S7F and S7G and Data S1.
pTau-induced NF-κB priming is MyD88 dependent
Proteopathic tau seeds have been shown to utilize heparan sulfate proteoglycans (HSPGs) on the cell surface/extracellular matrix to cross-seed pTau between neurons (Holmes et al., 2013). Studies have also suggested that damaged HSPGs are capable of activating toll-like receptors (TLRs, specifically TLR4) (Akbarshahi et al., 2011). Moreover, engagement of TLRs by disease-relevant protein aggregates has been reported by multiple studies (Caplan and Maguire-Zeiss, 2018; Costello et al., 2015; Gambuzza et al., 2014; Griffioen et al., 2018; Kang et al., 2016). We reasoned that pTau-mediated NF-κB priming and NLRP3-ASC inflammasome activation might be driven by the engagement of TLRs/PRRs. To explore this possibility, we first reviewed our RNA-seq data to determine whether TLR/MyD88/NF-κB pathway was altered in human microglia treated with hPHFs. Strikingly, mRNAs for two key TLRs (TLR2 and TLR8), MyD88, p62 (autophagy adaptor protein) and IRAK2/3, were also significantly upregulated in hPHF-treated human primary microglial cells (Figures 6A and 6B). Instead of testing individual TLRs/PRRs, we focused on myeloid differentiation primary response 88 (MyD88), which is a common adaptor protein for many TLRs, PRRs, and IL-1Rs (Warner and Núñez, 2013) (Figures 6A and 6C). We knocked down MyD88 with siRNA in bone-marrow-derived macrophages (BMMs) and stimulated them with mPHFs for 18 h and assessed the activation of NF-κB. We observed >70% MyD88 knockdown (Figures S8A and S8B). As expected, the levels of phospho(S536)-p65 (NF-κB) was significantly reduced in siMyD88 BMMs compared to siScr (Figures S8A and S8B). Next, to assess the functional role of MyD88, we crossed MyD88f/f mice to CD11bCre mice and generated CD11bCre+MyD88f/f mice. These mice express Cre recombinase under the CD11b promoter and delete floxed allele of MyD88 specifically in myeloid cells (Yu et al., 2014). As expected, fluorescence-activated cell sorting (FACS) purified microglia from CD11bCre+MyD88f/f mice (Figure S8C) showed significantly reduced MyD88 mRNA compared to MyD88f/f microglial cells (Figure 6D). Stimulation of CD11bCre+MyD88f/f microglia with LPS (1 ng/mL for 12 h) showed a significant reduction in the secreted MIP1α, IL-12, IL-6, and TNF-α in the cell supernatant compared to LPS-treated MyD88f/f microglia (Figures S8C and S8D), suggesting that microglia-restricted deletion of MyD88 show reduced cytokine secretion in response to TLR4 agonist LPS. Finally, to determine the effect of MyD88 deficiency in hTau mice, CD11bCre+MyD88f/f mice were bred to hTau mice to generate hTauCD11bCre+ MyD88f/f mice. As expected, the MyD88 level in the total hippocampal lysates was significantly reduced in hTauCD11bCre+ MyD88f/f mice compared to hTau mice (Figures 6E and 6F). Importantly, tau phosphorylation at both AT8 and PHF1 sites were significantly reduced in the hippocampus of hTauCD11bCre+MyD88f/f mice compared to age-matched hTau mice (Figures 6E and 6F). Most importantly, both IL-1β mRNA and protein levels were significantly reduced in the hippocampus of 6-month-old hTauCD11bCre+MyD88f/f mice (Figure 6G). The reduction in IL-1β was due to reduced levels of phosphorylated-p65 (NF-κB), pro-IL-1β, and increased levels of the inhibitor of κB (IκB) (Figures S8E and S8F). Finally, to confirm that pTau can directly induce NF-κB activation, we treated BMMs with hPHF and assessed nuclear localization of total p65 (a component of NF-κB) by Cellomics high-content automated microscopy. A significant number of macrophages with nuclear p65 were observed in hPHF-treated cells compared to those treated with vehicle control (Figures S9G and S9H). Taken together, these results suggested that pTau-induced priming of NF-κB, which is cell autonomous to myeloid cells (including microglia), is MyD88 dependent.
Figure 6. p-Tau-induced NF-κB priming and inflammasome activation is MyD88 dependent.
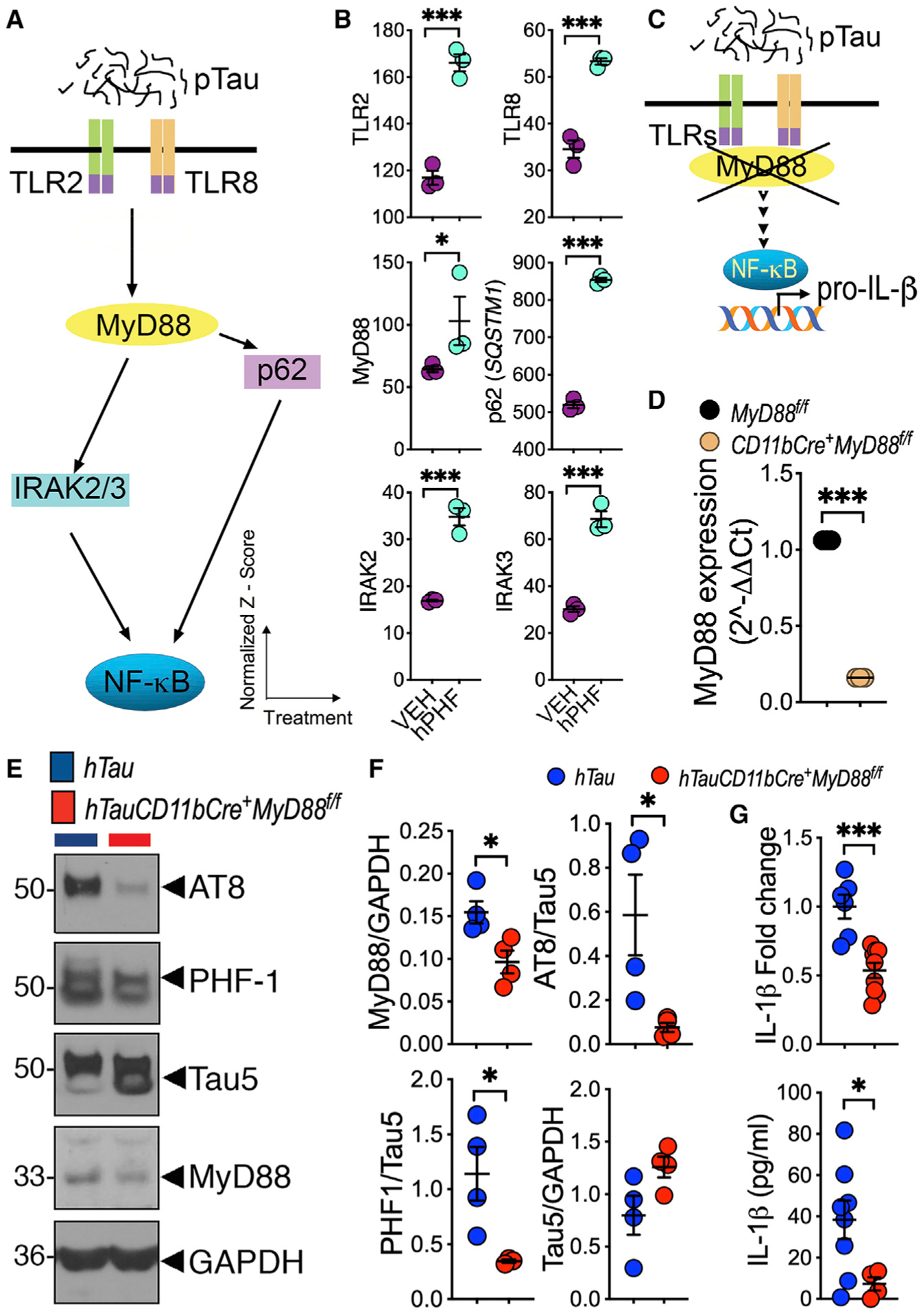
(A) A working model depicting the potential pathway by which pTau upregulates TLR2/8, MyD88, p62, and IRAK2/3 to lead to NF-κB activation.
(B) Significant upregulation of TLR2, TLR8, MyD88, p62, IRAK2, and IRAK3 mRNA in human primary microglia treated with 2 μg/mL hPHF for 18 h. Data shown are normalized Z score versus treatments (VEH or hPHF).
(C) Working model shows that microglia-restricted deletion of MyD88 blocks pTau-induced NF-κB activation and IL-1β expression.
(D) Purified microglia from CD11bCre+MyD88f/f mice shows significantly reduced MyD88 expression.
(E and F) Six-month-old hTauCD11bCre+MyD88f/f mice show reduced MyD88 and AT8/PHF1+ pTau in the hippocampus compared to age-matched hTau controls.
(G) Both IL-1β mRNA (qRT-PCR) and cleaved IL-1β (ELISA) are significantly lower in 6-month-old hTauCD11bCre+MyD88f/f mice compared to age-matched hTau mice.
Data displayed as mean ± SEM, *p < 0.05, ***p < 0.005, unpaired Student’s t test, n = 3 (B–D); n = 4 (E–G).
Evidence of inflammasome activation in human FTLD-tau
Next, to determine whether pTau could act as damage-associated molecular patterns (DAMPs) and induce microglial activation, we assessed levels of pTau, ASC, and the expression and activation of IL-1β. As previously reported (Hernández and Avila, 2007; Lee et al., 2001), temporal cortices of all FTLD-tau samples showed elevated levels of Sarkosyl-insoluble tau (Ksiezak-Reding et al., 1994) compared to age-matched healthy controls (Figures 7A and 7B). Levels of IL-1β, ASC, and Iba1 were also elevated in FTLD-tau samples, with PiD showing significant elevation of both ASC and IL-1β (Figures 7C and 7D). However, levels of pro- or cleaved-caspase 1 or NLRP3 were not altered (Figures 7C, 7D, S9A, and S9B). To confirm the elevated ASC in FTLD-Tau samples, we performed immunofluorescence analysis and observed numerous ASC-positive microglia that were also Iba1-positive and TMEM119-positive, specifically in the temporal white matter of FTLD-tau (tauopathy) samples compared to healthy controls (Figures 7E and S9C). In some instances, ASC appeared as a single bright speck inside the cells or multiple specks in the extracellular space in tauopathy samples, indicating the likelihood of inflammasome assembly (Figure 7E) and spreading, as previously described (Venegas et al., 2017). To confirm this possibility in tauopathies, we quantified secreted ASC-specks (ASC-positive inflammasomes that are ~5 μM wide) (Venegas et al., 2017) in the human healthy controls and tauopathy CSF using Amnis ImageStream imaging flow cytometry. We observed that extracellular ASC-specks can be detectable in the patient’s CSF and their numbers were significantly higher in tauopathy compared to healthy controls (Figures 7F and 7G). Strikingly, the tau phosphorylated at threonine 181 (pT181) levels were also higher in the CSF of patients with tauopathy compared to controls (Figure 7H). These results suggest that inflammasome activity and IL-1β levels are elevated in the brains and CSF of human tauopathies and complements with increased level of pTau.
Figure 7. Sarkosyl insoluble tau and inflammasome activation are evident in human FTLD-Tau brain.
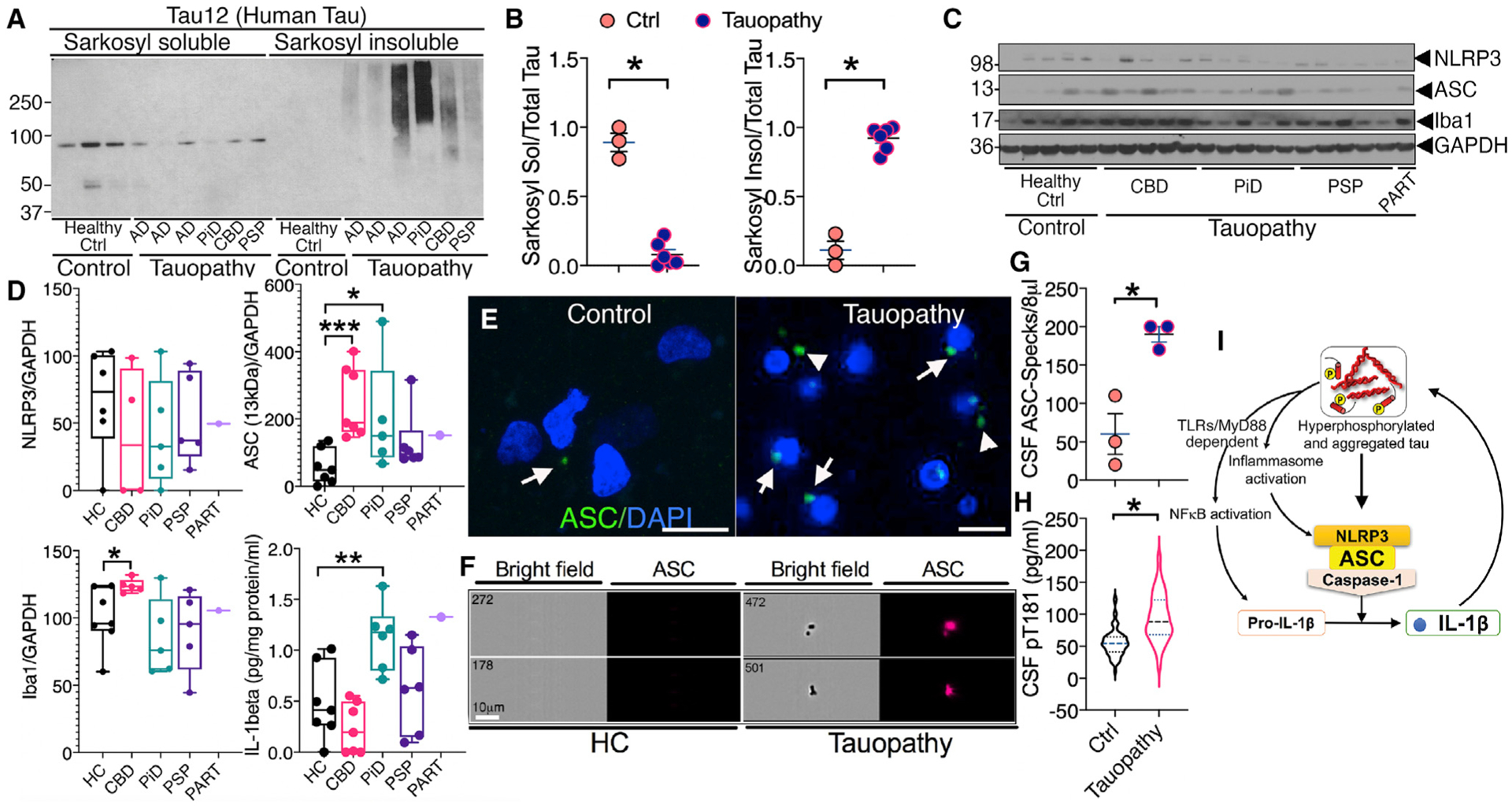
(A and B) Western blot and quantification showing significantly reduced Sarkosyl soluble and increased Sarkosyl insoluble human tau in the cortical lysates of human autopsy brains of Alzheimer’s disease (AD) (n = 3), Pick’s disease (PiD) (n = 1), corticobasal degeneration (CBD) (n = 1), and progressive supranuclear palsy (PSP) (n = 1), which were collectively called “Tauopathy” (total n = 6) compared to healthy controls (“Healthy Ctrl” or “Control”) (n = 3) with no history of dementia.
(C and D) Western blot and quantification show a significant increase in ASC–GAPDH and Iba1–GAPDH ratios as well as IL-1β levels in cortical lysates from human FTLD-Tau subjects (n = 4–5, except for primary age-related tauopathy or “PART”) (n = 1) compared to healthy controls (n = 7).
(E) Immunofluorescence showing increased ASC (green) in the temporal cortex of human subject with tauopathy compared to healthy control. Nuclear marker DAPI is in blue. ASC specks are intracellular (arrows) or possibly in extracellular space (arrowheads). Scale, 10 μm.
(F–H) Quantitative Amnis ImageStream analysis of the CSF samples from healthy controls or patients with tauopathy showing significantly higher levels of ASC-specks (~1–5 μm) in the bright field image (left panels) and immunoreactive to anti-ASC antibody (right panel, red) and pT181 (by ELISA in H) compared to control. Scale, 10 μm.
(I) Working model shows that pTau could prime myeloid cells including microglial cells via TLRs/MyD88/NF-κB to induce expression and activation of IL-1β and ASC-positive inflammasome complex. IL-1β in turn could exacerbate tau phosphorylation.
Data displayed as mean ± SEM (except violin plot in I); Student’s t test or one-way ANOVA (multiple groups), *p < 0.05, **p < 0.01, ***p < 0.005; n = 3–6 (B); n = 3 (G); n = 33–89 (H).
DISCUSSION
In the current study, we show that unbiased RNA-seq analysis of human primary microglia treated with human NFTs showed transcriptional upregulation of multiple proteins in the NF-κB signaling pathway along with increased IL-1β expression, suggesting pTau-induced IL-1β expression is occurring possibly through the NF-κB pathway. We further confirmed this link by showing microglial cells can internalize pTau from neuronal conditioned media or purified exosomes derived from neurons, which can lead to increased expression and/or activation of ASC, NLRP3, and IL-1β in microglia. We observed inflammasome activation and IL-1β secretion by hPHFs and mPHFs in microglia, which was ASC-dependent. Subsequent experiments suggested that suppression of human tau reduced both transcriptional and translational level of ASC and NLRP3 in the rTg4510 mouse model of tauopathy. Myeloid cells-/microglia-specific deletion of ASC and MyD88 reduced NF-κB activation, IL-1β expression and maturation, tau pathology, and improved memory in the hTau mouse model of tauopathy, suggesting that pTau induced IL-1β expression and activation is cell autonomous to myeloid cells (including microglia). Finally, pTau level is strongly associated with the levels of IL-1β, ASC, and inflammasome activation in the human FTLD brains. Our findings extend the observations made by few previous reports on the colocalization of NLRP3, IL-1β, and caspase-1 with pTau in the human AD brains (Ahmed et al., 2017) and significantly elevated levels of proinflammatory cytokines TNF-α, IL-1α, and IL-1β in the P301S mouse model of tauopathy (Sayed et al., 2018).
The DOX-mediated suppression of human P301L tau significantly reduced both mRNA and protein levels of NLRP3/ASC in the 3- to 6-month treatment groups, but only protein levels of NLRP3 and ASC in the 5- to 9-month treatment groups. Although the reason for such difference is unclear, especially when there is significant reduction in MAPT mRNA levels in both cases, it is conceivable that the longer period of mutant tau expression in case of the 5- to 9-month group may cause irreversible alterations in the induction of pro-inflammatory innate immune pathways, which are either non-responsive to MAPT suppression via DOX and/or independent of mutant P301L tau-induced activation. Indeed, a previous study has shown that inflammatory pathways are over-represented as a function of age in rTg4510 mice with over 139 genes related to immune pathways showing altered expression at 6.1 versus 1.9 months of age (Wes et al., 2014). Together, these results suggest that with age, there may be uncoupling of MAPT-dependent inflammasome activation in rTg4510 mice. At these later ages, the inflammasome activation may be driven by other DAMPs such as ATP, reactive oxygen species, and/or mitochondrial complex 1, which are affected in 8-month-old rTg450 mice (Barron et al., 2020). Nonetheless, suppression of human pTau by Qβ-pT181 vaccine did confirm that reduced pTau resulted in reduced ASC and NLRP3 in rTg4510 mice. Previous studies have shown that tau pathology is associated with neuroinflammation in AD and FTLD (Laurent et al., 2018). For instance, microglial primary culture released pro-inflammatory cytokines (IL-1β, IL-6, TNF-α, and TIMP1) after treatment with purified recombinant truncated tau, possibly through the activation of NF-κB (Kovac et al., 2011). However, it was unclear how truncated tau affects the upstream signaling to cause transcriptional changes and the downstream signaling that can result in the activation and secretion of the proinflammatory cytokines. Another study showed that tau oligomers co-localize with microglia, astrocytes, and a proinflammatory alarmin, HMGB1, in the frontal cortex of FTLD and AD (Nilson et al., 2017), suggesting a toxic relationship between oligomeric tau and neuroinflammation. Moreover, a separate study has shown that microglia can phagocytose pre-aggregated, oligomeric forms of tau and secrete them with exosomes, which leads to the trans-neuronal propagation of tau (Asai et al., 2015). However, how these internalized pTau affect the phenotype of microglia is unclear. We and others have previously shown that loss of tau was shown to rescue LPS-induced neurodegeneration (Maphis et al., 2015a), and active immunization with 0N4R WT or 0N4R P301L mutated tau protein was able to ameliorate the tau pathology in rTg4510 mice and reduce the number of activated microglia and astrocytes (Selenica et al., 2014).Yet, the molecular mechanisms and cell autonomous effects from these studies were unclear. In two recent studies, activation of NLRP3-ASC inflammations by pathological tau has been demonstrated. Notably, in Stancu et al., (2019) microtubule-binding K18 fragment (Mocanu et al., 2008; Shammas et al., 2015) with P301L mutation of human 4R tau activated inflammasome and IL-1β via ASC and NLRP3 in vitro and in P301S mouse model of tauopathy using specific inhibitors and global ASC knockout mice (Stancu et al., 2019). In a second study by Ising et al., (2019), global deficiency of ASC and NLRP3 in THY-Tau22 mouse model of FTDP-17 led to significant reduction in tau pathology and improved spatial memory via decreasing active forms of GSK3β, CamKIIα and the inactive form of protein phosphatase (PP2Ac) (Ising et al., 2019). Although these studies elegantly established the role of NLRP3-ASC in tau spreading model, the current study extends these prior observations by demonstrating that pTau-induced NLRP3 or ASC activation is cell-autonomous to myeloid cells including microglia in in vitro, rTg4510, and in non-mutant hTau mice, and it is MyD88-dependent. Our observation corroborates with another previous study, which suggested that Rho kinase inhibitor (Fasudil) reduced amyloid and tau pathology by blocking the activation of TLR-MyD88-NF-κB signaling axis (Yu et al., 2017). Limitations of this study are in the use of CX3CR1Cre and CD11bCre lines (the crosses that were started prior to availability of microglia-specific [TMEM119Cre] or conditional [CX3CR1-CreER] Cre lines), which may not distinguish the effect of microglia from that of peripheral immune cells. These experiments may need to be independently replicated using microglia-specific cre lines with human tau transgene.
Our study contrasts to other previous reports in a few different aspects. For example, previous reports have suggested that one of the major routes of extracellular secretion of pTau was via exosomes (Bellingham et al., 2012; Saman et al., 2012). In the present study, however, inhibition of exosomal secretion (that should have prevented secreted pTau from neuronal cells) showed a minimal effect in blocking NLRP3 expression in microglia, suggesting the possibility of non-exosomal pTau (soluble pTau) as a potential source of DAMP in inducing inflammasome activation. To our surprise, NLRP3 knockdown had only modest effects on IL-1β secretion unlike ASC or caspase-1 knockdowns. This indicates that pTau could be recognized by other inflammasome receptors (such as NLRP1 or AIM2) that also require ASC. These inflammasome receptors might play a synergistic role in the pTau-induced IL-1β maturation in microglia. Of note, pTau-induced IL-1β secretion is not as robust as the ones induced by other inflammasome agonists like ATP or nigericin. However, considering AD/Tauopathies are chronic neurodegenerative diseases that develop over many years to decades, this “low-grade” neuroinflammation is still of great importance because neuroinflammation can further worsen the tau pathology, forming a feed-forward loop (Bhaskar et al., 2010). It is important to note that although BV2 cells used in this present study have been utilized by numerous studies as a valid substitute for primary microglia (Henn et al., 2009) and consistent innate immune response to LPS (our unpublished observation), a clear limitation of using these cells is that they are cancerous cells and could change their phenotype. Therefore, confirming pTau and PHF’s effects on ASC, NLRP3, and IL-1β in iPSC-derived human microglia is warranted in future studies. On a related note, it is important to consider at what extent the amount of tau versus the type of tau (mutant) could trigger the expression of ASC/NLRP3/IL-1β in BV2 cells. Despite normalizing transfection amount, the volume of conditioned media used for treatment, etc., there appears to be slightly elevated expression of 0N3R-T231D/S235D compared to 0N3R WT tau by transfected N2a cells. Yet, the uptake of mutant tau by microglia was significantly higher than the WT tau. A controlled titration experiment may be needed to address this effect in future studies.
Although this study does provide a possible mechanistic insight into the IL-1β priming (via MyD88-NF-κB) and maturation (via ASC-containing inflammasomes) by different types of pathological tau (Table S3), there are still unexplored steps in this causal cascade. First, observed differences in inflammasome markers and IL-1β in different non-AD tauopathies (CBD, PSP, and PiD) needs to be confirmed in a separate and independent study with a larger sample size. Moreover, not all human samples used for various experiments (e.g., western blot, immunofluorescence, CSF ASC speck, and CSF pT181 levels) were matched because of limited availability of each sample. Future studies should be able to track the progress of different clinical stages and correlate it with alterations in pTau levels and inflammasome markers. Doing so should also help us address why CBD and PiD, but not PSP, samples showed significant difference in ASC and IL-1β. It could be possible that Pick’s bodies in PiD made up of 3R-Tau, with Lys254-Phe378 forming the core of Pick’s body, may serve as an efficient “danger” signal than 4R-dependent tau aggregates and drives inflammasome/IL-1β activation. A recent study demonstrated that ordered cores of tau filaments in PiD adopt a single novel fold of 3R-Tau, which is distinct from tau filaments of AD or other tauopathies (Falcon et al., 2018) and provide a possible explanation of 3R-Tau serving as an efficient DAMP for inflammasome activation.
Second, it will be interesting to define if TLR/NLR activation is direct or indirect (e.g., pTau aggregates causing lysosomal damage) (Papadopoulos et al., 2017), which in turn could activate TLR/NLR-MyD88. It is also of interest to determine if pTau contributes to other inflammasome-independent pathways (mediated by TNF-α or IL-6) and whether these are NF-κB dependent in FTLD-Tau brains. Based on our current understanding, we predict that in in vivo conditions, the microglia might get exposed to both soluble hyperphosphorylated (in Figure 2) and aggregated (in Figure 3) tau depending on the disease state (different sources of pTau used in the present study are summarized in Table S3). We speculate that early in the disease process, it is the extracellularly secreted hyperphosphorylated tau that likely encounters microglia and can lead to NF-κB and inflammasome activation via engaging MyD88. It will be tau oligomers and/or fibrils (PHFs) in the later stages of the disease, which further promotes this vicious cycle. Nevertheless, based on the present study, therapeutic strategies toward uncoupling MyD88, ASC, and pTau could block neuroinflammation, reduce pTau burden, and improve clinical outcome for FTLD-Tauopathies (Figure 7I).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Kiran Bhaskar (kbhaskar@salud.unm.edu).
Materials availability
This study did not generate new unique reagents. Commercially available reagents are indicated in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Alix | Millipore | Cat# ABC40; RRID: AB_10806218 |
| Rabbit monoclonal anti-total p65 | Cell Signaling | Cat# 4764; RRID: AB_823578 |
| Rabbit polyclonal anti-ASC | AdipoGen Life Sciences | Cat# AG-25B-0006-C100; RRID: AB_2885200 |
| Mouse monoclonal phospho-T231 tau (AT180) | Thermo Fisher Scientific | Cat# MN1040; RRID: AB_223649 |
| Mouse monoclonal phospho-S202 tau (AT8) | Thermo Fisher Scientific | Cat# MN1020; RRID: AB_223647 |
| Rabbit polyclonal anti-Caspase-1 | Santa Cruz Biotech | Cat# sc-514; RRID: AB_2068895 |
| Mouse monoclonal PE-Cy7 Rat Anti-CD11b | BD Bioscience | Cat# 561098; RRID: AB_2033994 |
| Mouse monoclonal anti-CD81 | Santa Cruz Biotech | Cat# sc-166029; RRID: AB_2275892 |
| Mouse monoclonal anti-GAPDH | Millipore | Cat# CB1001-500UG; RRID: AB_2107426 |
| Rabbit monoclonal anti-Iba1 | Wako | Cat# 019-19741; RRID: AB_839504 |
| Rabbit polyclonal anti-IκBα | Cell Signaling | Cat# 9242; RRID: AB_331623 |
| Goat polyclonal anti-IL-1β | R&D System | Cat# AF-401-NA; RRID: AB_416684 |
| Rabbit polyclonal anti-MyD88 | Santa Cruz Biotech | Cat# sc-11356; RRID: AB_2146724 |
| Mouse monoclonal anti-NLRP3 | AdipoGen Life Sciences | Cat# AG-20B-0014-C100; RRID: AB_2885199 |
| Mouse monoclonal anti-phospho-S396/S404 tau (PHF1) | Gift from Dr. Peter Davies | Greenberg et al., 1992 |
| Rabbit monoclonal anti-phospho-p65 | Cell Signaling | Cat# 3033s; RRID: AB_33128 |
| Mouse monoclonal anti-Tau5 | Thermo Fisher Scientific | Cat# AHB0042; RRID: AB_1502093 |
| Mouse monoclonal anti-Tau12 | Abcam | Cat# ab74137; RRID: AB_1281142 |
| Mouse monoclonal anti-Tau12 | Millipore | Cat# MAB2241; RRID: AB_1977340 |
| Goat anti-mouse IgG - HRP | Jackson Immuno Research | Cat# 115-035-146; RRID: AB_2307392 |
| Goat anti-rabbit IgG - HRP | Jackson Immuno Research | Cat# 111-035-144; RRID: AB_2307391 |
| Donkey anti-goat IgG-HRP | Santa Cruz Biotech | Cat# sc-2020; RRID: AB_631728 |
| Goat anti-mouse IgG - Alexa Fluor 488 | Invitrogen | Cat# A11029; RRID: AB_2534088 |
| Goat anti-rabbit IgG - Alexa Fluor 488 | Invitrogen | Cat# A11008; RRID: AB_143165 |
| Goat anti-mouse IgG - Alexa Fluor 555 | Invitrogen | Cat# A28180; RRID: AB_2536164 |
| Goat anti-mouse IgG - Alexa Fluor 568 | Invitrogen | Cat# A11036; RRID: AB_10563566 |
| Goat anti-rabbit IgG - Alexa Fluor 647 | Invitrogen | Cat# A21245; RRID: AB_141775 |
| Bacterial and virus strains | ||
| DH-5α Competent Cells | Thermo Fisher Scientific | Cat# 18265017 |
| Biological samples | ||
| Human Brain Tissues | NWNADC | N/A |
| Human CSF Samples | UNM Memory and Aging Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596026 |
| LPS | Sigma-Aldrich | Cat# L2880-25MG |
| ATP | Sigma-Aldrich | Cat# A1852-1VL |
| GW4869 | Sigma-Aldrich | Cat# D1692-5MG |
| IL-1β | Sigma-Aldrich | Cat# SRP8033-10UG |
| N-lauroylsarcosine sodium salt | Sigma-Aldrich | Cat# 61745-250G |
| 2-mercaptoethanol | Sigma-Aldrich | |
| 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) | Thermo Fisher Scientific | Cat# 28300 |
| Critical commercial assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368813 |
| TaqMan Gene Expression Assay (FAM) | Thermo Fisher Scientific | Cat# 4352339 |
| Amaxa® Cell Line Nucleofector® Kit V | Lonza Bioscience | Cat# VCA-1003 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher Scientific | Cat# L3000015 |
| Effectene Transfection Reagent | QIAGEN | Cat# 301425 |
| FAM FLICA® Caspase-1 assay | ImmunoChemistry | Cat# 97 |
| Mouse IL-1β ELISA Kit | R&D Systems | Cat# DY401 |
| Human IL-1β ELISA Kit | R&D Systems | Cat#DY201 |
| RNA-Sequencing Kit | KAPA Biosystems | Cat# KK8483 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| CSF pT181 tau Kit | Fujifilm | Cat# 298-81701 |
| Mouse Cytokine Magnetic 20-Plex Panel | Invitrogen | Cat# LMC0006M |
| Deposited data | ||
| RNA-seq data | European Bioinformatics Institute – Annotare® Title: RNA-Seq of human primary microglial cells treated with vehicle or human paired helical filaments (PHFs) purified from human tauopathy autopsy brain. | E-MTAB-5742, RRID: SCR_004727 |
| Deposited Data | This manuscript | http://dx.doi.org/10.17632/g3rfnf23cx.1 |
| Experimental models: cell lines | ||
| Neuro-2a | ATCC | Cat# ATCC® CCL-131; RRID: CVCL_0470 |
| BV2 cells | Gift from Dr. Gary Landreth | RRID: CVCL_0182 |
| ASC-mCerulean macrophages | Gift from Dr. Eicke Latz | Stutz et al., 2013 |
| Human primary microglia | ScienCell | Cat#1900 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson Laboratory | Cat# 000664. RRID: IMSR_JAX:000664 |
| rTg4510 mice | Jackson Laboratory | Santacruz et al., 2005 |
| hTau mice |
Andorfer et al., 2003; Dawson et al., 2001; Maphis et al., 2017 |
N/A |
| hTau/ASC−/− mice | This manuscript | N/A |
| hTauCX3CR1Cre+ASCf/f mice | This manuscript | N/A |
| hTauCD11bCre+MyD88f/f mice | This manuscript | N/A |
| CX3CR1Cre mice | Jackson Laboratory | Cat# 025524; IMSR_JAX:025524 |
| MyD88f/f mice | Gift from Dr. Xiaoxia Li | Yu et al., 2014 |
| CD11bCre | Gift from Dr. Xiaoxia Li | Yu et al., 2014 |
| ASCf/f mice | Gift from Dr. Amir Yazdi | Drexler et al., 2012 |
| ASC−/− mice | Gift from Dr. Vishwa Dixit (Genentech) | Mariathasan et al., 2004 |
| Oligonucleotides | ||
| ASC, PYD And CARD Domain Containing - Pycard | Thermo Fisher Scientific | # Mm00445747_g1 |
| Caspase-1 – Casp1 | Thermo Fisher Scientific | # Mm00438023_m1 |
| CD40, TNF receptor superfamily member 5 – CD40 | Thermo Fisher Scientific | # Hs01002913_g1 |
| Interleukin-1β – IL1B | Thermo Fisher Scientific | # Hs00174097_m1 |
| Interleukin-1α – Il1a | Thermo Fisher Scientific | # Mm00429620_m1 |
| Interleukin-1β – Il1b | Thermo Fisher Scientific | # Mm00434228_m1 |
| Interleukin-18 – Il18 | Thermo Fisher Scientific | # Mm00434225_m1 |
| Human tau - MAPT | Thermo Fisher Scientific | # Hs00902194_m1 |
| MyD88, myeloid differentiation primary response 88 – Myd88 | Thermo Fisher Scientific | # Mm00440338_m1 |
| NOD like receptor (NLR) family, pyrin domain containing 1 – NLRP1 | Thermo Fisher Scientific | # Hs00248187_m1 |
| NLR family, pyrin domain containing 1A Nlrp1a | Thermo Fisher Scientific | # Mm03047263_m1 |
| NLR family pyrin domain containing 3 – Nlrp3 | Thermo Fisher Scientific | # Mm00840904_m1 |
| p50 (NFKB1) nuclear factor of kappa light polypeptide gene enhancer in B cells 1 – NFKB1 | Thermo Fisher Scientific | # Hs00765730_m1 |
| Eukaryotic 18S rRNA Endogenous Control | Thermo Fisher Scientific | # 4319413E |
| Mouse GAPDH Endogenous Control | Thermo Fisher Scientific | # 4352339 |
| siRNA targeting Pycard | Horizon | # M-051439-01-0005 |
| siRNA targeting Casp1 | Horizon | # M-048913-01-0005 |
| siRNA targeting MyD88 | Horizon | # M-063057-00-0005 |
| siRNA targeting Nlrp3 | Horizon | # M-053455-01-0005 |
| siGENOME Non-Targeting siRNA Pool #1 - control | Horizon | # D-001206-13-20 |
| Recombinant DNA | ||
| pRC-CMV-n123c | Gift from Dr. Gloria Lee | Leugers and Lee, 2010 |
| pRC-CMV-n123c-T231D/S235D | Gift from Dr. Gloria Lee | Leugers and Lee, 2010 |
| Software and algorithms | ||
| ImageJ | Schindelin et al., 2012 | https://imagej.net/; RRID:SCR_003070 |
| AlphaEaseFC™ | AlphaInnotech | http://genetictechnologiesinc.com/alpha/alpha_ease_fc.htm |
| Zen Microscope software | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html; RRID: SCR_013672 |
| LAS Application Suite X | Leica | https://www.leica.com; RRID:SCR_013673 |
| Bowtie2 | Langmead, 2010 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID:SCR_016368 |
| RSEM v1.2.15. TMM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM; RRID:SCR_013027 |
| Ingenuity Pathway Analysis | Krämer et al., 2014 | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/content-exploration-and-databases/qiagen-ipa/; RRID:SCR_008653 |
| Amnis IDEAS® | Amnis | https://www.luminexcorp.com/eu/amnis-imaging-flow-cytometry-support/ |
| Adobe Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html; RRID: SCR_014199 |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| Other | ||
| Nigericin | Invivogen | Cat# 28643-80-3 |
| VECTASHIELD Mounting Medium with DAPI | Vector Labs | Cat# H1200 |
| Protease Inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Phosphatase Inhibitor cocktail | Sigma-Aldrich | Cat# P5726 |
| PhosSTOP® Phosphatase Inhibitor Cocktail Tablet | Roche | Cat# 4906845001 |
| Tissue Protein Extraction Reagent | Thermo Fisher Scientific | Cat# 78510 |
| lithium dodecyl sulfate (LDS) | N/A | Cat# B0007 |
| Reducing Agent | N/A | Cat# NP0009 |
| NuPAGE 4–12% Bis-Tris Protein Gels | N/A | Cat# NP0335BOX |
| CellMask Orange Plasma Membrane Stain | N/A | Cat# C10045 |
| SigmaFast® 3,3′-diaminobenzidine (DAB) | Sigma | Cat# D4418 |
| 10 nm gold particles | Aurion | Cat# 410.011 |
| Poly-L-Lysine | Sigma-Aldrich | Cat# P4707 |
| Percoll | GE Life Sciences | Cat# 17089102 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15713 |
| Qβ virus-like particle | Maphis et al., 2019 | N/A |
| Qβ-pT181 virus-like particle | Maphis et al., 2019 | N/A |
| M-CSF | R&D Systems | Cat# 416ML |
| PMSF | Sigma-Aldrich | Cat# 10837091001 |
Data and code availability
The RNA-seq data generated by this study is deposited to European Bioinformatics Institute – Annotare® (E-MTAB-5742, RRID:SCR_004727). Raw data for all figures and additional data for Figure 1 were deposited at Mendeley at http://dx.doi.org/10.17632/g3rfnf23cx.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental protocols involving animals were performed in accordance with US National Institutes of Health guidelines on animal care and were approved (16–200428-B-HSC; 15–200352-HSC) by the University of New Mexico Institutional Animal Care and Use Committees. Unless otherwise noted, animals of both sexes were used in this entire study. Doxycycline (DOX)-mediated regulatable rTg4510 (expressing human tau with P301L mutation) mice (Santacruz et al., 2005) were obtained from the Jackson Laboratory. One group of mice received DOX diet (TD.05125, 2.1 g DOX/day ad libitum in chow, Tekland/Envigo, USA) starting at 3 months of age until 6 months of age, while the other group received doxycycline treatment starting at 5 months of age until 9 months of age. Age-matched mice that didn’t receive DOX were used as controls. hTau mice were obtained through breeding as previously described (Maphis et al., 2017). These genomic hTau mice were generated by crossing the original 8c line (Duff et al., 2000), which expresses all six isoforms of human MAPT under the human MAPT promoter with complete endogenous mouse Mapt knockout lines (Dawson et al., 2001). We described this line as hTauMaptKO(Duke) mice in our recent report (Maphis et al., 2017). For convenience, we 5like to call hTauMaptKO(Duke) mice as ‘hTau’ mice, but these are slightly different (with complete tau knockout) from previously described hTau line (Andorfer et al., 2003), which expressed a short N-terminal fragment of endogenous mouse tau. hTau/ASC−/− mice were generated by crossing hTauMaptKO(Duke) mice with ASC−/− mice (provided by Dr. Vishwa Dixit from Genentech) (Mariathasan et al., 2004). hTau mice were crossed to CX3CR1Cre (Yona et al., 2013) (The Jackson Laboratory Stock # 025524) followed by ASCf/f mice (Drexler et al., 2012) (a kind gift from Dr. Amir Yazdi at University of Lausanne) to generate hTauCX3CR1Cre+ASCf/f mice. Similarly, hTau mice were also crossed to CD11bCre (ItgamCre) (Kang et al., 2010) and MyD88f/f mice (Yu et al., 2014) (a kind gift from Dr. Xiaoxia Li at the Cleveland Clinic Foundation) to generate hTauCD11bCre+MyD88f/f mice. All mice (except rTg4510 lines) were maintained in B6 background by backcrossing the original lines to C57BL/6j strain for more than ten generations.
Human subjects
All experiments performed with human cerebrospinal fluid (CSF) samples were approved by University of New Mexico Institutional Review Board (IRB) protocol # 04-294. The human subjects used in this study are listed in the Table S2. We used autopsy brain samples from healthy controls (Average age, n = 5 females, n = 3 males), AD, CBD, PSP, PiD, TPSD, PART. Furthermore, for CSF samples from live subjects, we used healthy controls (Average age, n = 5 females, n = 3 males) and AD (Average age, n = 5 females, n = 3 males).
Cell culture
Neuroblastoma (Neuro-2a or N2a, ATCC®, Manassas, VA) cells were maintained in Eagle’s minimum essential medium (ATCC) supplemented with 10% fetal bovine serum (FBS) and 10X antibiotic-antimycotic (Thermo Fisher Scientific, Waltham, MA). For conditioned media (CM) experiment, N2a cells were transfected with human tau plasmids (0N3R-Tau ‘WT’ tau; 0N3R-T231D/S235D-Tau (Lee et al., 1998; Sharma et al., 2007)) using Effectene transfection reagent as per company’s protocol (QIAGEN, Germany). After 48 hours post-transfection, cells lysate and supernatant were collected for western blotting. CM from N2a cells containing secreted tau was used to stimulate BV2 microglial cells. The BV2 microglial cells were a gift from Dr. Gary Landreth and were maintained in Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific) and 100X Penicillin-Streptomycin (Thermo Fisher Scientific). Mouse primary murine microglial cells were cultured as previously reported (Bhaskar et al., 2010) and utilized to confirm the effects observed on BV2 cells or macrophages. LPS (1 μg/ml) was used to prime BV2 cells, 6 hours prior to the collection. Bone-marrow derived macrophages were cultured by harvesting bone marrows from C67BL/6j mice as previously described (Dupont et al., 2011). For gene expression experiments, BV2 cells were not primed with LPS to see the priming effect of tau. Exosomes isolated from N2a cells transfected with Mock or human tau plasmids were used to treat BV2 cells. Lysates were collected for western blotting. NLRP3-deficient immortalized murine macrophages stably expressing ASC-mCerulean and NLRP3-FLAG were used as reporter cells for inflammasome activation as described before (Stutz et al., 2013). For conditioned media experiment, LPS-primed (250 ng/ml for 3h) ASC-mCerulean macrophages were stimulated with tau containing-CM from N2a cells. The controls included conditioned media derived from mock (vector only) transfected N2a cells. Cells were fixed for confocal microscopic analysis. For live cell imaging and confocal microscopic analysis, LPS-primed cells were further stimulated with ATP (5mM) or Nigericin (20 μM) or purified paired helical filaments (PHFs) (or the vehicle, which is buffer H – see below) for 30 min and then recorded with live cell microscopy or fixed for confocal microscopic analysis to observe inflammasome activation. For IL-1β secretion assay, cells were stimulated with PHFs for 18 h and supernatant was collected and tested for IL-1β secretion using ELISA kit (DY401, R&D systems, Minneapolis, MN). siRNA-mediated knockdown of target genes was performed as previously described (Chauhan et al., 2015). Specifically, the siASC, siCasp1 and siNLRP3 knockdowns were performed using nucleofection for 72 h as previously described (Chauhan et al., 2015). MyD88 knockdown was performed using siMyD88 and Lipofectamine 3000 for 48 h using the manufacturer’s protocol (Thermo Fisher Scientific).
METHODS DETAILS
Lipopolysaccharide (LPS) administration
A single dose of LPS (from E. coli 055:B5, Sigma, Catalogue # L2880–25MG, Lot # 102M4017V) was administered to two-month-old non-transgenic C57BL/6J or ASC−/− (also on C57BL/6J genetic background) mice as previously described (Bhaskar et al., 2010). Briefly, Vehicle (Veh, Hank’s Balanced Saline Solution) or LPS (Sigma-Aldrich, USA) was administered at 3.5 mg/kg body weight (intraperitoneal). After 24 h, the mice were sacrificed and the hippocampi from left hemisphere were processed for biochemical analysis.
Murine primary microglia, bone marrow macrophages and Cellomics® High-content microscopy assay
Primary mouse microglial cell cultures were prepared from postnatal day 14 (P14) pups as described before (Saura et al., 2003). Cells were seeded in a 96-well plate at 9 × 103 cells per well and allowed to adhere and grow for 72 hours prior to treatment. The cells were treated for 24 hours with 66% conditioned media from N2a cells transfected for 24 hours either with Mock, 0N3R-WT or 0N3R-T231D/S235D tau. The cell media was removed, and the cells were fixed with 4% paraformaldehyde in PBS for 10 minutes at room temperature. The paraformaldehyde was removed, and the cells were stained with Rabbit anti-ASC antibody (AL177, Adipogen, 1:200) and then Goat anti-rabbit IgG (H+L) Alexa Fluor 555 antibody (A27039, Thermo Scientific, 1:1000). Cells were stained with DAPI for 10 minutes at room temperature. The DAPI solution was removed, and 200 μL of PBS was added, and samples were stored at 4°C until analysis. The plate was transferred to the ThermoScientific Cellomics High Content Screening machine and the Spot Detector Bioapplication was used to detect ASC specks. DAPI-stained nuclei were detected in channel 1 with a 386 nm filter, and a mask fitted to define the area of the nucleus and predict the cell boundary. Alexa Fluor 555-labeled aggregates above an intensity threshold within the cytoplasmic area were counted in channel 2 with a 549 nm filter. 1000 cells/well were counted in randomly selected fields, with a maximum of 100 fields per well. “Mean object spot total area” (total area of all spots per cell, μm2) were compared among different treatments. Aggregates are pseudo-colored as green for better visualization and comparison.
Bone marrow macrophages (BMMs) isolated from wild-type mice were seeded in a 96-well plate at 105 cells per well and allowed to adhere and grow for 24 h prior to treatment. The cells were treated for 24 hours with vehicle or PHF (10ug/ml) and fixed with 4% paraformaldehyde in PBS for 10 minutes at room temperature. The paraformaldehyde was removed, and the cells were stained with rabbit anti-NF-κB total p65 antibody (4764S, Cell Signaling, 1:200) and then Goat anti-rabbit IgG(H+L) Alexa Fluor 568 antibody (A11036, Thermo Scientific, 1:1000). Cells were stained with DAPI for 10 minutes at room temperature. The DAPI solution was removed, and 200 μL of PBS was added, and samples were stored at 4 degrees until analysis. The plate was transferred to the Thermo Scientific Cellomics High Content Screening machine and the Spot Detector Bioapplication was used to detect nuclear translocation of NF-κB p65. DAPI-stained nuclei were detected in channel 1 with a 386 nm filter, and a mask fitted to define the area of the nucleus and predict the cell boundary. Alexa Fluor 555-labeled NF-κB p65 above an intensity threshold were counted in channel 2 with a 549 nm filter. 1000 cells/well were counted in randomly selected fields, with a maximum of 100 fields per well. A total of 5 wells were counted per each treatment. Nuclear translocation of NF-κB p65 measured by “Mean object spot average intensity” (average above-background intensity of anti-p65 staining in ROIs defined by nuclear labeling) were compared between vehicle and PHF.
Human brain tissue and CSF samples
The University of New Mexico Institutional Review Board approved the use of all human autopsy specimens under exempt status. CSF samples from healthy control and tauopathy were banked samples from MarkVCID cohort obtained from University of New Mexico Center for Memory and Aging with prior IRB approval. Human healthy control (HC), AD, Tauopathy brain tissue samples were kindly provided by Northwestern Cognitive Neurology & Alzheimer’s Disease Center (CNADC) Neuropathology Core (Table S2). Brain tissue samples were homogenized in 10% weight/volume Tissue Protein Extraction Reagent (T-PER®, Thermo Fisher Scientific) and soluble lysates were resolved via SDS-PAGE and immunoblotted. Soluble lysates were also processed for IL-1β ELISA (Cat# DY201; R&D Systems) as per manufacturers’ protocol. Levels of phosphorylated threonine 181 tau (pT181) were measured in human CSF samples using Fujifilm pT181 ELISA assay kit (Cat# 298-81701) as described by the manufacturer.
Microglial isolation
Mononuclear cells were isolated from a pool of two brains per group as previously described (Bergmann et al., 1999; Maphis et al., 2015b). Briefly, the mice were anaesthetized, transcardially perfused with phosphate buffer, brains removed and dissociated in 0.25% trypsin/RPMI media. Mononuclear cells were separated via a 30%/70% discontinuous isotonic percoll gradient. Isolated mononuclear cells (enriched in microglia) were immediately processed for either western blot or gene expression analysis.
Isolation of bone marrow derived macrophages (BMMs)
BMMs were prepared from the femurs and tibia of 3-month-old C57BL/6j mice. Briefly, the mice were sacrificed, femurs and tibia were dissected. BMMs were flushed with syringe with 24 G needle containing differentiation media, which include macrophage-colony stimulating factor (M-CSF) as previously described (Dupont et al., 2011). The cells were fed with fresh M-CSF media every 2 to 3 days. After this, the cells were utilized for the siRNA knockdown experiments.
Tissue Preparation for Biochemical Analysis
The mice were anesthetized and transcardially perfused with 0.125 M phosphate buffer (PB). Following perfusion, the brains were removed, the left hemisphere was immersion fixed in 4% paraformaldehyde in PB (4% PFA/PB), the right hemisphere was micro-dissected into the cortex and hippocampus, wet weights were recorded, and the tissues were snap frozen in liquid nitrogen for subsequent biochemical analysis. The rest of the right hemispheres were weighed and snap frozen in liquid nitrogen for subsequent mRNA extraction. Hippocampi from different mouse strains were used for biochemical (quantitative real-time polymerase chain reaction – qRT-PCR or western blot) analysis. 30 μm thick sagittal brain sections were used for the immunohistochemical analysis.
SDS-PAGE and Western immunoblotting
Cells were lysed in 1x LDS/RA buffer (Thermo Fisher Scientific) and sonicated for 30 s, boiled at 95°C for 15mins. Supernatant were also collected and mixed with 1x LDS/RA buffer and then boiled at 95°C for 15mins. Cell lysates and supernatant were resolved via SDS-PAGE and immunoblotted as previously described (Bhaskar et al., 2010). The dilutions of primary antibodies utilized are indicated in the antibody list table. Western blot data are quantified either using Bio-Rad Chemidoc system or developed autoradiographs. Raw blots are included in Data S1.
Gene expression analysis
RNA from cells and mouse brains was extracted using the TriZOL reagent as described by the manufacturer (Thermo Fisher Scientific). Total RNA (20 ng/μL) was converted to cDNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) and amplified using specific TaqMan assays (catalog # 4331182; Thermo Fisher Scientific) (additional details are in STAR methods). GAPDH (catalog # 4352339, Thermo Fisher Scientific) and 18 s rRNA (catalog # 4319413E, Thermo Fisher Scientific) were used as was used as a housekeeping gene for normalization. qRT-PCR assays were run on the StepOnePlus® Real-Time PCR System (Thermo Fisher Scientific) and the statistical analyses were performed using GraphPad Prism.
Sarkosyl soluble and insoluble pellet preparation
Hippocampus tissue (10–20 g) of mice or human brain tissue were homogenized in 10% (w/v) Tissue Protein Extraction Reagent (T-PER®, Thermo Fisher Scientific). The T-PER-insoluble pellet (P1) was sonicated with 10 volume of cold buffer H (10mM Tris-HCl, 1mM EGTA, 0.8 mM NaCl, 10% sucrose, pH 7.4) supplemented by 0.1mM PMSF (Sigma-Aldrich), PhosSTOP® Phosphatase Inhibitor Cocktail Tablet (Roche, 1 tablet per 10ml), Complete Protease Inhibitor Cocktail Tablet (Roche, 1 tablet per 10ml). After centrifugation at 28,000rpm in Beckman Ti TLA-120.2 rotor for 30min at 4°C, the supernatant (S1) was adjusted to 1% (w/v) N-laurylsarcosine (Sigma-Aldrich) and 1% (v/v) 2-mercaptoethanol (Sigma-Aldrich) and incubated at 37°C for 2 h with agitation (shaking). After centrifugation at 100,000rpm for 35 min at room temperature, the Sarkosyl-soluble supernatant (S2) was collected and resuspended in 1x lithium dodecyl sulfate (LDS) and sample reducing agent (RA) (Thermo Fisher Scientific). Sarkosyl-insoluble pellet (P2) was washed several times in 1% Sarkosyl in buffer H and the pellet was resuspended in 1x LDS/RA. Both samples (S2 and P2) were boiled at 95°C for 15 minutes for further western blot analysis.
Sarkosyl extraction of paired helical filaments (PHFs)
A slightly modified version of the previously published protocol (Greenberg and Davies, 1990) was used. Brains from rTg4510 mice or a patient with AD were homogenized in 10 volumes of buffer H (10mM Tris-HCl, 1mM EGTA, 800 mM NaCl, 10% sucrose, pH 7.4), briefly sonicated and centrifuged at 22,000 g for 30 min at 4°C. The supernatant was adjusted to 1% (w/v) N-laurylsarcosine and 1% β-mercaptoethanol and incubated at 37°C for 2 h with agitation (shaking). After centrifugation at 150,000 g for 35 min at room temperature, sarkosyl-insoluble pellet was collected and washed several times in 1% sarkosyl in buffer H. After the final wash, the pellet was resuspended in Buffer H with 1% CHAPS (w/v) and 1% β-mercaptoethanol (v/v). The PHF suspension was filtered through 0.45 μm cellulose acetate syringe filters and the filtration was centrifuged at 35,000rpm for 1 h at room temperature. PHF-containing pellet was resuspended with Buffer H with 1% β-mercaptoethanol (v/v) and layered over sucrose gradient (6 mL of 50% sucrose and 4 mL of 35% sucrose in 10 mM Tris/0.8 mM NaCl/1 mM EGTA/0.1% β-mercaptoethanol, pH 7.4) and centrifuged at 35,000 rpm for 2 h in Beckman SW 41Ti rotor at 4°C. PHFs were collected from the 35% layer and 35%–50% interface with a syringe.
PHFs were further purified using chloroform/methanol precipitation method to get rid of the sucrose. Briefly, four volumes of methanol were added to one volume of the protein sample, and the mixture was vortexed. One volume of chloroform was then added, and the mixture was vortexed. The sample was centrifuged at 10,000 g for 5 min and the aqueous methanol layer was removed from the top of the sample. PHFs remained at the phase boundary between the aqueous methanol layer and the chloroform layer. Finally, four volumes of methanol were then added, and the mixture was vortexed. The sample was spun at 10,000 g for 15 min. The supernatant was removed without disturbing the pellet, and the pellet was air-dried. The pellet, containing PHFs, was then resuspended in cell culture media. All procedures were performed at room temperature.
Live cell imaging
ASC-mCerulean macrophages were seeded in 8-well Nunc® Lab-Tek® II Chamber Slide system for 24 h prior to stimulation. Cells were primed with LPS (250ng/ml for 3 h) and stimulated with ATP (5mM) or PHFs (2 μg/ml) for 30min. Imaging was performed on the Olympus Disk Spinning Unit (DSU) confocal microscopy equipped with a temperature and CO2-controlled sample chamber for live-cell imaging, images were captured every 5 minutes starting at 0 min until 45 min after the stimulation with PHF. CellMask Orange Plasma Membrane Stain (Thermo Fisher Scientific) was used to label the plasma membrane.
FAM-FLICA Caspase-1 Assay and flow cytometry analysis
BV2 cells (treated with VEH or hPHFs 2 μg/ml for 18 h) were stained with FAM-FLICA Caspase-1 assay kit (ImmunoChemistry Technologies, #97) according to manufacturer’s instructions and also stained with CD11b PE-Cy7 surface marker (BD Bioscience #561098), then analyzed with BD LSRFortessa flow cytometry.
Exosome isolation
Exosomes were prepared from culture supernatants of N2a cells as previously described (Thery et al., 2006). Briefly, on the day of transfection using Effectene according to the manufacture’s protocol (QIAGEN), cell culture medium was replaced with medium supplemented with exosome-free serum (FBS was spun at 100,000 g for 2 hours, pellet omitted). For the exosome secretion inhibition experiment, the inhibitor GW4869 (Sigma, D1692-5MG, 3.5 μM) was used when replacing the exosome-free media. Cell culture supernatants were collected at two consecutive 48 h time points, combined and sequentially centrifuged at 3,000 g for 10 min to remove cells, dead cells, and debris. The conditioned medium was filter-sterilized by passing through a connected 0.22 μm filter syringe. Filtered medium was then spun at 100,000 g for 70 mins. The exosome pellet was resuspended in 40 mL of cold PBS and the exosome solution was again centrifuged at 100,000 g for 70 min at 4°C to obtain final purified exosome pellet.
Immunohistochemical/immunofluorescence Analysis
Free-floating sagittal sections (30 μm thick) from rTg4510 mice treated with or without DOX from 3 to 6 month, 6-month-old hTau and hTau/ASC−/− mice as well as formalin fixed autopsy brain sections from FTLD were processed for immunohistochemical (IHC) and immunofluorescence (IF) analysis as previously described (Bhaskar et al., 2010). Briefly, sections were first incubated in 10 mM sodium citrate buffer (pH 6.0) for 10 min at 95°C for antigen retrieval, washed in PBS with 0.1% Tween (PBST), quenched with 0.3% H2O2 in PBST for 15 min (only for IHC). Sections were blocked for 1 h at room temperature with the 5% normal sera (goat/donkey – from animal species in which the secondary antibodies were raised). The sections were incubated with rabbit polyclonal anti-ASC antibody (Adipogen, 1:200) or mouse monoclonal AT8 antibody (Millipore 1:250). After washing in PBST, the sections were incubated with biotinylated-secondary antibody (Jackson ImmunoResearch Laboratories Inc., 1:250, for IHC) or Alexa 488 conjugated goat anti-rabbit secondary antibody (1:250 in blocking solution; for IF). Sections were then incubated with ABC (Vector Laboratories) reagent for 1 h at room temperature (for IHC). The immunoreactive signals were revealed by developing sections in SigmaFast® 3,3′-diaminobenzidine (DAB) tablets (Sigma–Aldrich) (for IHC) or the sections were mounted on slides using Hardset (Vector Laboratories, Burlingame, CA) with DAPI (for IF). Images were acquired using Leica TCS-SP8 confocal microscope and Olympus bright field microscope. Quantitative morphometric analysis was performed by ImageJ software by manually scoring 5 random fields per sections; 3 sections per mouse brain; n = 7–21 mice per genotype (as indicated in the figure legend). Percentage of immunoreactive area was analyzed and plotted for each genotype.
Confocal Microscopy
Immunofluorescence confocal microscopy was carried out using Zeiss LSM 510 Meta and Leica TCS SP8 Confocal microscope. Antibody against Tau12 (Abcam, 1:200) and secondary antibody goat anti-mouse Alexa Fluor 555 (Invitrogen, 1:500) were used. CellMask Orange Plasma Membrane Stain (Thermo Fisher Scientific) was used at 0.5x dilution to label the plasma membrane.
Electron Microscopy
For Immunoelectron microscopy experiment, purified exosomes were pipetted onto carbon-coated TEM grids and incubated for 5 min at room temperature. The solution was then wicked off the grid with Whatman 1 filter paper until a thin film remained. Rabbit anti-Alix antibody (exosome marker) and mouse anti-Tau12 antibody were used to label exosomes and tau, respectively. Goat anti-rabbit secondary antibody conjugated to 10 nm gold particles (Aurion, Wageningen, the Netherlands) and goat anti-mouse secondary antibody conjugated to 5nm gold particles were used for detection. The grids were then negatively stained by placement on droplets of 2% uranyl acetate for 2 min. Samples were imaged in a Hitachi H7500 transmission electron microscope equipped with an Advanced Microscopy Sciences XR60 camera.
For PHF imaging, PHF derived from rTg4510 mice brain were pipetted onto carbon-coated TEM grids and incubated for 5 min at room temperature. The solution was then wicked off the grid with Whatman 1 filter paper until a thin film remained. The grids were then negatively stained by placement on droplets of 2% uranyl acetate for 2 min. Samples were imaged in a Hitachi H7500 transmission electron microscope equipped with an Advanced Microscopy Sciences XR60 camera.
ImageStream®X Mark II Imaging Flow Cytometry
Cells were trypsinized, neutralized and centrifuged at 3000rpm 5min. Cell pellet was resuspended in PBS containing 1mM EDTA and incubated at room temperature for 5min to avoid clumping. Cells were then fixed with 4% PFA, permeabilized with 0.1% Triton X-100 and blocked with 10% goat serum. Cells were stained with Tau12 antibody (Abcam, 1:500) for 1h and then with goat anti-mouse Alexa Fluor 555 (Invitrogen, 1:500) for 30min at room temperature, washed with 1x PBS and centrifuged at 3000rpm for 5min. Cells were resuspended in 50ul of PBS containing 2% FBS for polychromatic imaging flow cytometry analysis. Data were collected on an ImageStream® Mark II Imaging Flow Cytometer (Amnis® from EMD Millipore) and analyzed using IDEAS v6.1 software as previously described (Beum et al., 2006).
CSF samples from healthy controls (control) and tauopathy (mixed dementia) were obtained from the MarkVCID cohort. Briefly, CSF samples were stained with Rabbit anti-ASC antibody (AL177, Adipogen, 1:200) for 1 hour, ultra-centrifuged at 120,000rpm for 15 minutes. Pellets were resuspended in 1x PBS buffer and stained with Alexa Fluor 647-conjugated goat antibody to rabbit IgG for 30 minutes. Samples were ultra-centrifuged again at 120,000rpm for 15 minutes. Pellets were resuspended in 50 μL of 1x PBS buffer. Data were collected on an ImageStream® Mark II Imaging Flow Cytometer (Amnis® from Luminex) and analyzed using IDEAS v6.1 software as previously described (Beum et al., 2006).
RNA sequencing and analysis
Human microglial cells (ScienCell, 1900) were thawed and plated in a 6-well tissue culture plate pre-coated with 0.1 mg/ml of Poly-L-Lysine (Sigma, P4707). The culture media was changed 16 hours after thawing and cells remained undisturbed for another 24 hours. The human microglial cells were then treated with vehicle or 2 μg/ml of hPHF (paired helical filaments isolated from human AD brain) in triplicates for 18 hours. Cells were harvested, and RNA was extracted from cells using QIAGEN RNeasy mini kit with DNase treatment as per manufacturer’s instructions. RNA samples were shipped to Quick Biology (Pasadena, CA) for sequencing and analysis. Briefly, library construction was achieved using KAPA Stranded RNA-Seq Kits (KAPA Biosystems, KK8483) with RiboErase. RNA sequencing was performed using HiSeq 4000 system (Illumina) with 2×150bp pair-ended sequencing for 60 Million reads.
The reads were first mapped to the latest UCSC transcript set using Bowtie2 version 2.1.0. The gene expression level was estimated using RSEM v1.2.15. TMM (trimmed mean of M-values) was used to normalize the gene expression. LncRNA transcript set was extracted from LNCipedia (https://lncipedia.org). Differentially expressed genes were identified using the edgeR program. Genes showing altered expression with p < 0.05 and more than 2 fold changes were considered differentially expressed. The pathway and network analysis was performed using Ingenuity (IPA). IPA computes a score for each network according to the fit of the set of supplied focus genes. These scores indicate the likelihood of focus genes to belong to a network versus those obtained by chance. A score > 2 indicates a ≤ 99% confidence that a focus gene network was not generated by chance alone. The canonical pathways generated by IPA are the most significant for the uploaded dataset. Fischer’s exact test with FDR option was used to calculate the significance of the canonical pathway. MATS (Multivariate Analysis of Transcript Splicing) were used to perform splicing analysis. DCC was used to perform the circRNA analysis. RNA-seq data have been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-5742.
Novel object recognition task (NOR)
Novel object recognition task behavioral test was carried out as previously published (Maphis et al., 2015a). Briefly, on the first day of this 3-day paradigm animals were acclimated to an open 75-cm2 arena for 5 minutes. On the second day, they were exposed to two identical objects (2 glass jars) for 5 minutes. On the test day, they are exposed to one of the previous familiar objects (glass jar) and a novel object (plastic water bottle). The ratio of novel (on the bottom right side of the maze) or familiar (top right of the maze) object encounters to total object encounters was recorded as a measurement of recognition memory.
Morris Water Maze task (MWM)
The MWM test was performed as previously described (Maphis et al., 2015b) with minor modifications. Briefly, the mice were trained for 5 days (4 daily trials, approximately 25 min inter-trial interval) to find a hidden platform submerged beneath cloudy water (temperature maintained at 26°C) using spatial cues on the wall, as previously described (Morris, 1984; Vorhees and Williams, 2006). During the acquisition phase, mice navigate in a tank of opaque water for a period of 1 minute, and over 5 days (four trials daily) to learn to navigate to a submerged platform using visual cues on the wall (black shapes providing distinct visual cues). Mice were kept warm in cages between trials. On the sixth day, a probe trial was performed by removing the platform and the percentage of time spent in the target quadrant (where the platform was) versus the other quadrants was recorded as a measurement of spatial working memory.
Immunization of hTau mice Qβ-pT181
Qβ-virus-like particles (Qβ-VLPs or Qβ) were produced in Escherichia coli (E. coli) and conjugated to 175TPPAPKpTPPSSGEGGC190 (peptide encompassing threonine 181 in human tau) as previously described (Maphis et al., 2019). Eight-month-old hTau mice received unconjugated control vaccination Qß-VLP or Qß-VLP conjugated to a pTau peptide (pT181-Qß) at 5 μg/injection into the rear hind paw. Mice were injected bi-weekly, two doses and allowed to age for two months after the last injection. The mice were sacrificed, and the hippocampi were processed to assess the levels of AT180+ pTau, ASC, NLRP3, total human tau (Tau12) and GAPDH by SDS-PAGE and western blot as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, comparisons between the two groups were done via unpaired t test; comparisons between multiple treatment groups were done via one-way or two-way analysis of variance (ANOVA) with indicated multiple comparisons post hoc tests, p value greater than 0.05 is considered as statistically significant. p values can be found in the respective figure legends. All statistical analyses were performed using GraphPad Prism® (Version 6.0). All the statistical details of experiments can be found in the respective figure legends.
Supplementary Material
Highlights.
Pathological tau (pTau) primes NF-κB and IL-1β signaling in human primary microglia
Suppressing pTau reduces inflammasome activation in tauopathy mice
Myeloid-cell restricted deletion of ASC improves cognitive function in hTau mice
pTau co-exists with elevated IL-1β and ASC in human tauopathy brains and CSF
ACKNOWLEDGMENTS
We thank Dr. Vishwa Dixit (Genentech) and Dr. Vojo Deretic (UNM) for providing ASC−/− mice and various reagents, respectively. We thank Drs. Xiaoxia Li and Zizhen Kang (Cleveland Clinic) for providing CD11bCre/MyD88 flox mice. We thank Dr. Amir Yazdi (University of Lausanne) for providing ASC flox mice. We are thankful to Dr. Michael Paffett at the UNM Cancer Center Fluorescence Microscopy facility and CTSC T1 lab for help with confocal imaging and RNA quantification, respectively. We thank Dr. Paulus Mrass for his assistance in processing live image files. We thank Dr. Bryce Chackerian, Ms. Julianne Peabody, Mr. Jeff Thompson, and Ms. Sasha Hobson for assistance with tau immunization experiments and analyzing tau levels in the CSF samples. The graphical abstract was created with https://www.biorender.com. This study was supported by the NIH (RF1NS083704-05A1, R01NS083704, and R21NS077089; R21NS093442 to K.B.; and R01NS116051 to J.P.W.), AIM CoBRE Center (P20GM121176-04), the University of New Mexico (UNM) Health Sciences Center Bridge Funding, UNM Department of Molecular Genetics and Microbiology intradepartmental grant funding, Dr. Stephanie Ruby travel award (to N.M. and J.B.), and a T32 training grant (to L.W.). This study was also supported in part by Alzheimer’s Disease Core Center (P30AG013854) from the National Institute on Aging (NIA) to Northwestern University and New Mexico Alzheimer’s Disease Research Center (P20AG068077-01) from NIA to G.A.R. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109720.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmed ME, Iyer S, Thangavel R, Kempuraj D, Selvakumar GP, Raikwar SP, Zaheer S, and Zaheer A (2017). Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimers Dis 60, 1143–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarshahi H, Axelsson JB, Said K, Malmström A, Fischer H, and Andersson R (2011). TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J. Transl. Med 9, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, and Davies P (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem 86, 582–590. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, and Ikezu T (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G, Snellman A, Schöll M, Troakes C, Hye A, et al. (2021). Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol 141, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron AM, Ji B, Fujinaga M, Zhang MR, Suhara T, Sahara N, Aoki I, Tsukada H, and Higuchi M (2020). In vivo positron emission tomography imaging of mitochondrial abnormalities in a mouse model of tauopathy. Neurobiol. Aging 94, 140–148. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. ; Dominantly Inherited Alzheimer Network (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham SA, Guo BB, Coleman BM, and Hill AF (2012). Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Bugiani O, Ghetti B, and Spillantini MG (2011). Presence of Reactive Microglia and Neuroinflammatory Mediators in a Case of Frontotemporal Dementia with P301S Mutation. Neurodegener. Dis 8, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann CC, Yao Q, and Stohlman SA (1999). Microglia exhibit clonal variability in eliciting cytotoxic T lymphocyte responses independent of class I expression. Cell. Immunol 198, 44–53. [DOI] [PubMed] [Google Scholar]
- Beum PV, Lindorfer MA, Hall BE, George TC, Frost K, Morrissey PJ, and Taylor RP (2006). Quantitative analysis of protein co-localization on B cells opsonized with rituximab and complement using the ImageStream multispectral imaging flow cytometer. J. Immunol. Methods 317, 90–99. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Yen SH, and Lee G (2005). Disease-related modifications in tau affect the interaction between Fyn and Tau. J. Biol. Chem 280, 35119–35125. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, and Lamb BT (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan IF, and Maguire-Zeiss KA (2018). Toll-Like Receptor 2 Signaling and Current Approaches for Therapeutic Modulation in Synucleinopathies. Front. Pharmacol 9, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Kaushik DK, Gupta M, and Basu A (2010). Inflammasome signaling at the heart of central nervous system pathology. J. Neurosci. Res 88, 1615–1631. [DOI] [PubMed] [Google Scholar]
- Chauhan S, Ahmed Z, Bradfute SB, Arko-Mensah J, Mandell MA, Won Choi S, Kimura T, Blanchet F, Waller A, Mudd MH, et al. (2015). Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat. Commun 6, 8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, Carney DG, and Lynch MA (2015). α-TLR2 antibody attenuates the Aβ-mediated inflammatory response in microglia through enhanced expression of SIGIRR. Brain Behav. Immun 46, 70–79. [DOI] [PubMed] [Google Scholar]
- Couturier J, Stancu IC, Schakman O, Pierrot N, Huaux F, Kienlen-Campard P, Dewachter I, and Octave JN (2016). Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflammation 13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, and Vitek MP (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci 114, 1179–1187. [DOI] [PubMed] [Google Scholar]
- Drexler SK, Bonsignore L, Masin M, Tardivel A, Jackstadt R, Hermeking H, Schneider P, Gross O, Tschopp J, and Yazdi AS (2012). Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl. Acad. Sci. USA 109, 18384–18389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, et al. (2000). Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis 7, 87–98. [DOI] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, and Deretic V (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 30, 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidenmüller J, Fath T, Hellwig A, Reed J, Sontag E, and Brandt R (2000). Structural and functional implications of tau hyperphosphorylation: information from phosphorylation-mimicking mutated tau proteins. Biochemistry 39, 13166–13175. [DOI] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, and Goedert M (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambuzza ME, Sofo V, Salmeri FM, Soraci L, Marino S, and Bramanti P (2014). Toll-like receptors in Alzheimer’s disease: a therapeutic perspective. CNS Neurol. Disord. Drug Targets 13, 1542–1558. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K, and Brooks DJ (2004). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov. Disord 19, 1221–1226. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, and Brooks DJ (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord 21, 89–93. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, and Davies P (1990). A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 87, 5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, and Binder LI (1992). Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J. Biol. Chem 267, 564–569. [PubMed] [Google Scholar]
- Griffioen K, Mattson MP, and Okun E (2018). Deficiency of Toll-like receptors 2, 3 or 4 extends life expectancy in Huntington’s disease mice. Heliyon 4, e00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjärn M, Schrattenholz A, Pörzgen P, and Leist M (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26, 83–94. [DOI] [PubMed] [Google Scholar]
- Hernández F, and Avila J (2007). Tauopathies. Cell. Mol. Life Sci 64, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 110, E3138–E3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa K, and Dickson DW (2001). Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J. Neuropathol. Exp. Neurol 60, 647–657. [DOI] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, et al. (2010). Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Kim C, Cortez LM, Carmen Garza M, Yang J, Wille H, Sim VL, Westaway D, McKenzie D, and Aiken J (2016). Toll-like receptor-mediated immune response inhibits prion propagation. Glia 64, 937–951. [DOI] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, and Diamond MI (2012). Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem 287, 19440–19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, and Novak M (2011). Misfolded truncated protein τ induces innate immune response via MAPK pathway. J. Immunol 187, 2732–2739. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, Weidenheim K, and Dickson DW (1994). Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am. J. Pathol 145, 1496–1508. [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, and Dixit VM (2009). The inflammasomes. PLoS Pathog 5, e1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B (2010). Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics Chapter 11, Unit 11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Buée L, and Blum D (2018). Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J 41, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, and Panchamoorthy G (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci 111, 3167–3177. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, and Trojanowski JQ (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci 24, 1121–1159. [DOI] [PubMed] [Google Scholar]
- Leugers CJ, and Lee G (2010). Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J. Biol. Chem 285, 19125–19134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, and Bhaskar K (2015a). Loss of tau rescues inflammation-mediated neurodegeneration. Front. Neurosci 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, and Bhaskar K (2015b). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis NM, Jiang S, Binder J, Wright C, Gopalan B, Lamb BT, and Bhaskar K (2017). Whole Genome Expression Analysis in a Mouse Model of Tauopathy Identifies MECP2 as a Possible Regulator of Tau Pathology. Front. Mol. Neurosci 10, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis NM, Peabody J, Crossey E, Jiang S, Jamaleddin Ahmad FA, Alvarez M, Mansoor SK, Yaney A, Yang Y, Sillerud LO, et al. (2019). Qß Virus-like particle-based vaccine induces robust immunity and protects against tauopathy. NPJ Vaccines 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, and Dixit VM (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. [DOI] [PubMed] [Google Scholar]
- Meng XF, Wang XL, Tian XJ, Yang ZH, Chu GP, Zhang J, Li M, Shi J, and Zhang C (2014). Nod-like receptor protein 1 inflammasome mediates neuron injury under high glucose. Mol. Neurobiol 49, 673–684. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, et al. (2008). The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci 28, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, and Kayed R (2017). Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimers Dis 55, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, and Hall GF (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem 287, 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Tusell JM, and Serratosa J (2003). High-yield isolation of murine microglia by mild trypsinization. Glia 44, 183–189. [DOI] [PubMed] [Google Scholar]
- Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, et al. (2018). Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. USA 115, 10172–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossen-koppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, et al. (2016). PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 89, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenica ML, Davtyan H, Housley SB, Blair LJ, Gillies A, Nordhues BA, Zhang B, Liu J, Gestwicki JE, Lee DC, et al. (2014). Epitope analysis following active immunization with tau proteins reveals immunogens implicated in tau pathogenesis. J. Neuroinflammation 11, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, Mandelkow E, Knowles TP, Mandelkow E, and Klenerman D (2015). A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat. Commun 6, 7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VM, Litersky JM, Bhaskar K, and Lee G (2007). Tau impacts on growth-factor-stimulated actin remodeling. J. Cell Sci 120, 748–757. [DOI] [PubMed] [Google Scholar]
- Smith R, Puschmann A, Schöll M, Ohlsson T, van Swieten J, Honer M, Englund E, and Hansson O (2016). 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 139, 2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brône B, Huaux F, Octave JN, et al. (2019). Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137, 599–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz A, Horvath GL, Monks BG, and Latz E (2013). ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol 1040, 91–101. [DOI] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, and Clayton A (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, and Williams MT (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc 1, 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner N, and Núñez G (2013). MyD88: a critical adaptor protein in innate immunity signal transduction. J. Immunol 190, 3–4. [DOI] [PubMed] [Google Scholar]
- Wes PD, Easton A, Corradi J, Barten DM, Devidze N, DeCarr LB, Truong A, He A, Barrezueta NX, Polson C, et al. (2014). Tau overexpression impacts a neuroinflammation gene expression network perturbed in Alzheimer’s disease. PLoS ONE 9, e106050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem 288, 1856–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Zhou Q, Fan C, Zhao H, Wang Y, Qiu X, Yang K, and Ji Q (2019). Doxycycline inhibits NAcht Leucine-rich repeat Protein 3 inflammasome activation and interleukin-1β production induced by Porphyromonas gingivalis-lipopolysaccharide and adenosine triphosphate in human gingival fibroblasts. Arch. Oral Biol 107, 104514. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Zhou H, Zhao J, Xiao N, Roychowdhury S, Schmitt D, Hu B, Ransohoff RM, Harding CV, Hise AG, et al. (2014). MyD88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. J. Exp. Med 211, 887–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JZ, Li YH, Liu CY, Wang Q, Gu QF, Wang HQ, Zhang GX, Xiao BG, and Ma CG (2017). Multitarget Therapeutic Effect of Fasudil in APP/PS1transgenic Mice. CNS Neurol. Disord. Drug Targets 16, 199–209. [DOI] [PubMed] [Google Scholar]
- Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, and Novak M (2009). Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J. Neuroimmunol 209, 16–25. [DOI] [PubMed] [Google Scholar]
- Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, and Novak M (2012). Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflammation 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated by this study is deposited to European Bioinformatics Institute – Annotare® (E-MTAB-5742, RRID:SCR_004727). Raw data for all figures and additional data for Figure 1 were deposited at Mendeley at http://dx.doi.org/10.17632/g3rfnf23cx.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.