

Abstract
The gastrointestinal tract harbors trillions of microbial species, collectively termed the microbiota, which establish a symbiotic relationship with the host. Decades of research have emphasized the necessity of microbial signals in the development, maturation, and function of host physiology. However, changes in the composition or containment of the microbiota have been linked to the development of several chronic inflammatory diseases, including inflammatory bowel diseases. Intestinal epithelial cells (IECs) are in constant contact with the microbiota and are critical for maintaining intestinal homeostasis. Signals from the microbiota are directly sensed by IECs and influence intestinal health by calibrating immune cell responses and fortifying intestinal barrier function. IECs detect commensal microbes through engagement of common pattern recognition receptors or by sensing the production of microbial-derived metabolites. Deficiencies in these microbial-detecting pathways in IECs leads to impaired epithelial barrier function and altered intestinal homeostasis. This Review aims to highlight the pathways by which IECs sense microbiota-derived signals and the necessity of these detection pathways in maintaining epithelial barrier integrity.
Introduction
The mammalian gastrointestinal tract requires a constant balance of power between immune activation and tissue homeostasis. A monolayer of intestinal epithelial cells (IECs) separate the mucosal immune system from the external environment. Remarkably, trillions of bacteria, fungi, archaea, and viruses reside within the intestinal lumen and constantly interact with host mammalian cells. The sheer abundance and proximity of the microbiota, and their foreign antigens, create an immense source of potential immune stimuli. However, during homeostatic conditions host immune cells act in a restrained manner, balancing inflammatory and regulatory responses to prevent aberrant reactions to innocuous commensal antigens. Yet, during pathogenic infections, intestinal immune cells are poised to combat and eliminate invading microbes. Extensive work has been devoted to understanding the mechanisms by which the immune system discriminates between innocuous commensals and invading pathogens and how it mounts the appropriate immune response. Importantly, IECs are uniquely positioned and equipped to play a fundamental role in initial microbial sensing that directs downstream immune responses.
Intestinal Epithelium
In addition to providing a physical barrier, IECs coordinate numerous physiological processes including nutrient absorption, pathogen defense, and immune regulation. Epithelial stem cells reside at the base of intestinal crypts and undergo constant proliferation to give rise to diverse differentiated IECs (1). During homeostatic conditions, it is estimated that the intestinal epithelium is regenerated every 4 to 5 days (1). The diversity and constant renewal of IECs allows for their ability to modulate a variety of biological pathways. IECs are broadly separated into absorptive enterocytes, which are responsible for metabolic and digestive processes, or secretory lineages that are specialized to maintain digestive or barrier functions (1,2). Secretory lineages include goblet cells that produce mucin glycoprotein and form mucus, Paneth cells which reside at the base of intestinal crypts and secrete antimicrobial peptides (AMPs), microfold (M) cells which are critical for antigen capture and presentation to immune cells, enteroendocrine cells which secrete hormones that aid in digestion and communicate with the nervous system, and tuft cells that promote type 2 immune responses. Recent studies have employed single-cell RNA sequencing to further define the specific characteristics and behaviors of these distinct IEC subtypes (3,4). Together these IECs function to maintain intestinal barrier integrity and instruct downstream immune cell functions to regulate tissue homeostasis.
It is well appreciated that the microbiota influence various physiological functions including digestion, cellular metabolism, tissue development, and immune cell education. Germ-free (GF) mouse models, which lack all microbes, have demonstrated the requirement for the microbiota in calibrating IEC differentiation, proliferation, defense mechanisms, and immune cells (5). Interestingly, changes in the diversity or localization of the microbiota have been associated with several chronic diseases including cancer, diabetes, obesity, and inflammatory bowel diseases (IBD) (6). Therefore, it remains critical that immune responses to the microbiota be carefully controlled to avoid inappropriate stimulation or inflammatory reaction.
The intestinal epithelium provides both a physical and chemical barrier to separate mucosal immune cells from commensal microbial stimulation and invading pathogens. This complex and dynamic relationship between the host immune system and microbial signals hinges on IEC involvement. IECs must sense and decipher microbial stimulation and instruct immune cells how to respond. IECs recognize microbial stimuli through a number of different mechanisms including engagement of pattern recognition receptors (PRRs) including toll-like receptors (TLRs), nod-like receptors (NLRs), and inflammasomes (Figure 1A). In addition, IECs can sense microbial metabolites via receptors and enzymes that can lead to alterations in gene transcription (Figure 1B).
Figure 1. Microbial surveillance pathways in intestinal epithelial cells.
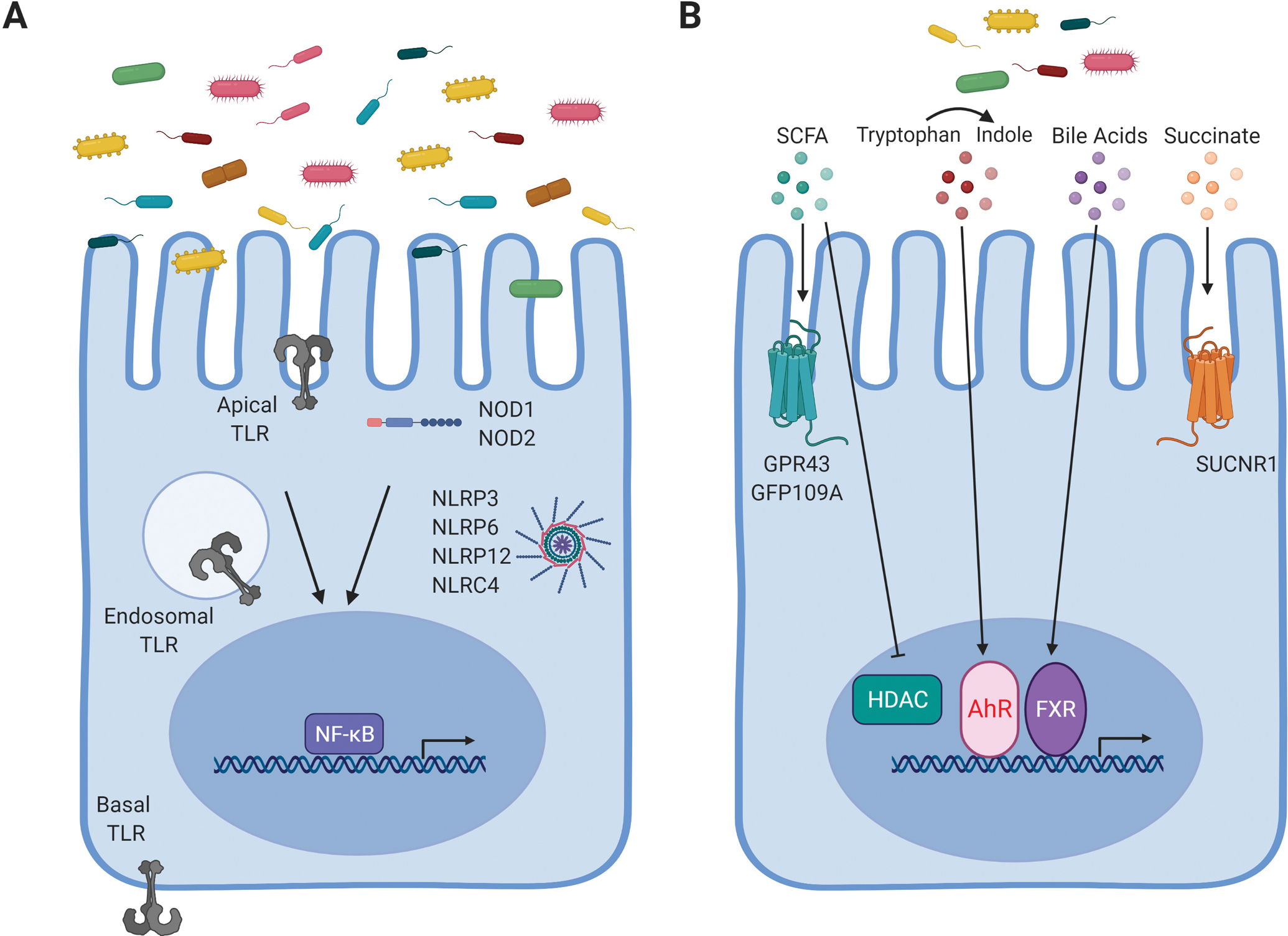
A) Common microbial/pathogen associated molecular patterns produced by the microbiota are detected by pattern recognition receptors, including TLRs and NLRs, expressed in IECs. B) Metabolites generated by microbial digestion of dietary components can be sensed through various pathways in IECs to alter intestinal health. Created with BioRender.com.
Microbe/Pathogen-associated molecular patters (PAMPs)
PRRs detect common microbial molecules, termed pathogen-associated molecular patterns (PAMPs), during infection and are critical for coordinating immune responses and protection against invading pathogens (7). Common PAMPs include bacterial cell walls components such lipopolysaccharide (LPS), peptidoglycan, muramyl-dipeptide (MDP), and D-glutamyl-meso-diaminopimelic acid (DAP), as well as flagella, dsRNA, and DNA molecules. However, ligands for PRRs are not exclusive to pathogenic microbes but are abundantly produced by the microbiota. Several studies have revealed that ligands from the microbiota signal through intestinal PRRs to promote healthy development of host tissues and maturation of the immune system (8). Interestingly, PRRs have been identified in both vertebrates, as well as invertebrates, suggesting these conserved molecules evolved to communicate with commensal microbes to maintain a symbiotic relationship between the microbiota and host cells (8).
Toll-like receptors.
Toll-like receptors (TLRs) were one of the first PRRs to be identified and TLR signaling has been shown to be critical for maintaining a healthy intestinal barrier. Polymorphisms or variants of TLR2, TLR4, TLR5, or TLR9 have been associated with increased incidence or severity of IBD (9–12). In mice, loss of TLR2, TLR4, TLR5, or TLR9 or the TLR signaling adapter, MyD88, contributed to intestinal inflammation in murine models of IBD (13–15). Interestingly, the exacerbated disease observed in these mouse models was not due to altered inflammatory responses, but instead, because of defects in IEC proliferation, survival, distribution of junction proteins, and overall barrier function (13,16,17). IECs are known to express TLR2, TLR3, TLR4, TLR5, and TLR9 (18). A recent study constructed five TLR fluorescent reporter mice to visualize the expression and localization of different TLRs throughout the gastrointestinal tract (18). This study revealed low expression of TLR2, TLR4, TLR5, TLR7 and TLR9 in small intestinal IECs but much higher expression in colonic IECs (18), suggesting higher expression of TLRs may be associated with the increased abundance of commensal microbes in the colon relative to the small intestine. Given the complex PAMPs found within the microbiota and the high potential for immune stimulation, the anatomical distribution of TLRs within polarized IECs has often been described as a potential mechanism for controlling overt stimulation. Indeed, several studies have reported that the majority of TLR expression is localized to the basolateral membrane, while TLR3 and TLR9 have also been shown at the apical surface (14,19,20). However, through the use of fluorescent reporter mice, TLR2, TLR4, and TLR5 were shown on both the apical and basolateral surface of colon IECs, and TLR4 was also observed intracellularly (18). Therefore, IECs can detect microbial PAMPs located within the lumen as well as the underlaying lamina propria through TLR signaling pathways (Figure 1A).
Once activated by ligand binding, TLR activation initiates a signaling cascade resulting in the nuclear translocation of NF-κB. This leads to the expression and secretion of various cytokines and chemokines including TNF-α, IL-6, IL-8, IL-18, CXCL2, CXCL2, and CCL20, which signal and calibrate underlying immune cells. Additionally, TLR signaling and NF-κB activation in IECs can result in induction of innate defense factors including antimicrobial peptides (AMPs), mucus production, and iNOS (21–23). Paneth cells are the main source of AMPs including α-defensins REG3β, and REG3γ (24–26). These molecules function to directly inhibit or lyse bacteria and are critical for preserving intestinal homeostasis. Further, disruption to their production is linked to elevated microbial translocation leading to exacerbated intestinal inflammation (25). Several studies have reported that induction of AMPs depends on TLR signaling downstream of a wide range of TLR agonists. For example, in vivo stimulation of TLR3 and TLR9 initiated rapid Paneth cell degranulation and secretion of AMPs (27). In addition, β-defensins were up regulated in IECs following TLR2, TLR3, and TLR4 engagement in an NF-κB-dependent manner (23,28). Deletion of the TLR signaling adaptor molecule, MyD88, resulted in significantly reduced or undetectable levels of AMPs (24,29–32). Furthermore, aged transgenic mice that employed an IEC-specific dominate negative MyD88 developed spontaneous intestinal inflammation due to a lack of AMP secretion and constant bombardment of bacterial antigens (32). However, other studies have not reported the development of spontaneous inflammation in other mouse models with MyD88 specifically deleted in IECs (33,34). Nevertheless, these papers each found that loss of MyD88 signaling in IECs resulted in dysfunctional AMP production and impaired barrier function, highlighting the necessity for TLR-MyD88 signaling in IECs. AMP secretion in the intestine is not exclusive to Paneth cells. Enteroendocrine cells, which are the main producer of hormones within the intestine, sense antigens from the microbiota through TLR4, TLR5, and TLR9. Ligand binding to these receptors triggered release of chemokines and β-defensin (35). In the colon, goblet cells also required TLR/MyD88 signaling to produce MUC2 and other mucin molecules (36,37). Separate from the direct effects on NF-κB activation and AMP production, TLR signaling in IECs also increased the expression of canonical and non-canonical inflammasome components (38). Together numerous studies have illustrated the importance of IEC-intrinsic TLR signaling pathways.
Nucleotide binding and oligomerization domain (NOD)-like receptors.
Recognition of microbial PAMPs by TLRs is critical for IEC development and intestinal barrier integrity. However, TLR signaling is usually restricted to external or phagosomal PAMPs. For detection of intracellular or cytosolic PAMPs, a large family of highly conserved proteins called Nucleotide binding and Oligomerization Domain (NOD)-like receptors (NLRs) have been described. While NLRs have been largely examined for their roles in detection and protection against invading pathogens, these molecules are also required for maintaining tissue homeostasis. NOD2 was identified early in gene association studies for IBD and remains one of the strongest genetic risks in the development of IBD (39–41). Consistently, in mice, deficiencies in NOD1, NOD2, or both NOD1 and NOD2 rendered mice extremely sensitive to models of IBD (42–46). In addition, deficiencies in NOD sensing pathways associate with alterations in the microbiota composition in IBD patients (46–48). NOD1 is constitutively expressed in IECs, while NOD2 expression is confined to Paneth cells in the small intestine (49). Activation of NOD1 and/or NOD2 by peptidoglycans and other bacterial cell wall components trigger downstream signaling cascades that elicit the production of a variety of antimicrobial peptides and pro-inflammatory cytokines and chemokines (48–52). In contrast to MyD88-deficient mice, animals null for NOD2 displayed normal levels of REG3β, REG3γ, and RELMβ, but exhibited reduced expression of α-defensins (30). However, deletion of both NOD1 and NOD2 ablated REG3γ expression in these mice thus resulting in elevated intestinal inflammation (45). Interestingly, administration of peptidoglycan or other NOD ligands in vivo suppressed TLR signaling and protected mice from the development of intestinal inflammation, suggesting that microbial sensing by NOD proteins in IECs is critical for intestinal homeostasis (43,53).
Aside from NOD1 and NOD2, other NLR molecules can associate and activate caspases to form an inflammasome complex with the end goal of cleaving the pro-forms of IL-1β and IL-18 into their mature active states. These inflammasome-forming NLRs are unique in their ability to sense and respond to diverse ligands thereby playing a critical role in regulation of intestinal homeostasis. NLRP3, a well-studied inflammasome-forming NLR member, responds to multiple stimuli, including commensal bacteria, as well as microbial products and metabolites (54). NLRP3 activation has been shown to play a protective role in intestinal homeostasis, as reduction in NLRP3 expression was linked with increased susceptibility to Crohn’s disease (55). In mouse models of IBD, deficiency in NLPR3 resulted in heightened intestinal inflammation, suggesting a protective role for NLRP3 activation (56,57). Mechanisms attributed to this protective role of NLRP3 activation include non-hematopoietic cell production of IL-18 which promoted epithelial barrier integrity (56) and secretion of AMP including β-defensin (57,58). Interestingly, mutations in NLRP3 that induce hyperactivity have been identified in humans with an autoimmune disease (59,60). Further, mice with this specific hyperactive NLRP3 mutation were resistant to mouse models of IBD (58). NLRP3 hyperactivity promoted IL-1β and AMP secretion and improved barrier function to protect mice from intestinal inflammation (58). Another well-studied inflammasome-forming NLR is NLRP6, which is predominately expressed in mucosal epithelial cells (61–63). Within the intestine, NLRP6 is preferentially expressed in enterocytes and goblet cells and is critical for regulating intestinal homeostasis and defense against invading pathogens. Deletion of NLRP6 disrupted secretion of mucin by goblet cells and rendered mice more susceptible to enteric infection (64). Furthermore, NLRP6 ablation has been shown to induce drastic changes to the microbiota composition which has been associated with dysregulated immune responses and induction of intestinal inflammation (62). Another NLR molecule, NLRP12, functions independent of the inflammasome and inhibits NF-κB signaling, and played a protective role in mouse models of colitis (65). Furthermore, the potent intracellular flagellin receptor, NLRC4, which is critical for host defense against pathogenic bacteria, protected against colitis models (66). Together, these studies highlight the importance of NLR signaling molecules on regulating the host-microbiota relationship (Figure 1A).
Microbiota-Derived Metabolites
In addition to interacting with host cells through PAMP-PRR engagement, the microbiota can influence host pathways through metabolites (Figure 1B). Metabolites are small molecules that are produced as intermediate or end products of microbial metabolism. These metabolites can derive from bacterial breakdown of dietary components, modification of host molecules, such as bile acids, or directly from bacteria. Signals from microbial metabolites can calibrate immune cell activation, host energy metabolism, IEC barrier integrity, and overall intestinal homeostasis. The importance of bacterial-derived metabolites in mediating host physiology has been illustrated by GF mice, and rodents exposed to broad-spectrum antibiotics, which have both shown dramatic alterations in the systemic and tissue profiles of metabolites (67–70). Mono-association studies where a single microbe was used to colonize GF mice have further demonstrated how specific microbes and their metabolites modulate intestinal homeostasis. For example, association of Clostridium sporogenes alone resulted in detectable levels of the metabolite, indole-3-propionic acid, which was absent in GF mice (67). Further, deletion of the gene responsible for indole-3-propionic acid production in C. sporogenes led to increased barrier permeability and intestinal inflammation compared to the wild-type C. sporogenes strain (71), highlighting the influence of a single metabolite on host physiology. Reintroduction of a single metabolite can have profound effects on the epithelial barrier integrity. Moreover, changes in the diversity or functionality of the microbiota can influence the metabolite profile, or metabolome, which has been associated with development of inflammatory conditions. Indeed, specific classes of metabolites, including bile acids, short-chain fatty acids (SCFAs), and tryptophan metabolites, have been implicated in the pathogenesis of IBD (72). Below we will discuss the role of bacterial-derived metabolites in mediating epithelial barrier integrity and intestinal homeostasis.
Short-chain fatty acids.
Short-chain fatty acids (SCFA), including acetate, propionate, and butyrate, are produced when dietary fiber is fermented by the microbiota and are among the most abundant microbial metabolite present within the intestine. SCFAs are a main energy source for colonocytes (73), and are crucial for intestinal epithelial homeostasis. In fact, dysbiosis observed in IBD patients was associated with the loss of SCFA-producing bacteria including Faecalibacterium prausnitzii (74) and Roseburia hominiswith (75). This was consistent with an overall trend of reduced intestinal levels of SCFA in IBD patients (72,76). Moreover, although still under investigation, some evidence suggests that increased intake of dietary fibers, or SCFAs, could be clinically beneficial in the treatment of IBD (77–80). In addition, supplementation of SCFAs improved chemically-induced intestinal inflammation in conventional as well as GF mice (81,82). SCFAs have been shown to have diverse effects on mucosal immune cell function and are essential for maintaining and fortifying epithelial barrier function. Stimulation with SCFAs enhanced expression of tight junction proteins and other claudin molecules (83–87), promoted AMP secretion in Paneth cells (88), and upregulated mucus production in goblet cells (89–91). Furthermore, SCFAs play an important role in regulating IEC proliferation and turnover. GF or antibiotic-treated mice exhibited reduced IEC proliferation, however, upon colonization with SCFA-producing bacteria or supplementation with SCFAs, IEC turnover was restored (92). Conversely, butyrate has also been shown to inhibit intestinal epithelial cell proliferation (73,93), suggesting SCFAs, and butyrate in particular, may exert cell type-specific effects on IECs that may be linked to local SCFA concentrations (73).
SCFAs mediate cellular functions through activation of cell surface G-protein coupled receptors such as GPR43 and GPR109A, which are expressed on IECs as well as immune cells. Deletion of GPR43 and GPR109A, specifically in non-hematopoietic cells, enhanced host susceptibility to mouse models of colitis (81,82), indicating an important role for GPR signaling in maintaining intestinal homeostasis. Furthermore, loss of GPR43 reduced Paneth cell production of AMPs including REG3γ and β-defensin (88), and GPR109A expression was required for butyrate to exert anti-inflammatory effects by suppressing LPS-induced NF-κB activation (94). In addition to signaling through cell surface GPRs, SCFAs can freely diffuse into cells or can be taken up through specific transporters (95). SCFAs, butyrate particularly, inhibit histone deacetylase (HDAC) enzymatic activity thereby promoting histone acetylation and regulating gene expression. SCFA inhibition of HDAC activity has been shown to influence immune cell function and promote anti-inflammatory or tolerogenic immune responses (95). In addition, the use of HDACs inhibitors reduced disease severity in experimental mouse colitis models (96). However, loss of HDAC activity in IECs resulted in dysregulated intestinal homeostasis. Specifically, mice with loss of IEC expression of the class I HDAC, HDAC3, exhibited reduced Paneth cells, impaired epithelial barrier function, and exacerbated intestinal inflammation (97), suggesting HDAC3 activity is essential for intestinal homeostasis. IEC expression of other HDACs, including HDAC1 and HDAC2, have also been found to be important for maintaining epithelial cell differentiation and intestinal barrier function (98–100). Given the necessity for IEC-intrinsic HDAC activity in promoting intestinal homeostasis, SCFA inhibition of HDAC activity must be carefully calibrated. A recent study demonstrated that wild-type mice containing abundant SCFAs, actually displayed increased HDAC activity in IECs relative to GF mice, suggesting that other microbiota-derived metabolites may counter SCFA-mediated inhibition of HDAC activity (93). Indeed, digestion of dietary phytate by the microbiota into inositol phosphate derivatives increased HDAC activity in IECs and promoted stem cell proliferation and epithelial repair (93). Thus, SCFAs and inositol phosphates fine-tune HDAC activity in IECs.
Secondary Bile Acids.
Bile acids are small molecules that are synthesized from cholesterol by liver hepatocytes. Primary bile acids are secreted into the small intestine after eating and are critical for lipid digestion and absorption. The vast majority of primary bile acids are reabsorbed by the time they reach the terminal ileum. In the colon, the remaining bile acids dynamically interact with commensal microbes where they exert mutual effects on each other. Bile acids can be toxic to some microbial species and therefore can directly influence microbiota composition and diversity (72). Bile acid signaling through the bile-responsive receptor, farnesoid X receptor (FXR), can prevent bacterial overgrowth and microbial translocation (101,102) as well as induce host production of AMPs (103–105). Moreover, direct FXR stimulation induced anti-inflammatory effects and protected mice against models of colitis (105,106), demonstrating the importance of bile acid signaling on regulating intestinal homeostasis. However, these results were obtained through the use of FXR-null mice, thus the specific contribution of bile acid sensing by IECs remains to be determined. Several commensal microbes have developed mechanisms to counteract bile toxicity (107), and can chemically modify bile acids into deconjugated secondary bile acids by expressing bacterial bile salt hydrolases (108,109). In fact, GF mice lack secondary bile acid production and mice mono-associated with a bile salt hydrolase-expressing Escherichia coli demonstrated improved host metabolism, increased production of AMPs, and altered epithelial barrier (110). Furthermore, intestinal biopsy samples from patients with active IBD demonstrated reduced FXR expression (106) and altered bile acid profiles characterized by increased fecal primary bile acids and reduced serum and fecal secondary bile acids (111).
Tryptophan metabolites.
Tryptophan, an essential amino acid acquired through the diet, is a precursor for the synthesis of several important molecules including serotonin, melatonin, and vitamin B3 (72). The intestine is the primary location for dietary tryptophan metabolism which can occur through one of three distinct pathways (72). One pathway depends on the microbiota to metabolize tryptophan into a variety of indole metabolites that can signal through the aryl hydrocarbon receptor (AhR). AhR is a widely expressed transcription factor that is required for immune and epithelial cell development and homeostasis. AhR signaling in T cells and innate lymphoid cells promotes intestinal barrier integrity and AMP production via regulation of IL-22 production (112,113). In IECs, AhR is required for the proliferation of colonic stem cells as well as tight junction integrity and IL-10 receptor expression (114–116). Indeed, IEC-specific loss of AhR rendered mice highly susceptible to mouse models of colitis (117), highlighting the importance of IEC-intrinsic AhR signaling. In addition, AhR expression was sensitive to the presence of microbiota in IECs (118). AhR expression was also reduced in inflamed mucosal tissues from IBD patients (119) and IBD patients display reduced indole derivates, including indole-3-propionic acid serum levels (120), which have been shown to promote intestinal barrier integrity (71).
In addition to breakdown directly by the microbiota, tryptophan can be metabolized through the kynurenine pathway, which is mediated by the enzyme, indoleamine 2,3-dioxygenase-1 (IDO1), to produce kynurenine. Kynurenine concentrations are known to increase along the intestinal tract and play barrier protective and immunoregulatory roles (112). IDO1 was upregulated by the presence of the microbiota and mice deficient for IDO1 expression were highly susceptible to colitis models (112), suggesting an important role for IDO1 in mediating intestinal inflammation. Lastly, tryptophan can be metabolized by tryptophan hydroxylase 1 to produce serotonin within specialized IECs called enterochromaffin cells. While serotonin can be produced within the brain, the majority of serotonin is actually generated in the intestine (112). Serotonin is critical for neuron signaling in the central nervous system as well as in the enteric nervous system where it coordinates intestinal motility, secretion, and nutrient absorption. The microbiota regulates serotonin production as GF mice have reduced intestinal and systemic serotonin levels (112); however, the mechanism of how the microbiota mediates serotonin production remains unknown. Together, these studies highlight the multifaceted effects of tryptophan metabolism by microbiota in the intestine.
Succinate.
Succinate is an intermediate of the citric acid cycle and is produced by host cells as well as the microbiota. Many microbial commensals and pathogens have evolved metabolic pathways to thrive in the nutrient-rich, oxygen-deprived environment of the intestine; thus, production of succinate is a frequent by-product. Recent studies have demonstrated the necessity of succinate signaling by epithelial tuft cells for induction of type 2 immunity and protection against parasitic infections (121,122). Interestingly, succinate metabolism has recently been linked with IBD. IBD patients exhibited elevated fecal and serum levels of succinate and increased expression of the succinate receptor within the intestine (123). Similarly, loss of succinate signaling in mice was protective against the development of colitis (123), suggesting succinate may promote pro-inflammatory immune responses. However, further research will be needed to unravel the role of succinate metabolism in intestinal homeostasis.
Conclusion and future perspectives
The microbiota plays a significant role in regulating health and disease. IECs are non-hematopoietic cells that are uniquely positioned to receive signals from the microbiota and direct intestinal homeostasis. Increasing evidence demonstrates that IECs are well equipped to detect and respond to microbial products, and defects in these sensing pathways are commonly associated with inflammatory conditions, stressing the importance of these mechanisms. This Review highlights pathways by which IECs sense microbial signals to enhance epithelial barrier integrity and promote intestinal homeostasis. Beyond sensing and fortifying barrier functions, IECs play critical roles in orchestrating downstream immune responses to the microbiota and evading pathogens. IECs can direct immune responses through the secretion of numerous cytokines and chemokines. However, the exact role IECs play in promoting immune education and tolerance to the microbiota remain under investigation. Several studies have focused on understanding microbiota-immune cell interaction in early life. However, few have investigated how IECs incorporate signals from the developing microbiota to educate immune cells. Further understanding of the contribution of IECs in promoting intestinal tolerance to the microbiota may have profound consequences for intestinal homeostasis.
While numerous sensing pathways have been uncovered in IECs, the field of microbiota-derived metabolites and their role in intestinal health is still evolving. Studies have commonly focused on the contribution of a single metabolite, or a class of metabolites, on intestinal homeostasis. However, how different metabolites, or the whole metabolome, function in concert to mediate health and disease remain unknown. Future studies investigating how fluctuations in the microbiota diversity and overall metabolome affect IECs and intestinal integrity, and how these pathways can be therapeutically targeted will have wide-reaching implications for human health.
Acknowledgements
We thank members of the Alenghat lab for discussions and reading this work. This is supported by the National Institutes of Health (DK114123, DK116868 to T.A; F32AI147591 to E.M.E.; P30 DK078392), Pew Charitable Trust, and a Kenneth Rainin Foundation award to T.A. T.A. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Van Der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60. [DOI] [PubMed] [Google Scholar]
- 2.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14(3):141–53. [DOI] [PubMed] [Google Scholar]
- 3.Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551(7680):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019;567:49–55. [DOI] [PubMed] [Google Scholar]
- 5.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19(2):59–69. [DOI] [PubMed] [Google Scholar]
- 6.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandey S, Kawai T, Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol. 2015;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14(7):668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, et al. Deficient host-bacteria interactions in inflammatory bowel disease? the toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53(7):987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gewirtz AT, Vijay-Kumar M, Brant SR, Duerr RH, Nicolae DL, Cho JH. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn’s disease. Am J Physiol - Gastrointest Liver Physiol. 2006;290(6):1157–63. [DOI] [PubMed] [Google Scholar]
- 11.Pierik M, Joossens S, Van Steen K, Van Schuerbeek N, Vlietinck R, Rutgeerts P, et al. Toll-like receptor-1, −2, and −6 polymorphisms influence disease extension in inflammatory bowel diseases. Vol. 12, Inflammatory Bowel Diseases. 2006. p. 1–8. [DOI] [PubMed] [Google Scholar]
- 12.Török HP, Glas J, Endres I, Tonenchi L, Teshome MY, Wetzke M, et al. Epistasis between toll-like receptor-9 polymorphisms and variants in NOD2 and IL23R modulates susceptibility to crohn’s disease. Am J Gastroenterol. 2009;104(7):1723–33. [DOI] [PubMed] [Google Scholar]
- 13.Rakoff-Nahoum S, Pglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of comensal microflora by toll-like receptors in required for intestinal homeostasis. Cell. 2004;118:229–41. [DOI] [PubMed] [Google Scholar]
- 14.Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagenllin in colonic inflammation. Proc Natl Acad Sci U S A. 2005;102(38):13610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ii WAR, Sakamoto K, Leifer CA. TLR9 is important for protection against intestinal damage and for intestinal repair. Sci Rep. 2012;2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cario E, Gerken G, Podolsky DK. Toll-Like Receptor 2 Controls Mucosal Inflammation by Regulating Epithelial Barrier Function. Gastroenterology. 2007;132(4):1359–74. [DOI] [PubMed] [Google Scholar]
- 17.Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23(1):107–13. [DOI] [PubMed] [Google Scholar]
- 18.Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, et al. A Map of Toll-like Receptor Expression in the Intestinal Epithelium Reveals Distinct Spatial, Cell Type-Specific, and Temporal Patterns. Immunity. 2018;49(3):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu S, Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci. 2015;72(17):3343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting Edge: Bacterial Flagellin Activates Basolaterally Expressed TLR5 to Induce Epithelial Proinflammatory Gene Expression. J Immunol. 2001;167(4):1882–5. [DOI] [PubMed] [Google Scholar]
- 21.Schlee M, Wehkamp J, Altenhoefer A, Oelschlaeger TA, Stange EF, Fellermann K. Induction of human β-defensin 2 by the probiotic Escherichia coli Nissle 1917 is mediated through flagellin. Vol. 75, Infection and Immunity. 2007. p. 2399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lotz M, König T, Ménard S, Gütle D, Bogdan C, Hornef MW. Cytokine-mediated control of lipopolysaccharide-induced activation of small intestinal epithelial cells. Vol. 122, Immunology. 2007. p. 306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vora P, Youdim A, Thomas LS, Fukata M, Tesfay SY, Lukasek K, et al. β-Defensin-2 Expression Is Regulated by TLR Signaling in Intestinal Epithelial Cells. J Immunol. 2004;173(9):5398–405. [DOI] [PubMed] [Google Scholar]
- 24.Cash HL, Whitham CV., Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science (80-). 2006;313(August):1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukherjee S, Hooper LV. Antimicrobial Defense of the Intestine. Immunity. 2015;42(1):28–39. [DOI] [PubMed] [Google Scholar]
- 26.Hooper LV., Stappenbeck TS, Hong CV., Gordon JI. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4(3):269–73. [DOI] [PubMed] [Google Scholar]
- 27.Rumio C, Sommariva M, Sfondrini L, Palazzo M, Morelli D, Viganò L, et al. Induction of Paneth cell degranulation by orally administered Toll-like receptor ligands. J Cell Physiol. 2012;227(3):1107–13. [DOI] [PubMed] [Google Scholar]
- 28.Omagari D, Takenouchi-Ohkubo N, Endo S, Ishigami T, Sawada A, Moro I, et al. Nuclear factor kappa B plays a pivotal role in polyinosinic-polycytidylic acid-induced expression of human β-defensin 2 in intestinal epithelial cells. Clin Exp Immunol. 2011;165(1):85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204(8):1891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105(52):20858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science (80-). 2007;317(5834):124–7. [DOI] [PubMed] [Google Scholar]
- 32.Gong J, Xu J, Zhu W, Gao X, Li N, Li J. Epithelial-specific blockade of MyD88-dependent pathway causes spontaneous small intestinal inflammation. Clin Immunol. 2010;136(2):245–56. [DOI] [PubMed] [Google Scholar]
- 33.Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, et al. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun. 2014;82(9):3753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA, et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012;5(5):501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, et al. Activation of Enteroendocrine Cells via TLRs Induces Hormone, Chemokine, and Defensin Secretion. J Immunol. 2007;178(7):4296–303. [DOI] [PubMed] [Google Scholar]
- 36.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;11(3):215–215. [DOI] [PubMed] [Google Scholar]
- 37.Grondin JA, Kwon YH, Far PM, Haq S, Khan WI. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front Immunol. 2020;11(September):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 2018;39(9):677–96. [DOI] [PubMed] [Google Scholar]
- 39.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hugot J, Chamaillard M, Zouali H, Lesage S, Cezard J, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. [DOI] [PubMed] [Google Scholar]
- 41.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshiftmutation in xyhP associated with susceptibility to Crohn’s disease. Nature. 2001;411(May):603–6. [DOI] [PubMed] [Google Scholar]
- 42.Chen GY, Shaw MH, Redondo G, Nunez G, Núñez G. Innate immune receptor nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68(24):10060–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118(2):545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barreau F, Meinzer U, Chareyre F, Berrebi D, Nilwas-Kawakita M, Dussailliant M, et al. CARD15 / NOD2 Is Required for Peyer ‘ s Patches Homeostasis in Mice. PLoS One. 2007;2(6):e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Natividad JMM, Petit V, Huang X, De Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−;Nod2−/− Mice. Inflamm Bowel Dis. 2012;18(8):1434–46. [DOI] [PubMed] [Google Scholar]
- 46.Couturier-maillard A, Couturier-maillard A, Secher T, Rehman A, Normand S, De Archangelis A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petnicki-Ocwieja T, Hrncir T, Liu Y, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine: Commentary. PNAS. 2009;166(37):15813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (80-). 2005;307(5710):731–4. [DOI] [PubMed] [Google Scholar]
- 49.Muniz LR, Knosp C, Yeretssian G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol. 2012;3(OCT):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32(6):597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YG, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L, et al. The Nod2 Sensor Promotes Intestinal Pathogen Eradication via the Chemokine CCL2-Dependent Recruitment of Inflammatory Monocytes. Immunity. 2011;34(5):769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parlato M, Yeretssian G. NOD-like receptors in intestinal homeostasis and epithelial tissue repair. Int J Mol Sci. 2014;15(6):9594–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernandez EM, Valenti V, Rockel C, Hermann C, Pot B, Boneca IG, et al. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut. 2011;60(8):1050–9. [DOI] [PubMed] [Google Scholar]
- 54.Guo H, Gibson SA, Ting JPY. Gut microbiota, NLR proteins, and intestinal homeostasis. J Exp Med. 2020;217(10):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villani A-C, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn ‘ s disease susceptibility. Nat Genet. 2009;41(1):71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 Inflammasome Protects against Loss of Epithelial Integrity and Mortality during Experimental Colitis. Immunity. 2010;32(3):379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17(6):1359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, et al. Remodelling of the gut microbiota by hyperactive homeostasis. Nat Commun. 2017;8:1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol. 2019;39(3):277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Menu P, Vince J. The NLRP3 inflammasome in health and disease: the good, the bad, and the ugly. Clin Exp Immunol. 2011;166:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen GY, Liu M, Wang F, Bertin J, Núñez G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J Immunol. 2011;186(12):7187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Normand S, Delanoye-crespin A, Bressenot A, Huot L, Grandjean T. Nod-like receptor pyrin domain-containing protein colorectal carcinogenesis upon injury. PNAS. 2011;108(23):9601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wlodarska M, Thaiss CA, Nowarski R, Henao-mejia J, Zhang J, Brown EM, et al. NLRP6 Inflammasome Orchestrates the Colonic Host-Microbial Interface by Regulating Goblet Cell Mucus Secretion. Cell. 2014;156(5):1045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis through the Negative Regulation of Noncanonical NF- k B Signaling. Immunity. 2012;36(5):742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 2012;5(3):288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106(10):3698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4(219):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yap IKS, Li JV., Saric J, Martin FP, Davies H, Wang Y, et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-lnduced gut microbiota modification in the mouse. J Proteome Res. 2008;7(9):3718–28. [DOI] [PubMed] [Google Scholar]
- 70.Swann JR, Tuohy KM, Lindfors P, Brown DT, Gibson GR, Wilson ID, et al. Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. J Proteome Res. 2011;10(8):3590–603. [DOI] [PubMed] [Google Scholar]
- 71.Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 2017;551(7682):648–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lavelle A, Sokoi H. Gut microbiota- derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17:223–37. [DOI] [PubMed] [Google Scholar]
- 73.Kaiko GE, Ryu SH, Koues OI, Pearce EL, Oltz EM, Stappenbeck TS. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell. 2016;165(7):1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63(8):1275–83. [DOI] [PubMed] [Google Scholar]
- 76.Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. 2007;6(2):546–51. [DOI] [PubMed] [Google Scholar]
- 77.Harig J, Soergel K, Komorowski R, Wood C. Treatment of diversion colitis with short-chain fatty acid irrigation. N Engl J Med. 1987;320(1):23–8. [DOI] [PubMed] [Google Scholar]
- 78.Kanauchi O, Suga T, Tochihara M, Hibi T, Naganuma M, Homma T, et al. Treatment of ulcerative colitis by feeding with germinated barley foodstuff: First report of a multicenter open control trial. J Gastroenterol. 2002;37(SUPPL. 14):67–72. [DOI] [PubMed] [Google Scholar]
- 79.Breuer RI, Buto SK, Christ ML, Bean J, Vernia P, Paoluzi P, et al. Rectal irrigation with short-chain fatty acids for distal ulcerative colitis - Preliminary report. Dig Dis Sci. 1991;36(2):185–7. [DOI] [PubMed] [Google Scholar]
- 80.Scheppach W, Bartram HP, Richter F, Müller JG, Greinwald K, Tauschel HD, et al. Treatment of distal ulcerative colitis with short-chain fatty acid enemas. A placebo-controlled trial. Dig Dis Sci. 1996;41(11):2254–9. [DOI] [PubMed] [Google Scholar]
- 81.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Di Yu, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6. [DOI] [PubMed] [Google Scholar]
- 83.Miao W, Wu X, Wang K, Wang W, Wang Y, Li Z, et al. Sodium butyrate promotes reassembly of tight junctions in Caco-2 monolayers involving inhibition of MLCK MLC2 pathway and phosphorylation of PKCβ2. Int J Mol Sci. 2016;17:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng L, Li Z, Green RS, Holzman IR, Lin J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco2 Cell Monolayer. J Nutr 2009;139:1619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valenzano MC, Diguilio K, Mercado J, Teter M, To J, Wertheimer J, et al. Remodeling of Tight Junctions and Enhancement of Barrier Integrity of the CACO-2 Intestinal Epithelial Cell Layer by Micronutrients. PLoS One. 2015;10(7):e0133926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zheng L, Kelly CJ, Battista KD, Lanis JM, Alexeev EE, Wang RX, et al. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor − Dependent Repression of Claudin-2. J Im. 2017;199:2976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang H, Wang P, Wang X, Wan Y, Liu Y. Butyrate Enhances Intestinal Epithelial Barrier Function via Up-Regulation of Tight Junction Protein Claudin-1 Transcription. Dig Dis Sci. 2012;57:3126–35. [DOI] [PubMed] [Google Scholar]
- 88.Zhao Y, Chen F, Wu W, Sun M, Bilotta AJ, Yao S, et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Nat Publ Gr. 2018;11(3):752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Willemsen LEM, Koetsier MA, Van Deventer SJH, Van Tol EAF. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E1 and E2 production by intestinal myofibroblasts. Gut. 2003;52(10):1442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaudier E, Jarry A, Blottière HM, De Coppet P, Buisine MP, Aubert JP, et al. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol - Gastrointest Liver Physiol. 2004;287(6 50–6):1168–74. [DOI] [PubMed] [Google Scholar]
- 91.Wrzosek L, Miquel S, Noordine M-L, Bouet S, Chevalier-Curt MJ, Robert V, et al. Bacteroides Thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. Gastroenterology. 2013;144(5):S–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park J, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria : Role of Short-Chain Fatty Acids. PLoS One. 2016;1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu S en, Hashimoto-Hill S, Woo V, Eshleman EM, Whitt J, Engleman L, et al. Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature. 2020;586(7827):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A Is a G-protein – Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res. 2009;69(7):2826–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Venegas DP, Fuente MK De, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short Chain Fatty Acids (SCFAs) -Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol. 2019;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F, et al. Histone Hyperacetylation Is Associated with Amelioration of Experimental Colitis in Mice. J Immunol. 2006;176:5015–22. [DOI] [PubMed] [Google Scholar]
- 97.Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504(7478):153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Turgeon N, Blais M, Gagné JM, Tardif V, Boudreau F, Perreault N, et al. HDAC1 and HDAC2 Restrain the Intestinal Inflammatory Response by Regulating Intestinal Epithelial Cell Differentiation. PLoS One. 2013;8(9):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zimberlin CD, Lancini C, Sno R, Rosekrans SL, Mclean CM, Vlaming H, et al. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. 2015;29:2070–80. [DOI] [PubMed] [Google Scholar]
- 100.Gonneaud A, Turgeon N, Fran C, Boudreau SOIS, Perreault N, Rivard N, et al. Distinct Roles for Intestinal Epithelial Cell-Speci fi c Hdac1 and Hdac2 in the Regulation of Murine Intestinal Homeostasis. J Cell Physiol. 2015;231:436–48. [DOI] [PubMed] [Google Scholar]
- 101.Lorenzo-Zúñiga V, Bartolí R, Planas R, Hofmann AF, Viñado B, Hagey LR, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37(3):551–7. [DOI] [PubMed] [Google Scholar]
- 102.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.D’Aldebert E, Biyeyeme Bi Mve MJ, Mergey M, Wendum D, Firrincieli D, Coilly A, et al. Bile Salts Control the Antimicrobial Peptide Cathelicidin Through Nuclear Receptors in the Human Biliary Epithelium. Gastroenterology. 2009;136(4):1435–43. [DOI] [PubMed] [Google Scholar]
- 104.Termén S, Tollin M, Rodriguez E, Sveinsdóttir SH, Jóhannesson B, Cederlund A, et al. PU.1 and bacterial metabolites regulate the human gene CAMP encoding antimicrobial peptide LL-37 in colon epithelial cells. Mol Immunol. 2008;45(15):3947–55. [DOI] [PubMed] [Google Scholar]
- 105.Gadaleta RM, Van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60(4):463–72. [DOI] [PubMed] [Google Scholar]
- 106.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J Immunol. 2009;183(10):6251–61. [DOI] [PubMed] [Google Scholar]
- 107.Ruiz L, Margolles A, Sánchez B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front Microbiol. 2013;4(DEC):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59. [DOI] [PubMed] [Google Scholar]
- 110.Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, et al. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci U S A. 2014;111(20):7421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, et al. Connecting dysbiosis, bile-acid dysmetabolism and Gut inflammation in inflammatory bowel diseases. Gut. 2013;62(4):531–9. [DOI] [PubMed] [Google Scholar]
- 112.Agus A, Planchais J, Sokol H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe. 2018;23(6):716–24. [DOI] [PubMed] [Google Scholar]
- 113.Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity review-article. Mucosal Immunol. 2018;11(4):1024–38. [DOI] [PubMed] [Google Scholar]
- 114.Stockinger B, Meglio P Di, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu Rev Immunol. 2014;32:403–32. [DOI] [PubMed] [Google Scholar]
- 115.Bansal T, Alaniz RC, Wood TK, Jayaraman A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc Natl Acad Sci U S A. 2010;107(1):228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Landis J, Alexeev E, Curtis V, Kitzenberg D, Kao D, Battista K, et al. Tryptophan Metabolite Activation of the Aryl Hydrocarbon Receptor Regulates IL10 Receptor Expression on Intestinal Epithelia. Mucosal Immunol. 2017;10(12):1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chinen I, Nakahama T, Kimura A, Nguyen NT, Takemori H, Kumagai A, et al. The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. Int Immunol. 2015;27(8):405–15. [DOI] [PubMed] [Google Scholar]
- 118.Wang J, Wang P, Tian H, Tian F, Zhang Y, Zhang L, et al. Aryl hydrocarbon receptor/IL-22/Stat3 signaling pathway is involved in the modulation of intestinal mucosa antimicrobial molecules by commensal microbiota in mice. Innate Immun. 2018;24(5):297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141(1):237–248.e1. [DOI] [PubMed] [Google Scholar]
- 120.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22(6):598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell. 2018;174(2):271–284.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Remodeling SI, Schneider C, Leary CEO, Moltke J Von, Pellizzon M, Ma A, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell. 2018;174:271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Macias-Ceja DC, Ortiz-Masiá D, Salvador P, Gisbert-Ferrándiz L, Hernández C, Hausmann M, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 2019;12(1):178–87. [DOI] [PubMed] [Google Scholar]