

Abstract
Distinct mutation signatures arise from environmental exposures and/or from defects in metabolic pathways that promote genome stability. The presence of a particular mutation signature can therefore predict the underlying mechanism of mutagenesis. These insults to the genome often alter dNTP pools, which itself impacts replication fidelity. Therefore, the impact of altered dNTP pools should be considered when making mechanistic predictions based on mutation signatures. We developed a targeted deep-sequencing approach on the CAN1 gene in Saccharomyces cerevisiae to define information-rich mutational profiles associated with distinct rnr1 backgrounds. Mutations in the activity and selectivity sites of rnr1 lead to elevated and/or unbalanced dNTP levels, which compromises replication fidelity and increases mutation rates. The mutation spectra of rnr1Y285F and rnr1Y285A alleles were characterized previously; our analysis was consistent with this prior work but the sequencing depth achieved in our study allowed a significantly more robust and nuanced computational analysis of the variants observed, generating profiles that integrated information about mutation spectra, position effects, and sequence context. This approach revealed previously unidentified, genotype-specific mutation profiles in the presence of even modest changes in dNTP pools. Furthermore, we identified broader sequence contexts and nucleotide motifs that influenced variant profiles in different rnr1 backgrounds, which allowed specific mechanistic predictions about the impact of altered dNTP pools on replication fidelity.
Keywords: DNA replication, replication fidelity, mutagenesis, dNTP pools, ribonucleotide reductase, next-generation sequencing
Introduction
Specific environmental exposures and/or genetic backgrounds generate distinct mutation signatures (Chan et al. 2012, 2015; Saini et al. 2020). Therefore, characterizing mutations provides insight into the underlying molecular mechanisms of mutagenesis in the absence of information about genotype or exposure. For example, C→T mutations have long been associated with UV exposure (Howard and Tessman 1964). This is the basis for the Catalog of Somatic Mutations in Cancer (COSMIC), which has curated somatic mutation signatures in cancers to infer mutagenic mechanisms underlying cancer development (Tate et al. 2019). Our goal in this study was to develop a broader analytic pipeline that allowed us to build genotype-specific mutation profiles from the ground up, using both canonical and signature mutations (Brash 2015), as well as sequence context.
In Saccharomyces cerevisiae, mutation spectra are frequently determined via one of two general strategies. The first requires selection of mutants, most often at the CAN1 locus (Xu et al. 2008; Kumar et al. 2011; Buckland et al. 2014), which encodes an arginine permease that also imports the toxic arginine analog, L-canavanine, leading to cell death (Fantes and Creanor 1984; Hoffmann 1985). Inactivating mutations in CAN1 block canavanine uptake and toxicity, allowing selection for mutations in CAN1. The CAN1 gene from individual resistant colonies is then amplified by PCR and subjected to Sanger sequencing. However, this approach is relatively low throughput (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2011) and will miss low-frequency variants (below 15–20%) that might exist within a resistant colony (Rohlin et al. 2009). The second approach has been mutation accumulation (MA) experiments followed by WGS, which avoids potential bias associated with focusing on a single genetic locus (i.e., CAN1). However, the depth of sequencing is limited because the entire genome is sequenced, complicating statistically significant comparisons among genotypes (Lujan et al. 2014; Zhu et al. 2014; Rentoft et al. 2016). Increased depth requires increased numbers of MA lines and many passages are required to accumulate sufficient mutations and low-frequency mutations that are potentially diagnostic of a genotype will be missed.
We developed a high-throughput sequencing approach and bioinformatic pipeline that allowed characterization and comparison of robust mutation profiles from different RNR1 genetic backgrounds. RNR1 encodes the large subunit of ribonucleotide reductase (RNR), the enzyme that catalyzes the rate-limiting step in dNTP synthesis. RNR expression and activity is tightly regulated to promote replication fidelity. Mutations that affect the allosteric regulation of RNR increase and/or skew dNTP pools and increase mutation rates to varying degrees (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010, 2011; Buckland et al. 2014; Watt et al. 2016). Altered dNTP pools compromise replication fidelity by: (1) increasing the frequency of misinsertion and misalignment by replicative DNA polymerases and (2) biasing those polymerases toward synthesis at the expense of proofreading, promoting extension beyond a mispair (Phear et al. 1987; Kunkel and Soni 1988; Kumar et al. 2010). Notably, altered RNR activity is associated with cancer and nucleobase analogs are frequently used as chemotherapeutics, commonly targeting RNR (Jordheim et al. 2013; Aye et al. 2015; Kohnken et al. 2015; Mathews 2015, 2018). Overexpression of Rrm2, encoding the small subunit of RNR, induced lung neoplasms in a mouse model, consistent with an elevated mutator phenotype (Xu et al. 2008).
We focused on three rnr1 alleles (rnr1D57N, rnr1Y285F, and rnr1Y285A) that decrease replication fidelity by altering dNTP pools (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010). We chose these rnr1 alleles because: (1) they have been demonstrated to have elevated mutation rates and (2) there was both Sanger and MA WGS sequencing data available for comparison and validation of our analytic approach (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010, 2011; Buckland et al. 2014; Watt et al. 2016). We paired selection for mutations at CAN1 with next-generation sequencing (NGS) to define mutation profiles for RNR1, rnr1D57N, rnr1Y285F, and rnr1Y285A. Importantly, mutations in CAN1 have been found to be representative of mutations occurring genome-wide when Sanger and WGS approaches were compared (Kumar et al. 2011; Buckland et al. 2014; Watt et al. 2016). While the majority of mutations identified were CAN1 inactivating mutations, the variant types (single nucleotide changes and small insertions and deletions) are not necessarily inactivating in other genomic contexts. The variants and the sequence contexts in which they occurred revealed mutations that could occur throughout the genome. While we recognize that some mutations may be missed via selection, pairing NGS with selection for mutations significantly increased sequencing depth compared with previous studies and allowed us to innovate in our computational analysis.
In this study, we defined and characterized mutation profiles in wildtype yeast and in isogenic strains bearing the rnr1D57N, rnr1Y285F, and rnr1Y285A alleles (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010). We built comprehensive mutation profiles from first principles, which consisted of analysis of the specific types and positions of variants observed, the trinucleotide context in which the variants occurred and the broader sequence context of the variants, identifying motifs that were specifically enriched or depleted in distinct rnr1 backgrounds. Importantly, and in contrast to previous work (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010, 2011; Buckland et al. 2014; Watt et al. 2016), our analysis defined novel, distinct profiles in all three rnr1 backgrounds, with rnr1Y285A exhibiting the most unique profile. We noted many of the same rnr1Y285A mutation motifs determined by MA WGS (Watt et al. 2016), while also revealing new high and low-frequency variants for rnr1Y285A and unique profiles in rnr1Y285F and rnr1D57N, indicating that even small changes in dNTP pools contribute to mutagenesis. Therefore, we propose that genotype-specific mutation profiling at CAN1 is complementary to MA WGS.
Materials and methods
Strains and plasmids
All strains in this study were derived from the W303 RAD5+ background (Supplementary Table S1). Strains containing rnr1Y285F/A in the pGAL-RNR1 background and integration plasmids to re-create these strains (Kumar et al. 2010) were kindly provided by Andrei Chabes. To integrate rnr1 alleles at the endogenous RNR1 locus in the absence of pGAL-RNR1, we created a new set of rnr1 integration plasmids. First, we amplified pRS416 (Sikorski and Hieter 1989) with SO261 and SO262, which each contain an AatII recognition site (Supplementary Table S2). The resulting PCR product, which consists of pRS416 without the ARS/CEN region, was digested with AatII and ligated to generate the yeast integration plasmid, pEM3, which carries a counter-selectable URA3 marker (Supplementary Table S3). RNR1 was amplified from JSY13 with SO263 (Supplementary Table S2; encodes a XhoI site) and SO265 (Supplementary Table S2; encodes a SpeI site) to amplify a product that extends ∼1 kb upstream of the endogenous RNR1 start site to ∼3 kb downstream of the RNR1 start site, to capture the full RNR1 gene. This fragment was digested with XhoI and SpeI and ligated into pEM3 to generate the RNR1 integration plasmid pNL1 (Supplementary Table S3). To generate a truncated version of RNR1 in the same plasmid, RNR1 was amplified from JSY13 with SO263 (encodes a XhoI site) and SO264 (Supplementary Table S2; encodes a SpeI site) to amplify a product that extends ∼1 kb upstream of the endogenous RNR1 start site to ∼2 kb into the RNR1 open reading frame. The PCR product was digested with XhoI and SpeI and ligated into pEM3 to generate pNL2 (Supplementary Table S3).
We generated versions of pNL1 and pNL2 that encode rnr1D57N, rnr1Y285F and rnr1Y285A using the Q5 site-directed mutagenesis kit (New England Biolabs) (Supplementary Table S3). Oligonucleotides used for the mutagenesis are described in Supplementary Table S2. Plasmids were sequenced to confirm the presence of the desired mutations and absence of secondary mutations. To integrate each rnr1 allele into the endogenous RNR1 locus, both the full-length and truncated integration plasmid for a given allele were linearized with Sph1 and used to co-transform JSY13 using the standard lithium acetate approach (Gietz et al. 1992). Transformants were selected on synthetic complete medium lacking uracil (SC-URA). Single colonies were patched onto YPD to allow for homologous recombination and transplacement of one copy of rnr1. After 2 days growth at 30 °C, cells from these patches were struck out to obtain single colonies on plates containing 5-FOA (0.1%), selecting for a loss of URA3. Plasmid integration was confirmed by PCR using SO266 (Supplementary Table S2; specific to the integration plasmid) and SO230 (Supplementary Table S2; specific to the genomic locus). Sanger sequencing was used to confirm retention of a mutant rnr1 allele and a loss of RNR1. rnr1 was amplified in two fragments for sequencing, using the SO32/SO21 and SO25/SO22 primer pairs (Supplementary Table S2).
CAN1 mutation rate analysis
Mutation rates were determined through canavanine resistance assays as described previously (Xu et al. 2008). Strains were struck out to single colonies on complete media. Individual 2 mm colonies were carefully measured and selected to assay. Colonies were suspended in 100 µl in TE (10 mM Tris-HCl, pH 7.5; 1 mM EDTA); 75 µl of undiluted colony suspension was plated on SC-ARG +canavanine plates. The cell suspension was diluted 1:10,000 and 20 µl was plated under permissive conditions on SC-ARG. At least two independent isolates of 11 colonies were assayed for each genotype. Isolates were first analyzed independently before grouping data to calculate rates. Mutation rates and confidence intervals were calculated utilizing FluCalc fluctuation analysis software (Radchenko et al. 2020).
Pooling canavanine resistant colonies
Strains were patched on SC-ARG plates and grown at 30 °C for 3 days, ∼30–35 generations. A quarter of the patch was used to inoculate a 25 mL SC-ARG liquid culture. A patch was used to inoculate the culture instead of a single colony to reduce the potential effects of jackpot mutations in the cultures. Cells were grown for approximately 3–4 additional generations and then plated on SC-ARG + canavanine plates, to select for approximately 2,000 canavanine resistant (CanR) colonies. It required another ∼30 generations to generate colonies, for a total of ∼65–70 generations of accumulated mutations, although mutations that confer canavanine resistance should occur in the first 35–40 generations. CanR colonies were selected in at least four independent experiments per genotype. For each genotype, at least two independent isolates were used (Supplementary Table S4).
Colonies were counted and collected by adding TE (100 mM Tris-HCl, pH 7.4, 10 mM EDTA) to the plate and using a sterile glass spreader to scrape off the cells. Colonies from multiple plates were pooled and resuspended in TE (pH 7.4) to a final volume of 10–12 mL. One mL of the colony suspension was used to extract genomic DNA (gDNA). Briefly, cells were lysed by vortexing in 200 µl 1:1 phenol: chloroform, 200 µl chromosome preparation buffer (10 mM Tris,-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1% SDS, 2% Triton X-100) and 0.3 grams acid-washed glass beads (Sigma; 425–600 microns). Two-hundred microliter TE pH 8.0 was added and reactions were centrifuged for 5 minutes at 16,000 × g. The resulting supernatant was collected. Three additional phenol: chloroform extractions were performed to increase DNA purity. gDNA was precipitated by addition of ammonium acetate to a final concentration of 100 mM followed by 2 volumes of 95% ethanol. The gDNA was collected by centrifugation at 16,000 × g for 10 minutes, followed by a wash with 70% ethanol. The gDNA pellets were resuspended in 50 µl nuclease free water with RNase A (final concentration of 50 µg/ml) and incubated at room temperature for at least 1 hour after which gDNA was stored at −20°C.
Pooling colonies growing on nonselective media
In addition to canavanine resistant colonies, we also pooled ∼2,000 colonies grown on nonselective media from RNR1, rnr1D57N, rnr1Y285F and rnr1Y285A backgrounds as permissive controls. These colonies were grown as described above, except that in the final step, cells were grown on SC-ARG in the absence of canavanine. Colonies were pooled and genomic DNA extracted as described above.
Library preparation and sequencing
CAN1 was amplified from gDNA in 6 overlapping, 349-350 base pair fragments using primers listed in Supplementary Table S2. KAPA HiFi ReadyMix (Roche) was used to amplify these fragments in 25 μl reactions for each CAN1 region for each of the 150 samples (total of 906 reactions). Two-microliter of DNA was added to each reaction, in the range of 50–500 ng. Two-microliter of each PCR reaction was electrophoresed on a 1% agarose gel to confirm amplification. For each sample, 20 μl of PCR product from each of the 6 regions (each ∼ 300 bp) were pooled in a 96-well plate and purified using the Zymo ZR-96 DNA Clean-up Kit. A total of 65 pooled sample sets (Supplementary Table S4) were generated for paired-end sequencing (2 × 300), including technical replicates. PCR products from the same genomic preparation of pooled samples were independently amplified and sequenced.
Library barcoding and QC
Nextera barcode adapters were added to CAN1 amplicons and were then minimally PCR amplified (8 cycles) for attachment of Illumina Nextera XT index primers set A (Illumina). After PCR, excess adapters were removed using Ampure XP beads (Beckman Coulter) and samples were eluted into EB buffer. Barcoded amplicons were checked for quality using an Advanced Analytical Fragment Analyzer and Qubit Fluorescence (Invitrogen). Amplicons were pooled to 10 nM in EB buffer and the final concentration was determined using the Illumina Universal qPCR Amplification kit from Kapa Biosystems. All pooled samples were diluted to 4 nM, denatured using NaOH and loaded onto an Illumina MiSeq sequencing platform (PE300, V3) with 20% PhiX control. The sequencing was performed in two separate runs to increase coverage and as a check for reproducibility.
Upstream sequencing analysis
Reads were trimmed using a variable length trimmer (CutAdapt version 1.14) specifying a quality score of Q30. Trimmed reads were further processed using CLC Genomics Workbench Version 11. Paired-end reads were merged, primer locations were trimmed, and processed reads were aligned to the SacCer3 reference genome. Variants were then called using the CLC low-frequency variant caller with required significance of 0.01%. Variant files were exported from CLC as VCF files and downstream analysis was performed in RStudio (version 1.2.1335), paired with custom python scripting (Figure 1).
Figure 1.

A schematic of (A) experimental design and (B) pipeline for data analysis.
The variant classes included 6 possible single nucleotide variants (SNVs), single base A/T or G/C insertions and deletions, complex insertions and deletions, as well as multinucleotide variants (MNVs) and replacements (Replac.). MNVs are dinucleotide SNVs, where two neighboring nucleotides are both mutated, e.g., CC>AT. Replacements are complex insertions or deletions, where the deleted or replaced base is a variant. Two examples include AAC>G and C>AT. Both MNVs and replacements are extremely low-frequency events and rarely occurred in our data set; neither had a significant impact on clustering. Our initial analysis assessed the frequency of each variant type as a function of genotype.
Mutation spectra visualization
Variants across biological replicates were analyzed in two different ways. The first, was a more conservative approach based on presence or absence of a variant at a particular position within CAN1 for a given genotype, referred to as “unique counts.” Each position-specific, unique variant was counted only once per replicate and the fraction of biological replicates in which it was observed was scored. In our NGS approach, we cannot easily distinguish between a mutation appearing early in the growth of the culture and a mutation occurring independently multiple times. By not considering the variant frequency in this analysis, we eliminated this concern. It also mitigated the effects of any “jackpot” mutations that might skew variant frequencies. The second approach incorporated the frequency of each unique variant across CAN1 (sum of frequencies). This approach added the sum of the variant frequencies if a particular variant occurred in multiple biological replicates. This analysis generated a genotype-specific mutation profile that incorporates variant type, frequency, and position. Overall, the results from both types of analysis were consistent when mutation spectra were visualized.
Permissive variant analysis and filtering
We applied a filter based on our permissive samples (RNR1, rnr1D57N, rnr1Y285F, rnr1Y285A grown in the absence of selection) to remove background mutations from CanR samples (Figure 2). The permissive filter removes any variant that occurs below the average permissive sample unique variant frequency of 0.109% (Figure 3C). We customized the permissive filter because we also observed position-specific variants at a frequency higher than the overall average permissive variant frequency of 0.109%. Thus, these systematically higher frequency variants were removed from the selected samples if they occurred at a frequency below the highest frequency variant that occurred at the same position in the permissive data set. Figure 3, E–G shows an example of the permissive filter applied. This is a conservative filter, and undoubtedly low-frequency events that are biologically relevant may be removed but are below the sensitivity of this assay. The filtering parameters can be adjusted accordingly, for other applications of this targeted sequencing approach.
Figure 3.

Permissive controls show consistent mutation spectra and are filtered from our data set. (A) The SNV spectra for all permissive samples. Plotted are the number of unique variants in a sample normalized out of 100% for comparison purposes. Individual biological replicates for each genotype are shown. (B) The deletion spectra for each permissive sample. Shown are single G/C (blue), A/T (green) base deletions and deletions greater than 1 base (pink). Individual biological replicates for each genotype are shown. (C) A histogram plotting the number of variants (y-axis) and the frequency (x-axis) at which they occur in both permissive and (D) selected samples. The red vertical line represents the average variant frequency in permissive and selected samples respectively. (E) A 100 bp window from 32,000 to 32,100 displaying the different types of variants (deletions [blue circles]; insertions [magenta circles]; MNV [green circles]; replac. [red circles]; SNV [orange circle]) and the frequency at which they occur in 4 different permissive genotypes. (F) The same window displaying the variants that occur in the same four genotypes where mutants were selected in the presence of canavanine. (G) Variants that remain in selected samples post-permissive variant filter.
Note: Figure Replacement Requested.
Determining SNV trinucleotide context
The trinucleotide context surrounding the SNV was determined by taking a 3 bp window surrounding the reference position within CAN1. We cannot definitively determine which strand incurred a mutation and therefore all SNVs were categorized as C or T changes for this analysis. There are a total of 96 different possible SNV changes in unique sequence context (Alexandrov et al. 2015). For a given sample, the number of SNVs in each of these 96 contexts was totaled. This analysis does not take frequency into account and only scores the presence or absence of a particular type of SNV. The data was further condensed by taking the average of each of the 96 different contexts for all the biological replicates in one genotype.
The number of trinucleotide sequence contexts in CAN1 was calculated using a sliding window approach utilizing python scripting. For each of the 96 different SNV changes in triplet context, the average number of SNVs in a genotype was divided by the number of times the triplet sequence context occurs in CAN1. This data set was imported into R-studio and plotted via the barplot() function.
Hierarchical cluster analysis and motif enrichment
To identify genotype-specific variants and mutation profiles and to eliminate frequency bias from variants that occurred early on in the growth of a particular culture, we condensed unique variants based on the number of biological replicates sequenced for that genotype. While we were hesitant to include variant frequency in this analysis, we reasoned that observing a variant in multiple replicates increased the probability that it was specific to that genotype. If a variant occurred in 4 out of 4 biological replicates it was represented as 1, if it occurred in 3 out of 6 replicates it was represented as 0.5. This strategy provided an unbiased way to assess the probability that a given variant was genotype-specific. These data were clustered on rows (or unique variants), after applying a row sum cut-off of >-2 to eliminate low-frequency variants that are less likely to be driving the observed differences in mutation spectra. Clustering the data based on unique variants allows us to identify different types of mutations in specific sequence contexts that are potentially predictive of a genotype. We performed motif enrichment on the different variant classes (i.e., G/C deletion, CG>AT SNVs) independently, with a 12 base window surrounding the variant. Heatmaps were plotted using the pheatmap library in RStudio and motif enrichment was performed using Berkely web logos (Crooks et al. 2004).
Data availability
All data and reagents are available upon request. All variant sequences are provided in Supplementary Table S5. The Supplemental Material is available at figshare: https://doi.org/10.25387/g3.14187080.
Results
Rates of canavanine resistance in different genetic backgrounds
To characterize and compare mutation profiles between different isogenetic backgrounds, we genetically altered dNTP pools using previously characterized mutations in RNR1, the large subunit of RNR. We chose RNR because alterations in dNTP pool levels generate distinct mutation profiles and RNR is associated with cancer (Xu et al. 2008; Kumar et al. 2010, 2011; Buckland et al. 2014; Aye et al. 2015; Mathews 2015, 2018; Watt et al. 2016). The rnr1D57N mutation, in the activity site, leads to a balanced twofold increase in each of the four dNTPs (Chabes et al. 2003). Two mutations in the RNR specificity site, rnr1Y285F and rnr1Y285A, lead to threefold and 20-fold skewed increases in dCTP and dTTP, respectively (Kumar et al. 2011). The previously characterized rnr1Y285F and rnr1Y285A strains also encoded a wild-type copy of RNR1 under control of the inducible pGAL1 promoter (pGAL-RNR1) (Kumar et al. 2010). All strains were grown in glucose, which restricts expression of RNR1 from the pGAL promoter, but leaky expression remained a possibility. Therefore, we also constructed strains that encoded only rnr1Y285F and rnr1Y285A for comparison and for use in this study (Supplementary Table S1).
We determined mutation rates of all strains at the CAN1 locus using a canavanine resistance assay (see Materials and Methods). The rnr1D57N and rnr1Y285F strains exhibited low mutation rates (∼threefold increase), while the rnr1Y285A strain exhibited a larger ∼10–20-fold increase in mutation rate (Kumar et al. 2010; Supplementary Table S6), consistent with previous work (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2010, 2011). The pGAL-RNR1 construct did not affect mutation rates in rnr1Y285F, but did modulate levels of mutagenesis in the rnr1Y285A background despite growth in glucose (Supplementary Table S6).
Selection at CAN1 paired with next generation sequencing (NGS) to define mutation profiles
To generate mutation sequence data in RNR1/rnr1 backgrounds, we selected ∼ 2,000 canavanine resistant colonies (“selected samples”), each of which contained at least one mutation within CAN1. Resistant colonies were pooled, genomic DNA was extracted and CAN1 was amplified in 6 overlapping regions. To amplify the 5′ end of CAN1, which includes a highly repetitive promoter element, we used a primer that annealed to that element, to avoid replication slippage during PCR or sequencing (Supplementary Table S2: CAN1 reg1 Forward_anchored). Amplicons from each pooled sample, representing a single replicate for a given genotype, were barcoded, combined and sequenced using 2 × 300 paired-end sequencing (see Materials and Methods) (Figure 1A).
Identifying genotype-specific trends in mutation profiles is complicated by the stochastic nature of mutations. To help account for this, we sequenced at least four independent samples for each genotype, using at least two independently generated isolates (Supplementary Tables S1 and S4). In parallel, we sequenced pooled samples (∼1,000 colonies each) grown in the absence of canavanine selection, which we called “permissive” samples (Figure 1A). These permissive controls allowed us to develop a threshold for background mutations at each position along CAN1 that are a result of low frequency, stochastic mutations and sequencing and/or PCR polymerase bias (see below).
We developed a custom bioinformatic pipeline for sequence analysis. Upstream analysis was performed in CLC Genomics Workbench 11, determining variants utilizing a low-frequency variant caller, and downstream analysis was performed in R Studio and through custom python scripts to determine the effect of sequence context and to compare genotypes (Figure 1B; see Materials and Methods and below for details). All sequence data were scored for: (1) SNVs, (2) single base (A/T or G/C) insertions or deletions, (3) complex (>1 base) insertions or deletions, (4) dinucleotide SNVs at adjacent nucleotides, i.e., MNVs, and (5) complex replacements (see Materials and Methods). We could not definitively determine which strand incurred a mutation; a C to A transition could also be a G to T change and was represented as such, i.e., CG>AT. NGS allowed deep sequencing of pooled samples at each position along CAN1, providing: (1) large sample sizes for each pooled group, (2) sequencing depth sufficient to uncover low-frequency variants, and (3) novel insight into mutation profiles and positional effects on mutations, all of which would be unattainable via a whole genome approach because the sequencing depth/coverage is insufficient for this type of analysis.
On average CAN1 was sequenced at a depth of 16,000x coverage per sample. The total variant frequency for each sample was calculated by taking the sum of the number of variants and dividing by the total number of reads sequenced for that sample. The variant frequencies for all biological replicates within a genotype were averaged. The total variant frequency for all selected samples ranged from ∼80 to >100%, with an average of 99.35% (Figure 2A). Frequencies above 100% indicated more than 1 mutation within CAN1 in a CanR colony, and were observed in rnr1Y285A strains, which had higher mutation rates (Supplementary Table S6).
Figure 2.

Absolute variant frequency is consistent with selection at CAN1. The average absolute variant frequency was calculated for each genotype by dividing the total number of variants by the total number of reads sequenced. The average was then taken for all biological replicates sequenced in a genotype. Error bars represent the standard deviation between biological replicates within each genotype. The number of total biological replicates sequenced varied by genotype; numbers are displayed in Supplementary Table S4. (A) The average total variant frequency differed significantly between selected and permissive samples, regardless of genotype. (B) The average variant frequency decreased following application of the permissive variant filter.
Permissive sample filtering removes background mutations
Under permissive conditions (no canavanine selection), the average variant frequency for all four genetic backgrounds (wildtype, rnr1D57N, rnr1Y285F, and rnr1Y285A) was 8.7% compared to >90% in the samples selected in the presence of canavanine (Figure 2A). While the variant frequencies in permissive controls were lower, they were higher than expected in the absence of selection, indicating that these background mutations were being introduced as part of the PCR/sequencing pipeline. Consistent with this prediction, the variant distribution in permissive controls from wildtype, rnr1D57N, rnr1Y285F, and rnr1Y285A were virtually identical between genotypes, despite differences in mutation rates and mutation spectra in selected samples (Chabes et al. 2003; Xu et al. 2008) (Supplementary Table S6, Figure 3, A and B). Of the 296 unique variants observed in all permissive samples, 82 (27.7%) were observed in all 4 genotypes; over half were observed in at least 2 genetic backgrounds. Moreover, the SNV spectra from permissive samples, independent of genotype (Figure 3A), closely resembled the SNV spectrum observed for KAPA HiFi polymerase (Oyola et al. 2012; Potapov and Ong 2017), with a large bias toward CG>TA changes that represented ∼70% of all SNVs. The small number of unique variants observed and the significant overlap in variant and position among genotypes, precluded a genotype-specific analysis of the permissive samples. This overlap is illustrated in Figure 3E which shows the variant profile from a section of CAN1 for all four genotypes grown in the absence of selection. Note that the variant patterns are low frequency and are largely superimposable.
To correct for these effects, we developed a permissive variant filter to remove potentially artefactual variants as part of the analytical pipeline. We determined the average frequency of each variant at each position along CAN1 observed in permissive samples, which was 0.109% compared to 0.407% in the selected samples (Figure 3, C and D). Any variant that occurred below the average permissive variant frequency of 0.109% was removed by the permissive filter, which represents ∼1 variant called for every 1,000 reads sequenced. Some position-specific variants systematically occurred at average frequencies above 0.109%, in multiple biological replicates and genotypes. We incorporated this into the permissive filter, setting the threshold for each position-specific variant to its highest observed frequency in a permissive sample. The position-specific variant frequencies were consistent across genotypes.
This conservative approach ensured that our analysis of mutation profiles in selected samples (see below) was not driven by background variants. We set a blanket cutoff at 0.109%, rather than making the filter exclusively position-specific, because we sequenced significantly fewer permissive samples (18) than selected (47). Therefore, the permissive filter likely underestimates stochastic mutations. The higher the number of samples sequenced, the greater the probability that low-frequency mutations due to “noise” will be sequenced. Figure 3E illustrates the application of this filter, where the filter cutoff is 0.109%, except for positions 32057, 32081, and 32092, where variants occurred at position-specific higher average frequencies in the permissive samples. The application of this filter reduced the noise in the selected data (Figure 3, F and G), decreased variant frequency in selected samples by an average of 7.5% (Figure 2B) and resulted in only minor changes to mutation spectra overall (Supplementary Figure S1).
Alterations in dNTP pools change mutation profiles
We determined the mutation profiles for each individual replicate (Supplementary Figure S2) and then compared the relative levels of transitions, transversions, in/dels and other variants from previous analyses of rnr1D57N, rnr1Y285F pGAL-RNR1 and rnr1Y285A pGAL-RNR1 (Xu et al. 2008; Kumar et al. 2011) with our data sets (Figure 4), using pooled biological replicates (Figure 5). The distributions for the same genotypes were very similar, indicating that our sequencing approach and analysis pipeline replicated these previous results. The most pronounced differences were observed in the rnr1D57N spectra, likely a result of the small sample size (n = 16 CanR colonies) used in the previously published results (Xu 2008). We also compared the relative rates of each type of variant that we observed with previous work (Xu et al. 2008; Kumar et al. 2011; Buckland et al. 2014), which were also similar (Supplementary Table S6). Because of the depth of sequencing in our data, we were able to perform a more nuanced analysis (see below).
Figure 4.
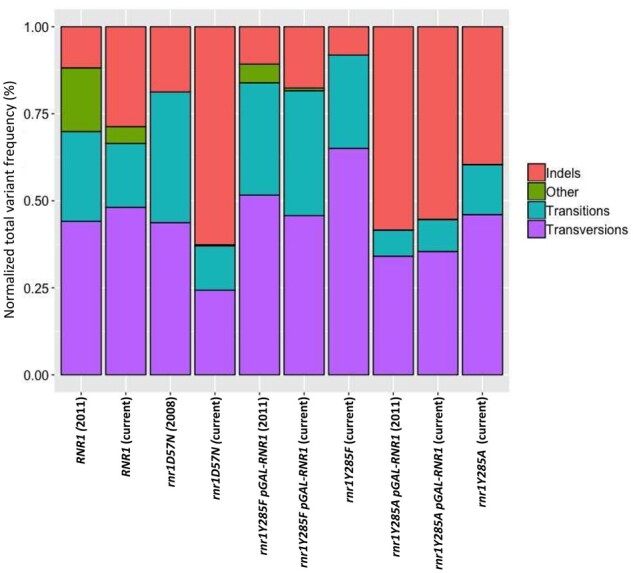
A comparison to previous studies which utilized Sanger sequencing. Variant counts determined using Sanger sequencing from previous studies (Xu et al. 2008; Kumar et al. 2011) were compared with the normalized sum of frequencies from genotypes in our study. Relative frequencies were similar across studies.
Figure 5.

Mutation spectra vary by rnr1 allele. (A) The SNV spectra normalized to total SNVs (upper panel) and normalized out of total variants (lower panel). (B) The deletion spectra normalized to total deletions (upper) and normalized to total variants (lower). (C) The insertion spectra normalized to total insertions (upper) and normalized out of total variants (lower).
After the initial analysis, we characterized mutation events in two ways: (1) the presence of a particular variant at a unique position, i.e., the number of different CG>AT changes within CAN1 (“unique variants”) (Counts in Supplementary Table S7) and (2) the frequency at which each of these unique variants occurred, i.e., the combined frequency of all CG>AT observed changes along CAN1 (“sum of frequencies”) (Freq. in Supplementary Table S7) (see Materials and Methods). The latter essentially provides mutation spectra but is structured to incorporate position information. The former prevented potential “jackpot” mutations from dominating and skewing the mutation spectra. These analyses allowed us to determine whether different types of mutations occurred in a genotype-dependent manner, independent of frequency, and whether variant frequencies were altered in a significant way (Counts/Freq. in Supplementary Table S7). For example, a decreased number for “unique variants” combined with unchanged or increased “sum of frequencies” indicated that variant type is more localized, possibly indicating a mutational hotspot. Independent biological replicates with the same genotype closely resembled each other (Supplementary Figure S2); variant frequencies were more variable than unique counts among biological replicates of the same genotype (Supplementary Figure S2), consistent with the stochastic nature of MA during the growth of a culture. Distinct mutagenic events were observed in multiple, independent, experiments, consistent with systematic, genotype-specific changes in mutation profiles.
The relative frequency of SNVs, insertions and deletions, normalized by the number of sequence reads, varied significantly by RNR1/rnr1 allele (Supplementary Table S6, Figure 5), as did the absolute variant frequencies (Figure 5). In rnr1D57N, deletions increased substantially compared to RNR1, while insertions remained unchanged (Figure 5, B and C). The SNV profile of rnr1D57N was very similar to wildtype, although the overall SNV frequency was reduced (Supplementary Table S7, Figure 5A), consistent with more stochastic rather than systematic mutation events. In contrast, SNVs dominated the rnr1Y285F profile. Despite its lower mutation rate (Supplementary Table S6), the normalized variant profiles of rnr1Y285F were very similar to those of rnr1Y285A, although CG>GC changes were essentially eliminated in rnr1Y285A. The proportion of variant type was skewed toward SNVs in rnr1Y285F and deletions in rnr1Y285A. While the rnr1Y285A and rnr1Y285A pGAL-RNR1 strains resulted in almost indistinguishable mutation profiles, rnr1Y285F and rnr1Y285F pGAL-RNR1 showed more variation compared to one another (Supplementary Table S7 and Figure S2), likely because there is a higher proportion of stochastic versus systematic mutations when mutation rates are lower. All three rnr1 backgrounds exhibited a significant increase in G/C −1 bp deletions compared to wildtype, although the absolute frequency varied (Supplementary Table S7, Figure 5B). Overall, very few insertions were observed, but we noted that the G/C + 1 bp insertions were extremely rare in rnr1Y285A cells compared to other genotypes (Supplementary Table S7, Figure 5C). The depth of sequencing coverage in the current study revealed more distinct and detailed mutation profiles than previously identified in rnr1D57N and rnr1Y285F (Xu et al. 2008; Kumar et al. 2010, 2011; Buckland et al. 2014; Watt et al. 2016), with clear shifts in the types and frequency of mutations that accumulate in the presence of balanced versus skewed elevations in dNTP levels.
Unique variants occur within CAN1 in a genotype-specific manner
Mutations occurred across the 1,773 bp CAN1 in all genotypes tested. For each genotype, we identified unique variants in each replicate and then calculated the average variant frequency of each unique variant (Supplementary Figure S3). Combined, we identified 860 unique variants in all tested genotypes; 288 in rnr1Y285A genotypes (rnr1Y285A and rnr1Y285A pGAL-RNR1), 570 in rnr1Y285F genotypes (rnr1Y285F and rnr1Y285F pGAL-RNR1), 452 in wildtype and 322 in rrn1D57N (Supplementary Figure S3). On average this is over 5 times greater than the number of unique mutational events picked up by previous Sanger sequencing approaches.
Many unique variants were observed in a single isolate at low frequency, while others occurred in multiple biological replicates at increased variant frequency. The majority of average variant frequencies were below 5%; 44/860 variants occurred at an average frequency greater than 5% (Supplementary Figure S4). When a unique variant occurred at a frequency above 25% in only one biological replicate, we defined it as a “jackpot” mutation; a mutation that arose after two generations of growth would result in >25% of the cells (colonies) harboring that mutation. This is distinct from high-frequency unique variants, which had an average frequency above 5% in multiple replicates, which we analyzed to identify systematic, genotype-specific variants.
We compared the average variant frequency of high-frequency variants (>5% average frequency) that were enriched within a particular genotype (Figure 6), which could represent positions within CAN1 that are particularly susceptible to mutation in that genetic background. In RNR1 (9 biological replicates), we identified 15 unique high-frequency variants, 9 of which occurred in more than one RNR1 biological replicate (Figure 6A). The majority occurred in a small proportion of biological replicates (Figure 6A). When a high-frequency variant was present in multiple RNR1 replicates, it typically occurred at variant frequencies of less than 1% for the remaining replicates. Most of the RNR1 high-frequency variants were not observed in any of the rnr1 backgrounds. The exception of the SNV at position 32114 which was observed in all four rnr1Y285F/A genotypes and was previously identified as a “hotspot” in rnr1Y285A pGAL-RNR1 (Buckland et al. 2014). In our data set, this was also the most significantly mutated position in RNR1; it was found in over half the biological replicates.
Figure 6.

High-frequency variants occur systematically in rnr1Y285F/A genotypes. Unique variants with an average variant frequency >5% for (A) wildtype, (B) rnr1D57N, (C) rnr1Y285F-pGAL RNR1, (D) rnr1Y285F, (E) rnr1Y285A-pGAL-RNR1, (F) rnr1Y285A. For comparison, the average frequency of each variant in other genotypes is also plotted. Below each plot, the fraction of biological replicates in which the variant occurred is observed and the surrounding sequence context are indicated. *, unique variant occurred in ≥50% of biological replicates for the genotype analyzed; **, unique variant occurred in 100% of biological replicates for the genotype analyzed.
Note: Figure Replacement Requested.
In rnr1D57N, 3 high-frequency unique variants were systematically mutated in multiple rnr1D57N biological replicates, and were specific to rnr1D57N (Figure 6B, mustard bars). For example, the G deletion at position 31971 occurred in 4/7 biological replicates (Figure 6B) and drives the observed overall mutation spectrum for this genotype (Figure 5). These susceptible positions have not been previously observed. Similarly, rnr1Y285F (periwinkle) and rnr1Y285F pGAL-RNR1 (pink bars) exhibited very few high-frequency unique variants. There was some overlap between rnr1Y285F and rnr1Y285F pGAL-RNR1 at about half of these positions (32449, 32940, 31986, 32008; Figure 6, C and D), which were unique to these genetic backgrounds. Susceptible positions in rnr1D57N and rnr1Y285F genotypes have not been previously observed. In contrast, we observed overlapping, systematic high-frequency unique variants in multiple replicates of rnr1Y285A (green) and rnr1Y285A pGAL-RNR1 (turquoise) (Figure 6, E and F), including CG>AT and GC>TA SNVs as well as G/C single base deletions. Notably, several of these high-frequency unique variants were also observed in both rnr1Y285F genotypes (periwinkle and pink bars) at lower frequencies, but in multiple biological replicates. This indicates that these types of mutations occur in the same sequence contexts when dCTP and dTTP levels are skewed, whether by a modest threefold or the more significant 20-fold increase. Variants at these positions, commonly G/C −1 deletions and CG>AT changes, are increasingly probable when dNTP pools are further elevated and skewed.
Several of the high-frequency unique variants that we observed in rnr1Y285A-pGAL-RNR1 and rnr1Y285A overlapped with previously defined “hotspots” in rnr1Y285A pGAL-RNR1 by Sanger sequencing, i.e., occurring at 10x higher frequency than in wildtype (Kumar et al. 2011; Buckland et al. 2014). Specifically, CAN1 positions 32027, 32556, 32608, 32658, 32670, 32842, 32917, and 33151 were susceptible to mutation in the current study and in previous studies (Figure 6, E and F) (Kumar et al. 2011; Buckland et al. 2014). In addition, our targeted sequencing approach identified two additional susceptible positions in rnr1Y285A backgrounds; the TA>GC SNV at position 32823 and the CG>AT SNV at position 33114 (Figure 6, E and F). Three positions in CAN1 were highly susceptible to mutation in the rrn1Y285A backgrounds in our work and in previous work: (1) G/C deletion at position 32658, (2) GC>TA SNV at position 32027, and (3) the CG>AT SNV at position 32917 (Figure 6, E and F) (Buckland et al. 2014). Therefore, these three high-frequency variants may be diagnostic of this genotype.
All of the “hotspots” identified previously (Buckland et al. 2014) were observed in our rnr1Y285A samples and the majority were observed at higher frequencies in rnr1Y285A than in wildtype (Supplementary Table S5) Several previously identified “hotspots” were observed as systematic low-frequency variants (see below). We also noted a shift in the type of variant observed at some of these positions in rnr1Y285A relative to RNR1 (Supplementary Table S5). For example, at position 33168, RNR1 variants include CG>TA, CG>AT, CG>GC substitutions, single C deletions as well as MNVs and replacements. In contrast, in rnr1Y285A, variants at this position were dominated by CG>AT changes, with some CG>TA changes. Similarly, at position 33154, variants were evenly distributed between CG>GC and CG>AT changes in RNR1 while CG>AT changes dominated in rn1Y285A.
Low-frequency variant analysis identifies new mutational hotspots in rnr1Y285A strains
The sequencing depth across CAN1 achieved in our targeted NGS approach also allowed us to analyze low-frequency variants for systematic, genotype-specific changes. For this analysis, the inclusion of multiple biological replicates was critical. We analyzed low-frequency variants, defined as less than 5% average variant frequency enriched in a genotype-specific manner. RNR1, rnr1D57N, and rnr1Y285F mutation profiles exhibited a large number of low-frequency variants (56–123) found in only one replicate and/or at low variant frequency, consistent with a high proportion of stochastic mutations. In contrast, both rnr1Y285A genotypes exhibited a much smaller number of unique low-frequency events (13 deletions, 16 SNVs; Figure 7). The concentration of low-frequency events in rnr1Y285A backgrounds indicates a more systematic, rather than stochastic, pattern of mutagenesis. Notably, there were many G/C deletions (Figure 7A) and CG>AT SNVs (Figure 7B) unique to, and therefore characteristic of, rnr1Y285A backgrounds. Furthermore, these genotype-specific mutations appeared to drive the mutation profiles of rnr1Y285A backgrounds (Figure 5). Many of these low-frequency variants were previously observed in rnr1Y285A, including several that were categorized as “hotspots,” e.g., deletions at positions 33356, 33078, and 33747 and base substitutions at positions 33153 and 32929 (Figure 7) (Buckland et al. 2014). We further investigated these variants by systematically analyzing sequence context.
Figure 7.

Low-frequency variants specific to rnr1Y285A genotypes. Low-frequency deletions (A) and SNVS (B) in rnr1Y285A pGAL-RNR1 (turquoise) and rnr1Y285A (green) genotypes. None of these variants were observed in wildtype, but some occurred at low frequencies in rnr1D57N (brown) and rnr1Y285F (blue and pink) genotypes. *, variant occurred in ≥50% of rnr1Y285A or rnr1Y285A pGAL-RNR1 biological replicates; **, variant occurred in 100% of the rnr1Y285A or rnr1Y285A pGAL-RNR1 biological replicates analyzed.
Note: Figure Replacement Requested.
SNVs in trinucleotide context reveal unique mutation signatures
The analysis of both high- and low-frequency variants above indicated specific positions within CAN1 that were susceptible to mutation in a genotype-specific manner. The sequencing depth achieved in our study provided the opportunity to analyze genotype-specific variants and sequence context more systematically. We took two distinct approaches, which have not been previously applied to these rnr1 alleles: (1) trinucleotide context analyses and (2) hierarchical cluster analyses paired with motif enrichment.
Assessing the trinucleotide context of mutations is an increasingly common approach to extract mutation signatures, especially in human cancers (Alexandrov et al. 2016; Haradhvala et al. 2018). For our analysis, each unique SNV was categorized with respect to the nucleotide immediately 5′ and 3′ to the variant, with 96 possible triplet contexts. We determined the average number of times an SNV was observed in a particular triplet context per genotype, normalized to the number of times the triplet context occurs in CAN1 (Figure 8). Pearson correlation coefficients of each SNV in unique trinucleotide context were calculated to evaluate patterns of SNV mutagenesis (Supplementary Figure S5 and Table S8). Notably, in all genotypes, C→T changes (red bars, Figure 8), particularly in GCC and GCG sequence contexts, dominated. The proportion of GCC and GCG changes increased with altered dNTPs, most dramatically in rnr1Y285F and rnr1Y285A samples, which were highly correlated within a genotype (rnr1Y285A:rnr1Y285A pGAL-RNR1 rs = 0.930, rnr1Y285F:rnr1Y285F pGAL-RNR1 rs = 0.947) and between rnr1Y285F and rnr1Y285A genotypes (rnr1Y285F:rnr1Y285A rs = 0.924, rnr1Y285F pGAL-RNR1:rnr1Y285A pGAL-RNR1 rs = 0.909). RNR1 and rnr1Y285A genotypes showed the weakest correlations (RNR1:rnr1Y285A rs = 0.693, RNR1:rnr1Y285A pGAL-RNR1 rs = 0.658), with many variants absent in rnr1Y285A C>G (black), T>A (gray), T>C (green), and T>G (pink) variants. For example, C>G errors in ACG, ACT, CCG, GCA, GCG, GCT, TCC, and TCT contexts were completely absent in both rnr1Y285A genotypes. Many of these missing variants occurred in repetitive sequences, while errors in different repetitive contexts dominated in rnr1Y285A.
Figure 8.

The average number of each SNV as it occurs in unique triplet nucleotide context is distinct between rnr1 alleles. Bars are colored according to the six different types of SNVs. (A) The 16 different triplet contexts are lettered for display purposes. The variant change (C>A, blue bar) occurs at the middle nucleotide marked X in each triplet context for (B) RNR1, (C) rnr1D57N, (D) rnr1Y285F pGAL-RNR1, (E) rnr1Y285F, (F) rnr1Y285A pGAL-RNR1, and (G) rnr1Y285A.
Note: Figure Replacement Requested.
RNR1 and rnr1D57N or rnr1Y285F were more strongly correlated (RNR1: rnr1D57N rs = 0.774, RNR1: rnr1Y285F rs = 0.736), consistent with more subtle differences between SNV spectra in trinucleotide context (Figure 8). In rnr1D57N, the most apparent difference is a loss of C>G variants in CCA, CCC, CCG, and CCT; both rnr1D57N and rnr1Y285F also exhibited some variability in T>A (gray), T>C (green), and T>G (pink) variants.
Motif enrichment reveals CC dinucleotides are commonly mutated in rnr1Y285F/A genotypes
We performed hierarchical cluster analysis of the unique variants in our data set to determine differences between mutation profiles and to determine which variants in CAN1 were driving these differences. This analysis was paired with motif enrichment to determine broader sequence contexts prone to mutagenesis in a particular genetic background. The rnr1Y285A and rnr1Y285A pGAL-RNR1 genotypes clustered, indicating shared common features that were underrepresented in RNR1 and rnr1D57N (Figure 9), as did the rnr1Y285F genotypes. Both rnr1Y285F and rnr1Y285A clustered away from rnr1D57N and RNR1, which were more closely related in this analysis. We observed three main clusters of unique variants, labeled I, II, and III.
Figure 9.

Unique variants in enriched motifs form distinct clusters. (A) Hierarchical cluster analysis of the highest occurring variants reveals distinct clusters, annotated I, II and III. (B) The different classes of variants enriched in each cluster were subset, and 12 basepairs surrounding the variant was used to perform motif enrichment. The corresponding motifs from the different variant classes in each boxed cluster are displayed on the right and labeled accordingly (boxes I, II, and III). The mutated position is indexed at 0 and the 6 bases on either side of the variant are labeled as such.
In the first two clusters (Figure 9, clusters I and II) RNR1 and rnr1D57N were very similar, while rnr1Y285F/A exhibited distinct differential enrichment profiles. The variants in cluster I were more significantly overrepresented in rnr1Y285A backgrounds, relative to rnr1Y285F. The reverse was true of variants in cluster II. Both clusters I and II were underrepresented in RNR1 and rnr1D57N. Within cluster II, there were several variants that were more significantly underrepresented in rnr1D57N than in RNR1. In contrast, the third cluster (III) did not exhibit clear trends, with different variants under- or over-represented in different genotypes. Combined, these data indicate genotype-specific mutagenesis.
From each of these three clusters, we performed motif enrichment on the sequence surrounding the variants and identified unique contexts differentially enriched in rnr1Y285F/A backgrounds, consistent with the trinucleotide context data (Figure 8) as well as high- (Figure 6) and low-frequency (Figure 7) variant analysis. C>A and C>T changes in cluster I occurred at CC dinucleotides, while those in cluster III, which were underrepresented in rnr1Y285F/A, did not. This is consistent with the prediction that repetitive GC sequences are more prone to mutation in the presence of skewed elevations in dCTP and dTTP. Similarly, G/C deletions in repetitive G/C context were differentially enriched in rnr1Y285A genotypes (Figure 9, cluster II), while A/T insertions and deletions were enriched across all samples (Figure 9, cluster III). Similar logos were previously identified via MA WGS only when rnr1Y285A pGAL-RNR1 was combined with msh2Δ (Watt et al. 2016).
Discussion
To build mutation profiles from first principles, we developed a CAN1 selection-based next generation sequencing approach to determine robust, information-rich, genotype-specific mutation profiles. We focused on rnr1 alleles that reduced replication fidelity and increased mutation rates (Chabes et al. 2003; Xu et al. 2008; Kumar et al. 2011). While CAN1 is a widely used mutational reporter gene in yeast, the application of deep-sequencing allowed streamlined sample preparation and more accurate determination of mutation spectra, by increasing both the number of colonies and biological replicates analyzed at one time. This approach allowed us to (1) identify low-frequency sequence variants, (2) develop mutational fingerprints that resulted from compromised replication fidelity as a result of altered dNTP pools, and (3) identify broader sequence contexts significantly enriched in specific genotypes. While some noninactivating mutations will have been missed, we did observe ∼1–3% of variants resulted in synonymous changes (data not shown). It is also worth noting that the inactivating mutations at CAN1 we observed were primarily SNVs and small in/dels; other rearrangements may be selected in different genetic backgrounds (e.g., rad27 alleles, Xie et al. 2001). Nonetheless, we effectively expanded our understanding of the impact of altered dNTP pools on mutagenesis and developed an approach that can be applied to study other genetic backgrounds or environmental exposures.
Our sequencing/analytic approach allowed us to define genotype-specific mutation profiles, including trinucleotide sequence context for mutations and broader sequence motifs surrounding mutations. Deep sequencing and analysis of only CAN1 mutations identified mutation sequence motifs similar to those generated through WGS (Watt et al. 2016), uncovered genotype-specific variants and motifs not identified by WGS or previous CAN1 sequencing, and revealed new details for mutation profiles in genotypes with lower mutations rates. From this information, we can infer mechanisms of mutagenesis (see below). Notably, we identified sequence motifs in rnr1Y285A similar to those identified previously by WGS in rnr1Y285A msh2Δ, which eliminates MMR (Watt et al. 2016), indicating that the errors generated when dNTPs are elevated and skewed are substrates for MMR.
Mechanisms of mutagenesis from distinct elevations in dNTP levels
Despite a twofold increase in mutation rate above wildtype, the rnr1D57N mutation spectrum closely resembled wildtype (Figure 5), consistent with previous studies (Xu et al. 2008; Kumar et al. 2011). This indicated that the twofold balanced increase in dNTPs causes errors to accumulate in a stochastic manner, similar to wildtype. Given the low-mutation rate in rnr1D57N (Supplementary Table S6), the majority of these errors are likely corrected by the MMR system, which identifies replication errors and targets them for repair (Xu et al. 2008; Kunkel and Erie 2015). Nonetheless, we observed a significant increase in G/C single base deletions in this genetic background (Figure 5B) and were able to identify new sequence contexts specific to mutations in rnr1D57N. This includes CG>TA, CG>AT changes, A/T deletions in repetitive A/T sequences (Figures 5, 6B, and 9), which appeared diagnostic of rnr1D57N mutagenesis. We predict that the regions in which these mutations occur are inherently more prone to mutation; the twofold increase in dNTP levels exacerbates this by favoring replicative polymerase synthesis over proofreading activity.
The high frequency of G/C deletions observed in rnr1D57N was driven by a single base G deletion at position 31971 that was mutated in 4 out of 7 rnr1D57N replicates and occurs at a high frequency, on average ∼60% (Figures 5B and 6B, S2C & S2D—compare unique counts vs. frequency of variants). The high frequency indicated the mutation arose early in the growth of the cultures, but the fact that it occurred and was enriched in multiple biological replicates indicated that this position is uniquely susceptible to deletion, specifically in rnr1D57N. This variant was flanked by repeats on both the 5′ side (repetitive A/T motif) and 3′ side (CC dinucleotide) (ATAAGCCA). Therefore, both the excess of dTTP incorporated opposite the AA dinucleotide and the excess of dCTP incorporated on the opposite strand could result in transient misalignment and misincorporation at this position and it is therefore likely this variant occurred during both leading and lagging strand replication. Increased dNTP levels alter replication fork dynamics (Davidson et al. 2012; Poli et al. 2012), potentially enhancing the next nucleotide effect and resulting in increased variant frequencies at this position in rnr1D57N.
The rnr1Y285F allele exhibited a modest effect on mutation rate and a distinct mutation profile (Figure 5). Meta-analysis of the types and positions of variants in rnr1Y285F significantly overlapped with that of rnr1Y285A (Figures 6–9), despite the differences in mutation rate (Supplementary Table S6) and dCTP/dTTP levels (Kumar et al. 2010). There was a strong correlation between the trinucleotide context for variants in rnr1Y285F and rnr1Y285A backgrounds (Supplementary Table S8; Figure S5). Similarly, hierarchical cluster analysis indicated that similar variants were enriched in the two genetic backgrounds in clusters I and II, although the degree of enrichment varied (Figure 9). This was particularly noticeable with G/C variants, i.e., G/C deletions (e.g., C deletion at position 33356 and G deletion at 32670) and CG>AT variants (e.g., CG>AT SNV at position 33168 and CG>AT change at 32037). Therefore, even modestly increased dCTP and dTTP pools resulted in distinct error accumulation. We predict an increased probability that replicative polymerases incorporated the excess nucleotides (dCTP, dTTP) during synthesis and more efficiently extended mismatches at the expense of proofreading (Kumar et al. 2011; Watt et al. 2016).
We noted a substantial increase in G/C deletions specific to rnr1Y285A backgrounds (Figure 6, E and F), which may be explained by limiting levels of dGTP in rnr1Y285A. dGTP levels are already limiting in both yeast and mammalian cells (Chabes et al. 2003; Håkansson et al. 2006); in rnr1Y285A, the proportion of dGTP relative to the total dNTP pool is extremely small, reducing the probability of its incorporation. The reduced probability of dGTP incorporation and the concomitant increased probability of dCTP and dTTP incorporation in rnr1Y285A allow us to predict the strand that has sustained the initial misincorporation event (Figure 9) (Buckland et al. 2014). We propose that the predicted reduction in dGTP incorporation led to the observed distinct pattern of deletion in rnr1Y285F/A (Mathews 2015), within specific sequence contexts, i.e., CC and CCC runs (Figure 9). Notably, rnr1 alleles that dramatically increased dGTP levels were also severely mutagenic (rnr1K243E and rnr1I262V, N291D), increasing SNVs and frame shift mutations in repetitive contexts (Schmidt et al. 2019). This highlights the importance of determining the absolute and relative abundance of dGTP when assessing mutation profiles.
Our data indicated that the mutation profiles generated using our sequencing and analytical approach were diagnostic of the balanced or unbalanced nature of the dNTP pools, i.e., which dNTPs were elevated. The mutation rate and variant frequencies reflected the absolute levels of dNTPs; far more in/dels than SNVs were observed for rnr1Y285A while the reverse was true for rnr1Y285F. Despite this difference, the proportions were similar and many of the same errors in the same sequence context accumulated in both these genetic backgrounds. This hypothesis could be tested by comparing the rnr1D57N mutation profile with that of RNR1 galactose-induced overexpression, which similarly leads to balanced dNTP pools, but elevated ∼10-fold above wild-type levels (Chabes and Stillman 2007), as well as other rnr1 alleles.
Implications for understanding mutation signatures in human cancers
Mutation signatures of human tumors are used to identify molecular drivers of carcinogenesis (Alexandrov et al. 2013; Nik-Zainal et al. 2016; Haradhvala et al. 2018; Alexandrov et al. 2020), which have clear implications for diagnosis, prognosis and treatment options for patients (Van Hoeck et al. 2019). However, in general, elevated dNTP levels (balanced or skewed) have not been considered when evaluating mutation signatures from human tumors, although they almost certainly contribute to mutagenesis in cancer (Aye et al. 2015; Mathews 2015, 2017; Pai and Kearsey 2017; Degasperi et al. 2020). We noted distinct similarities between rnr1 SNV triplet mutation profiles (Figure 8) and specific COSMIC signatures, most notably signatures 6 and 15. Signature 6 occurs most commonly in colorectal and uterine cancers and is associated with defective MMR. We previously noted synergistic effects on mutation rate between rnr1D57N and MMR deletions (Xu et al. 2008). The contribution of elevated dNTP pool levels to mutagenesis in combination with MMR is intriguing.
While elevated dNTP levels have been implicated in supporting rapidly proliferating cancer cells, altered dNTP pools may be tumor-specific (Wilson et al. 2011; Kohnken et al. 2015; Mathews 2015; Purhonen et al. 2020). Different skewed elevations result in distinct mutation spectra (Schmidt et al. 2019) and thus more studies are necessary to determine what dNTP imbalances are relevant to different types of cancers. In the meantime, certain indicator mutations, such as a high number of G/C single base deletions, can point towards specific dNTP imbalances i.e., high dCTP and dTTP levels seen in rnr1Y285A.
Acknowledgments
The authors are grateful to Dr. Andrei Chabes for providing strains and plasmids. They are grateful to Dr. Don Yergeau for his advice in next generation sequencing. They are grateful to members of the Surtees lab (past and present) for helpful discussions and input. They are grateful to Dr. Mark Sutton and Dr. John Panepinto for input and helpful discussions.
Funding
N.A.L. is a University at Buffalo Presidential Scholar. This work was supported by the American Cancer Society (RSG-14-2350-01) to J.A.S. J.A.S. is an ACS Research Scholar. J.A.S. is also grateful for support from the University at Buffalo’s Genome, Environment and Microbiome (GEM) Community of Excellence.
Conflicts of interest
None declared.
Literature cited
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, et al. 2015. Clock-like mutational processes in human somatic cells. Nat Genet. 47:1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, et al. 2016. Mutational signatures associated with tobacco smoking in human cancer. Science 354:618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, et al. 2020. The repertoire of mutational signatures in human cancer. Nature 578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, et al. 2013. Signatures of mutational processes in human cancer. Nature 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y, Li M, Long MJ, Weiss RS.. 2015. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34:2011–2021. [DOI] [PubMed] [Google Scholar]
- Brash DE. 2015. UV signature mutations. Photochem Photobiol. 91:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckland RJ, Watt DL, Chittoor B, Nilsson AK, Kunkel TA, et al. 2014. Increased and imbalanced dNTP pools symmetrically promote both leading and lagging strand replication infidelity. PLoS Genet. 10:e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, et al. 2003. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112:391–401. [DOI] [PubMed] [Google Scholar]
- Chabes A, Stillman B.. 2007. Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 104:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, et al. 2015. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 47:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, et al. 2012. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 8:e1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE.. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, et al. 2012. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J. 31:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degasperi A, Amarante TD, Czarnecki J, Shooter S, Zou X, et al. 2020. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancer 1:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantes PA, Creanor J.. 1984. Canavanine resistance and the mechanism of arginine uptake in the fission yeast Schizosaccharomyces pombe. J Gen Microbiol. 130:3265–3273. [DOI] [PubMed] [Google Scholar]
- Gietz D, St Jean A, Woods RA, Schiestl RH.. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson P, Hofer A, Thelander L.. 2006. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem. 281:7834–7841. [DOI] [PubMed] [Google Scholar]
- Haradhvala NJ, Kim J, Maruvka YE, Polak P, Rosebrock D, et al. 2018. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun. 9:1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W. 1985. Molecular characterization of the CAN1 locus in Saccharomyces cerevisiae. A transmembrane protein without N-terminal hydrophobic signal sequence. J Biol Chem. 260:11831–11837. [PubMed] [Google Scholar]
- Howard BD, Tessman I.. 1964. Identification of the altered bases mutated in single-stranded DNA. 3. Mutagenesis by ultraviolet light. J Mol Biol. 9:372–375. [DOI] [PubMed] [Google Scholar]
- Jordheim LP, Durantel D, Zoulim F, Dumontet C.. 2013. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 12:447–464. [DOI] [PubMed] [Google Scholar]
- Kohnken R, Kodigepalli KM, Wu L.. 2015. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer 14:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, et al. 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Viberg J, Nilsson AK, Chabes A.. 2010. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 38:3975–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA.. 2015. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Soni A.. 1988. Mutagenesis by transient misalignment. J Biol Chem. 263:14784–14789. [PubMed] [Google Scholar]
- Lujan SA, Clausen AR, Clark AB, MacAlpine HK, MacAlpine DM, et al. 2014. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 24:1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews CK. 2015. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat Rev Cancer 15:528–539. [DOI] [PubMed] [Google Scholar]
- Mathews CK. 2017. Oxidized deoxyribonucleotides, mutagenesis, and cancer. Faseb J. 31:11–13. [DOI] [PubMed] [Google Scholar]
- Mathews CK. 2018. Still the most interesting enzyme in the world. Faseb J. 32:4067–4069. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyola SO, Otto TD, Gu Y, Maslen G, Manske M, et al. 2012. Optimizing illumina next-generation sequencing library preparation for extremely at-biased genomes. BMC Genomics 13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai CC, Kearsey SE.. 2017. A critical balance: dNTPs and the maintenance of genome stability. Genes (Basel). 8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phear G, Nalbantoglu J, Meuth M.. 1987. Next-nucleotide effects in mutations driven by DNA precursor pool imbalances at the aprt locus of Chinese hamster ovary cells. Proc Natl Acad Sci USA. 84:4450–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, et al. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 31:883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov V, Ong JL.. 2017. Examining sources of error in PCR by single-molecule sequencing. PLoS One 12:e0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purhonen J, Banerjee R, McDonald AE, Fellman V, Kallijärvi J.. 2020. A sensitive assay for dNTPs based on long synthetic oligonucleotides, EvaGreen dye and inhibitor-resistant high-fidelity DNA polymerase. Nucleic Acids Res. 48:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radchenko EA, McGinty RJ, Aksenova AY, Neil AJ, Mirkin SM.. 2018. Quantitative analysis of the rates for repeat-mediated genome instability in a yeast experimental system. Methods Mol Biol. 1672:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentoft M, Lindell K, Tran P, Chabes AL, Buckland RJ, et al. 2016. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci USA. 113:4723–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlin A, Wernersson J, Engwall Y, Wiklund L, Björk J, et al. 2009. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat. 30:1012–1020. [DOI] [PubMed] [Google Scholar]
- Saini N, Sterling JF, Sakofsky CJ, Giacobone CK, Klimczak LJ, et al. 2020. Mutation signatures specific to DNA alkylating agents in yeast and cancers. Nucleic Acids Res. 48:3692–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TT, Sharma S, Reyes GX, Gries K, Gross M, et al. 2019. A genetic screen pinpoints ribonucleotide reductase residues that sustain dNTP homeostasis and specifies a highly mutagenic type of dNTP imbalance. Nucleic Acids Res. 47:237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P.. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, et al. 2019. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoeck A, Tjoonk NH, van Boxtel R, Cuppen E.. 2019. Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer 19:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt DL, Buckland RJ, Lujan SA, Kunkel TA, Chabes A.. 2016. Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res. 44:1669–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PM, Labonte MJ, Russell J, Louie S, Ghobrial AA, et al. 2011. A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 39:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Liu Y, Argueso JL, Henricksen LA, Kao HI, et al. 2001. Identification of rad27 mutations that confer differential defects in mutation avoidance, repeat tract instability, and flap cleavage. Mol Cell Biol. 21:4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, et al. 2008. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 68:2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA.. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci USA. 111:E2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and reagents are available upon request. All variant sequences are provided in Supplementary Table S5. The Supplemental Material is available at figshare: https://doi.org/10.25387/g3.14187080.