

Abstract
Experience-dependent formation and removal of inhibitory synapses are essential throughout life. For instance, GABAergic synapses are removed to facilitate learning, and strong excitatory activity is accompanied by the formation of inhibitory synapses to maintain coordination between excitation and inhibition. We recently discovered that active dendrites trigger the growth of inhibitory synapses via CB1 receptor-mediated endocannabinoid signaling, but the underlying mechanism remained unclear. Using two-photon microscopy to monitor the formation of individual inhibitory boutons in hippocampal organotypic slices from mice (both sexes), we found that CB1 receptor activation mediated the formation of inhibitory boutons and promoted their subsequent stabilization. Inhibitory bouton formation did not require neuronal activity and was independent of Gi/o-protein signaling, but was directly induced by elevating cAMP levels using forskolin and by activating Gs-proteins using DREADDs. Blocking PKA activity prevented CB1 receptor-mediated inhibitory bouton formation. Our findings reveal that axonal CB1 receptors signal via unconventional downstream pathways and that inhibitory bouton formation is triggered by an increase in axonal cAMP levels. Our results demonstrate an unexpected role for axonal CB1 receptors in axon-specific, and context-dependent, inhibitory synapse formation.
SIGNIFICANCE STATEMENT Coordination between excitation and inhibition is required for proper brain function throughout life. It was previously shown that new inhibitory synapses can be formed in response to strong excitation to maintain this coordination, and this was mediated by endocannabinoid signaling via CB1 receptors. As activation of CB1 receptors generally results in the suppression of synaptic transmission, it remained unclear how CB1 receptors can mediate the formation of inhibitory synapses. Here we show that CB1 receptors on inhibitory axons signal via unconventional intracellular pathways and that inhibitory bouton formation is triggered by an increase in axonal cAMP levels and requires PKA activity. Our findings point to a central role for axonal cAMP signaling in activity-dependent inhibitory synapse formation.
Keywords: cAMP/PKA, endocannabinoids, G-protein-coupled receptors, inhibitory synapses, synapse formation
Introduction
Synaptic plasticity, the strengthening and weakening of existing synapses, is often considered the physiological basis for learning and adaptation. In addition, the experience-dependent formation and removal of synapses is equally important (Bailey and Kandel, 1993; Caroni et al., 2012). Changes in the number of synaptic connections have been shown to be critical during learning in vivo (Bailey and Chen, 1989; Hofer et al., 2009; Ruediger et al., 2011; Caroni et al., 2012; Kozorovitskiy et al., 2012) and strongly determine postsynaptic function (Scholl et al., 2021). Plasticity of GABAergic synapses is particularly important for shaping and controlling brain activity throughout life (Flores and Méndez, 2014; Maffei et al., 2017; Chiu et al., 2019; Herstel and Wierenga, 2021) and GABAergic dysfunction is associated with multiple brain disorders, including schizophrenia and autism (Lewis et al., 2005; Mullins et al., 2016; Tang et al., 2021). For example, the number of inhibitory synapses is rapidly adjusted during learning (Bourne and Harris, 2011; Donato et al., 2013, 2015; Chen et al., 2015) or when sensory input is lost (Keck et al., 2011) to facilitate plasticity at nearby excitatory synapses. Vice versa, the potentiation of excitatory synapses can trigger the formation of inhibitory synapses to maintain a local balance (Knott et al., 2002; Bourne and Harris, 2011; Hu et al., 2019). The formation, stabilization, and removal of synapses likely requires local context-dependent signaling mechanisms (Kleindienst et al., 2011; Nishiyama and Yasuda, 2015; Oh et al., 2016; Niculescu et al., 2018; Hu et al., 2019; Kirchner and Gjorgjieva, 2021), but our current understanding of these processes, especially at inhibitory synapses, is far from complete.
We recently discovered that strong, clustered activation of excitatory synapses along dendrites of hippocampal CA1 pyramidal neurons can trigger the formation of a new inhibitory bouton onto the activated dendrite (Hu et al., 2019). We proposed that this dendritic mechanism serves to maintain local balance between excitatory and inhibitory inputs during ongoing synaptic plasticity. Inhibitory bouton formation required dendritic endocannabinoid synthesis and activation of CB1 receptors (Hu et al., 2019). Dendritic endocannabinoids are well known to serve as retrograde signals to regulate synaptic plasticity (Alger, 2002; Chevaleyre and Castillo, 2003; Kano et al., 2009; Castillo et al., 2012; Katona and Freund, 2012), but it is unclear how CB1 receptors can trigger new inhibitory bouton formation.
CB1 receptors are G-protein-coupled receptors and are widely abundant in the brain. They are expressed in both excitatory and inhibitory neurons, as well as in glia cells (Navarrete et al., 2014; Hebert-Chatelain et al., 2016; Maroso et al., 2016; Bonilla-Del Río et al., 2021). Perhaps the most prominent CB1 expression is in a subset of inhibitory axons in the dendritic layer of the hippocampal CA1 area (Dudok et al., 2015; Bonilla-Del Río et al., 2021). Axonal CB1 signaling plays an important role during axon guidance (Berghuis et al., 2007; Argaw et al., 2011; Roland et al., 2014; Njoo et al., 2015), but axonal CB1 receptor expression remains high during adulthood. The best described actions of CB1 receptors in adulthood are in suppressing neurotransmitter release (Alger, 2002; Kano et al., 2009; Castillo et al., 2012). However, CB1 receptors are not enriched in boutons, but freely diffuse within the entire axonal membrane (Dudok et al., 2015). It is possible that axonal CB1 receptors may function as replacement pool for internalized synaptic receptors at boutons as recently suggested for opioid receptors (Jullié et al., 2020), although synaptic enrichment would still be expected. In addition, GABA release at dendritic inhibitory synapses is not strongly modulated by CB1 receptors (Lee et al., 2010, 2015), and coupling between CB1 receptors and the active zone is weak (Dudok et al., 2015). This suggests that CB1 receptors in inhibitory axons serve an additional purpose. Interestingly, it was recently described that CB1 receptors can also mediate synaptic potentiation (Cui et al., 2016; Monday and Castillo, 2017; Wang et al., 2018). Although CB1 receptors typically signal via Gi/o-proteins, many additional downstream pathways, both dependent and independent of G-proteins, have been described (Glass and Felder, 1997; Berghuis et al., 2007; Flores-Otero et al., 2014; Roland et al., 2014; Cui et al., 2016; Zhou et al., 2019; Marti-Solano et al., 2020).
Here, we demonstrate that the activation of axonal CB1 receptors can trigger the initial formation of inhibitory synapses. Using two-photon time-lapse imaging, we observed the formation of inhibitory boutons on brief application of the CB1 receptor agonist WIN. We demonstrate that this requires the presence of CB1 receptors on inhibitory axons. Furthermore, we found that CB1-mediated inhibitory bouton formation is independent of Gi/o-protein signaling and neuronal activity. We find that new inhibitory boutons are formed in response to elevated cAMP levels or activation of Gs-protein signaling in inhibitory axons. Our data indicate that the activation of axonal CB1 receptors triggers inhibitory synapse formation via an atypical signaling pathway via Gs-proteins. Furthermore, our data identify an increase in axonal cAMP as a crucial second messenger for mediating inhibitory bouton formation.
Materials and Methods
Animals.
All animal experiments were performed in compliance with the guidelines for the welfare of experimental animals issued by the Federal Government of The Netherlands. All animal experiments were approved by the Animal Ethical Review Committee of Utrecht University.
Mouse hippocampal slice culture.
Organotypic mouse hippocampal slices were acquired from female and male GAD65-GFP mice at 6–7 d after birth. In these mice, ∼20% interneurons are labeled by GFP from early embryonic developmental stage into adulthood (López-Bendito et al., 2004). Most GFP-labeled interneurons target dendrites of CA1 pyramidal cells and express VIP (vasoactive intestinal polypeptide) or reelin, while parvalbumin- and somatostatin-positive neurons are not labeled (Wierenga et al., 2010). Slice culture preparation details have been described previously (Frias et al., 2019; Hu et al., 2019). Mice were killed and the isolated hippocampus was placed in ice-cold HEPES-Gey's balanced salt solution (containing 1.5 mm CaCl2 · 2H2O, 0.2 mm KH2PO4, 0.3 mm MgSO4 · 7H2O, 5 mm KCl, 1 mm MgCl2 · 6H2O, 137 mm NaCl, 0.85 mm Na2HPO4, and 12.5 mm HEPES) supplemented with 12.5 mm HEPES, 25 mm glucose, and 1 mm kynurenic acid, with pH set at ∼7.2, osmolarity set at ∼320 mOsm, and sterile filtered. Slices were vertically chopped along the long axis of hippocampus at a thickness of 400 µm. They were then quickly washed with culturing medium (consisting of 48% MEM, 25% HBSS, 25% horse serum, 30 mm glucose and 12.5 mm HEPES, with pH set at 7.3–7.4 and osmolarity set at 325 mOsm), and transferred to Millicell Cell Culture Inserts (Millipore) in six-well plates. Slices were cultured in an incubator (35°C, 5% CO2) until use. Culturing medium was completely replaced twice a week. Slices were used after 2–3 weeks in vitro, when the circuitry is relatively mature and stable (De Simoni et al., 2003).
Pharmacological treatments.
The following drugs were used: 20 μm WIN 55212–2 (WIN; Tocris Bioscience), 100 μm 2-arachidonoylglycerol (2-AG; Tocris Bioscience), 25 μm forskolin (Abcam), 1 µg/ml pertussis toxin (PTX; Tocris Bioscience), 10 μm clozapine-N-oxide (CNO; Tocris Bioscience), 5 μm AM251 (Tocris Bioscience), 1 μm TTX (Abcam), and 1 μm PKI 14-22 (Tocris Bioscience). For acute treatments, artificial CSF (ACSF) containing the drug or 0.1% DMSO vehicle was bath applied for 5 min. AM251 and CNO were bath applied after a baseline period (5 time points) and continued until the end of the experiment. TTX or PKI 14-22 was added to the ACSF and were present during the entire two photon-imaging period. Pertussis toxin was added to the slice culture medium, and a small drop was placed on top of the slice 24 h before the start of the experiment. Treated slices were kept in the incubator.
CB1 receptor activation can result in different downstream signaling pathways, which depend on ligand concentration and duration (Cui et al., 2015, 2016). We used a relatively high concentration of WIN (20 μm) to aim for the strong activation of CB1 receptors. We used relatively short applications to mimic CB1 receptor activation under physiological conditions (Hu et al., 2019) and to avoid the induction of synaptic depression, which have been observed with longer applications (Cui et al., 2016; Monday et al., 2020). We expect that CB1 receptor activation under physiological conditions will be shorter, and therefore perhaps more efficient in triggering inhibitory bouton formation.
For repeated treatment with 2-AG, normal culturing medium was replaced by medium containing 100 μm 2-AG or 0.1% DMSO for 20 min. This was repeated three times with 2 h intervals. At the start of each medium replacement, a small drop was placed on top of the slices to facilitate exchange. A treatment duration of 20 min (rather than 5 min) was chosen to ensure penetration in the entire slice as solution exchange may be slower in the incubator compared with the microscope bath. After the last treatment, the medium was replaced three times with fresh medium to ensure washout. During and after the treatment, slices were kept in the incubator and experiments (immunocytochemistry or electrophysiology) were performed 24 h after the start of the first treatment.
Electrophysiology recording and analysis.
Slices were transferred to a recording chamber, which was continuously perfused with carbogenated ACSF (containing 126 mm NaCl, 3 mm KCl, 2.5 mm CaCl2, 1.3 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, 20 mm glucose, and 1 mm Trolox) at 32°C. Whole-cell patch-clamp measurements were recorded with a MultiClamp 700B amplifier (Molecular Devices) and stored using pClamp 10 software. Recordings were filtered with a 3 kHz Bessel filter. Thick-walled borosilicate pipettes of 4–6 MΩ were filled with pipette solution containing (in mm): 70 K-gluconate, 70 KCl, 0.5 EGTA, 10 HEPES, 4 MgATP, 0.4 NaGTP, and 4 Na2-phosphocreatine. Cells were discarded if series resistance was above 35 MΩ or if the resting membrane potential exceeded −50 mV. Recordings were excluded when the series resistance after the recording deviated >30% from its original value. To isolate miniature IPSCs (mIPSCs), TTX, AP5, and DNQX were added to the ACSF. The mIPSCs were analyzed in pClamp and MATLAB with homemade scripts (Ruiter et al., 2020). The rise times of mIPSCs were determined as the time between10% and 90% of the peak value. The distribution of the rise times of mIPSCs recorded in control conditions (generated from 150 mIPSCs/cell) were fitted with two Gaussians, and their crossing point determined the separation between fast and slow mIPSCs (Ruiter et al., 2020). A double Gaussian fit for the rise time distribution in 2-AG conditions gave a similar separation value (control, 0.9 ms; 2-AG, 1.1 ms), and we verified that our conclusions did not change by taking the 2-AG separation value.
Two-photon time-lapse imaging.
Time-lapse two-photon imaging was performed in carbogenated, continuously perfused ACSF at 32°C. Slices were transferred in a 3 cm dish containing ACSF. Two-photon imaging was performed on a customized two-photon laser scanning microscope (Femto2D, Femtonics) with a Ti-Sapphire femtosecond pulsed laser (MaiTai HP, Spectra-Physics) with a 60× water-immersion objective (numerical aperture, 1.0; NIR Apochromat, Nikon). A 4× objective (Plan Apochromat, Nikon) was used to determine the location of the dendritic layer of the CA1 region. GFP was excited at 910 nm to visualize GFP-labeled axons. 3D image stacks were acquired at a size of 93.5 × 93.5 µm (1124 × 1124 pixels) with 50–63 z-steps (0.5 µm step size). The acquisition time per image stack was ∼7 min. We acquired image stacks every 10 min, with a total of 15 time points (140 min). After a baseline of five time points, drugs were bath applied.
For slices in which we performed post hoc immunostaining, an overview of the imaging region was made after the last time point (203 × 203 µm; ∼50 z-steps of 1.0 µm step size), and a line scar was made using high-intensity laser power at 910 nm at the edge of the zoomed out imaging area to facilitate alignment with post hoc confocal microscopy.
Two-photon image analysis.
Individual axons with at least 50 μm length were traced using the CellCounter plugin imbedded in Fiji for all time points (TPs). Individual boutons were identified with custom-built semiautomatic MATLAB software, as described previously (Frias et al., 2019). In short, a 3D axonal intensity profile was reconstructed at each TP, and individual boutons were selected based on a local threshold (0.5 SD above mean axon intensity). Only boutons containing at least 5 pixels above threshold were included. All boutons at all time points were visually inspected, and manual corrections were made if deemed necessary.
Persistent (P) boutons were defined as boutons that were present at all TPs. Nonpersistent (NP) boutons were absent at one or more TPs. Boutons that were present for only one time point were considered transport events and were excluded (Schuemann et al., 2013; Frias et al., 2019). Based on their presence or absence during baseline (first 5 TP) and after treatment, NP boutons were further classified into five subgroups (Frias et al., 2019; Ruiter et al., 2021): new boutons (only present after baseline), lost boutons (only present during baseline), stabilizing boutons (nonpersistent during baseline, persistent after treatment), destabilizing boutons (persistent during baseline, nonpersistent after treatment), and intermittent boutons (nonpersistent during baseline and after treatment).
Bouton density was calculated per axon as the average number of boutons at each TP divided by the 3D axon length. NP bouton density was determined for each TP as the number of NP boutons that were present divided by the 3D axon length. NP bouton densities were normalized to the average baseline value (first five TPs) to allow comparison between axons. The maximum change in NP bouton density change was calculated as the maximum NP bouton density (average over three TPs) divided by the baseline NP bouton density (average over TPs 2–4). NP presence was determined as the fraction of NP boutons that were present at each time point, and these values were averaged for the first, second, and third period of five TPs each. Changes in NP presence reflect changes in the density of NP bouton subgroups, as well as in bouton duration. However, differences in bouton duration (percentage of TPs present) of NP bouton subgroups were never observed in any of the conditions, and we therefore only report NP bouton densities.
Immunocytochemistry and confocal microscopy.
Fixation of the slices was performed in 4% paraformaldehyde for 30 min at room temperature covered by aluminum foil. Following washing with PBS (3 × 10 min), slices were permeabilized with 0.5% Triton X-100 (15 min), followed by PBS washing (3 × 5 min), and 1 h incubation in blocking solution (0.2% Triton X-100 and 10% goat serum). The application of primary antibodies in blocking solution was performed overnight at 4°C. After PBS washing (3 × 15 min), secondary antibodies were applied for 4 h. After PBS washing (2 × 15 min), slices were mounted in Vectashield solution.
We used the following primary antibodies: rabbit α-VGAT (vesicular GABA transporter; 1:1000; Synaptic Systems; RRID:AB_887869), mouse α-gephyrin (1:1000; catalog #147011, Synaptic Systems; RRID:AB_887717), mouse α-CB1R (1:1000; catalog #258011, Synaptic Systems; RRID:AB_2619969), and rat α-HA (1:500; catalog #11867423001, Roche; RRID:AB_390918). The following secondary antibodies were used: anti-mouse Alexa Fluor 647 (1:500; catalog #A21241, Thermo Fisher Scientific; RRID:AB_2535810) and anti-rabbit Alex Fluor 405 (1:250; catalog #A31556, Thermo Fisher Scientific; RRID:AB_221605) for VGAT and gephyrin staining; anti-mouse Alexa Fluor 647 (1:500; catalog #A21236, Thermo Fisher Scientific; RRID:AB_2535805) and anti-mouse Alexa Fluor 568 (1:500; catalog #A11031, Thermo Fisher Scientific; RRID:AB_144696) for CB1R staining; and anti-rat Alexa Fluor 568 (1:500; catalog #11077, Thermo Fisher Scientific; RRID:AB_2534121) for HA staining.
Confocal imaging was performed using a LSM-700 microscope system (Zeiss) with a 63× oil-immersion objective. A 20× objective was used to find back the two-photon imaging area based on the line scar. Image size was 101.3 × 101.3 µm (1024 × 1024 pixels) with 0.3 µm z-steps for synapse quantification, and up to 203 × 203 µm for post hoc axon identification. Confocal images were analyzed in Fiji and corresponding axons in the confocal and two-photon images were identified using the line scar as a guide. Expression of CB1R or HA was determined by visual inspection. In some cases, the image was mirrored to confirm or reject positive staining. Negative axons were always chosen close to positive axons in the same imaging area, assuring that the absence of CB1R or HA expression was not because of low immunostaining quality. In addition, we verified that CB1R expression or staining levels did not affect our conclusion as we found the same results when we split CB1R+ axons in two separate groups with high and low CB1R levels. Per slice, two to six axons per group were included in the analysis.
For synapse quantification, images were analyzed in Fiji using a custom macro (Ruiter et al., 2020). An average projection image was made from five z-planes, images were median filtered (radius, 1 pixel), and individual puncta were identified using watershed segmentation. VGAT and gephyrin puncta were analyzed separately, and overlap was determined afterward. Four independent experiments were performed with one or two image areas per slice. To compare between treatments, data were normalized per experiment.
Adeno-associated virus application in VGAT-Cre slices.
Hippocampal slice cultures were prepared as described above from VGAT-Cre mice (stock #028862, The Jackson Laboratory) at 6–7 d after birth. Floxed adeno-associated virus 5 (AAV5) viruses to express GFP [ssAAV-5/2-hSyn1-dlox-EGFP(rev)-dlox-WPRE-hGHp(A); catalog #v115-5, Viral Vector Facility, Zurich University; RRID:Addgene_50457] and HA-tagged Gs-DREADDs [designer receptors exclusively activated by designer drugs; pssAAV-2-hSyn1-dlox-HA_rM3D(Gs)_IRES_mCitrine(rev)-dlox-WPRE-hGHp(A); catalog #v111-5, Viral Vector Facility, Zurich University; RRID:Addgene_50456] were applied at day in vitro 1 (DIV1) on top of the hippocampal CA1 region by a microinjector (FemtoJet, Eppendorf) aided by a stereoscopic microscope (model M80, Leica). This resulted in widespread, but sparse, GFP and Gs-HA expression in GABAergic neurons, which partially overlapped. Two-photon time-lapse imaging was performed when slices were kept for 2–3 weeks in vitro. After a baseline period (5 time points), Gs signaling was activated by bath application of 10 μm CNO (Tocris Bioscience), which was continued until the end of the experiment. Post hoc immunostaining was performed using rat anti-HA primary antibodies (1:500; catalog #11867423001, Roche; RRID:AB_390918), and anti-rat Alexa Fluor 568 (1:500; catalog #A11077, Thermo Fisher Scientific; RRID:AB_2534121) as secondary antibodies. We selected slices with good GFP labeling in the dendritic layer for the two-photon experiments, and in 4 of 13 slices we were able to identify up to 5 axons of each type within the imaging area. Identification of HA+ and HA– axons was performed in Fiji, and bouton dynamics analysis was performed in MATLAB, as described above.
Experimental design and statistical analyses.
All experiments were performed and analyzed blindly. Live-imaging experiments for bouton dynamics analysis were performed in paired slices from the same animal and the same culture. Statistical analysis was performed using GraphPad Prism software. Data are reported as mean ± SE, unless stated otherwise. The variance between axons was larger than the variance between slices, indicating that individual axons are independent measurements. Results from treatment and control experiments were compared using the nonparametric Mann–Whitney U (MW) test. Distributions were compared with the Kolmogorov–Smirnov (KS) test. Multiple comparisons were made using two-way ANOVA (two-way ANOVA) followed by Sidak's test. Repeated-measures two-way ANOVA was used for comparing NP bouton density and NP presence over time. The p values (not adjusted for multiplicity) are indicated in the figure legends. Differences were considered significant at p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Repeated CB1 receptor activation increases functional presynaptic terminals
We previously demonstrated that new inhibitory boutons can form in response to brief CB1 receptor activation (Hu et al., 2019). Newly formed boutons often did not persist (Hu et al., 2019), suggesting that additional or repeated signaling is required to eventually form functional inhibitory synapses (Wierenga, 2017; Frias et al., 2019). It was recently reported that strong, but brief, CB1 receptor activation can induce synaptic potentiation, while longer CB1 activation induces synaptic depression (Cui et al., 2015, 2016). This suggests that the CB1 activation pattern is an important factor in determining its downstream signaling. We therefore sought to use repeated, short activation of CB1 receptors to induce the formation of inhibitory synapses. We activated CB1 receptors in hippocampal slice cultures by repeated short exposure to the CB1 receptor ligand 2-AG (100 μm; 3× 20 min with a 2 h interval; Fig. 1A). We verified that this treatment did not affect the distribution of CB1 receptors in these slices (data not shown). We recorded mIPSCs in CA1 pyramidal neurons to assess functional inhibitory synapses 24 h after the start of the first 2-AG exposure (Fig. 1B). Repeated CB1 receptor activation resulted in an increase of the mean mIPSC frequency by 38% (control, 3.9 ± 0.3 Hz; 2-AG, 5.5 ± 0.4 Hz, p = 0.013; Fig. 1C), while mIPSC amplitudes were not affected (Fig. 1D). Continuous exposure to 2-AG for 24 h did not alter the frequency or amplitude of spontaneous IPSCs (Fig. 1E,F), which is consistent with the notion that activation pattern determines CB1 downstream signaling. Interestingly, mIPSCs after repeated 2-AG exposure appeared to have longer rise times (Fig. 1G), while decay times were not different (Fig. 1H). We separated mIPSCs with slow and fast rise times based on a double Gaussian fit of the distribution of rise times (Fig. 1I). When we then analyzed the interevent intervals of fast and slow mIPSCs separately, we observed that the interevent intervals of slow mIPSCs were decreased after repeated CB1 activation, while the interevent intervals of fast mIPSCs were not affected (Fig. 1J,K). This analysis revealed that the observed increase in mIPSC frequency was because of a specific increase in the frequency of slow mIPSCs with long rise times (Fig. 1L). The rise times of mIPSCs depend on synaptic maturation (Lazarus and Josh Huang, 2011; Gonzalez-Burgos et al., 2015; Pardo et al., 2018), but are also strongly influenced by subcellular location, as dendritic filtering attenuates mIPSCs originating from dendritic inhibitory synapses (Rall, 1967; Bekkers and Clements, 1999; Wierenga and Wadman, 1999). This suggests that the increased mIPSC frequency after CB1 receptor activation may reflect an increase of inhibitory currents from dendritic locations or from immature synapses.
Figure 1.
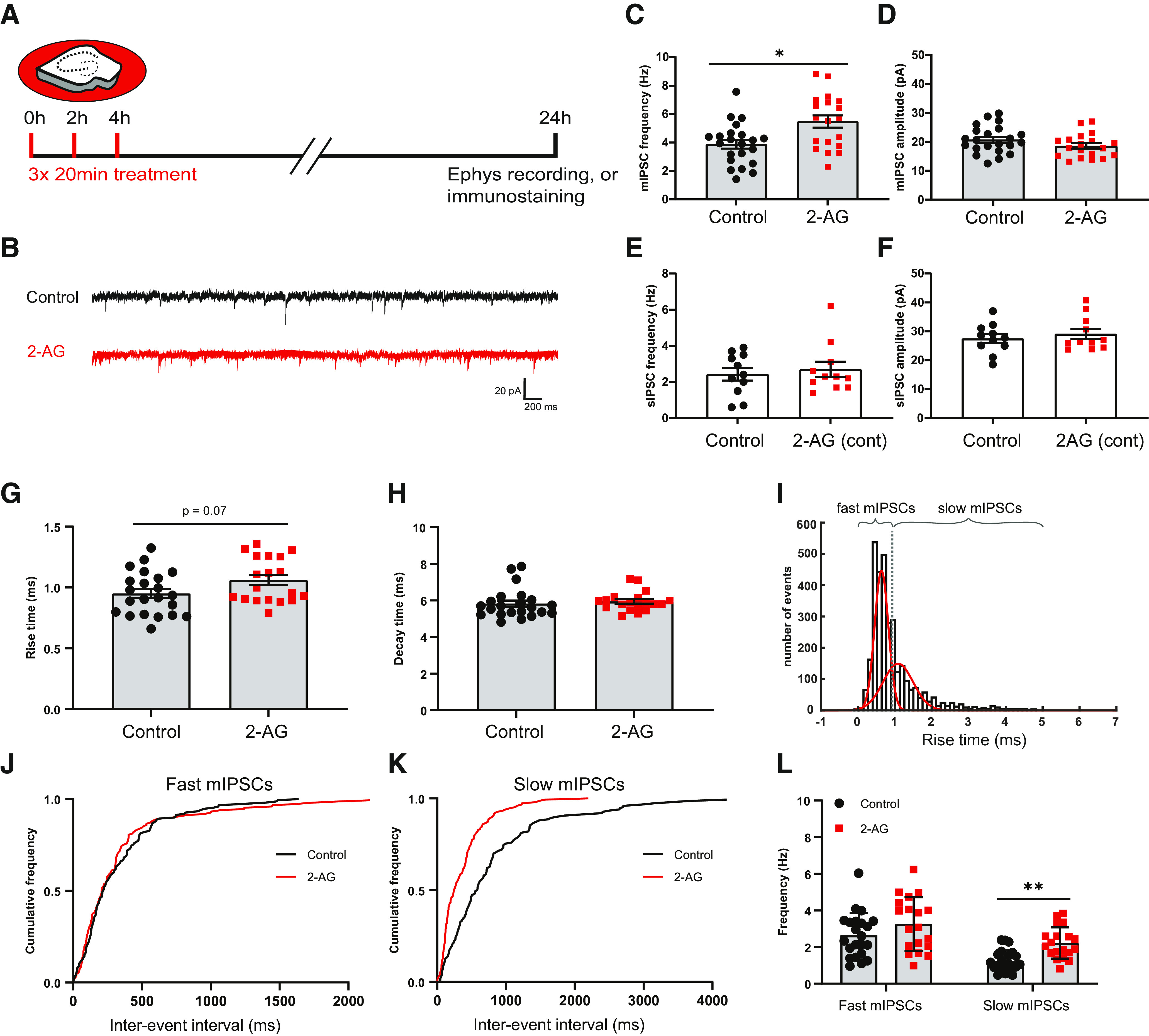
Repeated CB1 receptor activation results in increased mIPSC frequency. A, Organotypic hippocampal cultures were treated three times with culturing medium containing 100 μm 2-AG or DMSO (control) for 20 min with 2 h intervals. After 24 h, slices were used for electrophysiology and immunostaining experiments. B, Example traces of mIPSC recordings from control (black) and 2-AG-treated slice (red). C, D, Mean frequency (C) and amplitude (D) of mIPSCs in control and 2-AG-treated slices (MW test: p = 0.013 in C; p = 0.16 in D). Data from 22 cells in six control slices and 19 cells in six 2-AG-treated slices. E, F, Mean frequency (E) and amplitude (F) of sIPSCs (spontaneous IPSCs) in control and 2-AG-treated slices, when 2-AG was continuously present for 24 h (MW test: p = 0.99 in E; and p = 0.95 in F). Data from 11 cells in five control slices and 11 cells in six 2-AG-treated slices. G, Mean rise time of mIPSCs in control and 2-AG-treated slices (MW, p = 0.073). H, Mean of mIPSC decay time in control and 2-AG-treated slices (MW test, p = 0.19). I, The distribution of the rise times of mIPSCs was fitted with a double Gaussian to separate fast and slow mIPSCs. J, K, Cumulative distribution of interevent intervals of mIPSCs with fast (J) and slow (K) rise times (KS test: p = 0.65 in J; p < 0.0001 in K). L, Mean frequency of mIPSCs with fast and slow rise times (two-way ANOVA Sidak test: fast, p = 0.14; slow, p = 0.0095). Data in G–L, C, and D are from the same dataset. Asterisks indicate significant differences (*p < 0.05; **p < 0.01).
To determine whether the observed increase in mIPSCs was associated with an increase in the number of inhibitory synapses, we analyzed presynaptic VGAT and postsynaptic gephyrin puncta in the dendritic region of the CA1 area in parallel immunohistochemistry experiments (Fig. 2A). We observed that the density of VGAT puncta was slightly increased after repeated 2-AG application (Fig. 2B), while the VGAT puncta size was decreased (Fig. 2C). Gephyrin puncta density and size were not affected by repeated 2-AG exposure (Fig. 2D,E), and the density of inhibitory synapses, defined as VGAT–gephyrin associations, was also not different from control slices (Fig. 2F,G). We therefore made a distinction between VGAT puncta that were associated with gephyrin and VGAT puncta without gephyrin (Fig. 2A, last panels at the right). We observed that the increase in VGAT density was because of a specific increase in VGAT puncta that were not associated with gephyrin (Fig. 2H). In contrast, the reduction in VGAT puncta size was mostly because of a reduction in the size of VGAT puncta with a gephyrin association (Fig. 2I). This suggests that repeated short activation of CB1 receptors has two separable effects on inhibitory synapses: on the one hand, it leads to the shrinkage of VGAT clusters at inhibitory synapses, possibly reflecting synaptic depression (Monday et al., 2020), while at the same time new VGAT clusters are formed that are not associated with the postsynaptic scaffold gephyrin. Live-imaging experiments have shown that VGAT is rapidly recruited when new boutons are formed in inhibitory axons, and that gephyrin normally follows within a few hours (Wierenga et al., 2008; Dobie and Craig, 2011; Frias et al., 2019). Our data suggest that repeated CB1 receptor activation induces the formation of presynaptic VGAT clusters, likely reflecting immature inhibitory synapses.
Figure 2.
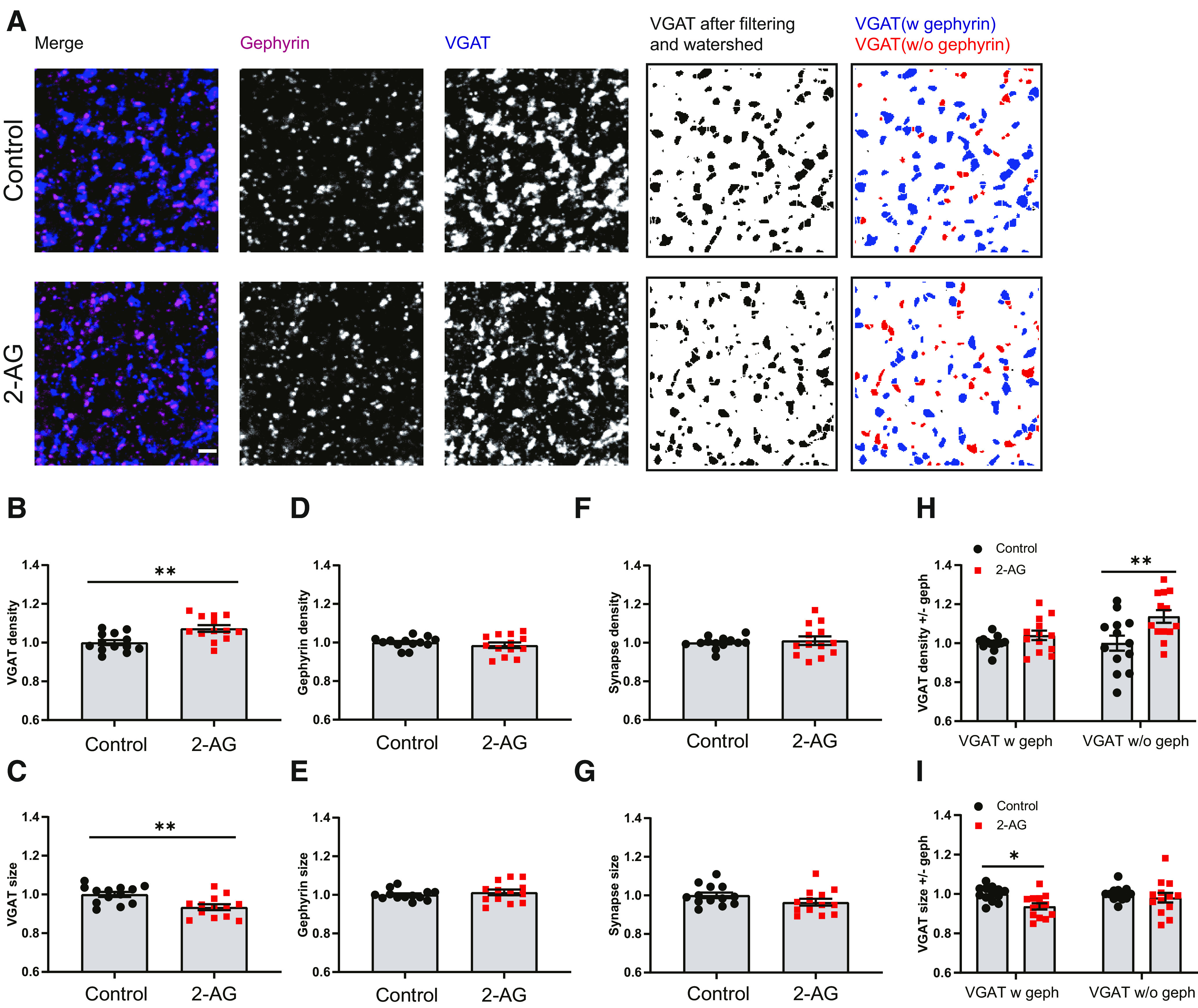
Repeated CB1 receptor activation induces the formation of partial inhibitory synapses. A, Representative immunostaining images showing the presynaptic VGAT (blue) and postsynaptic gephyrin (purple) in control (top) and 2-AG (bottom) slices. Individual VGAT puncta were identified using watershed segmentation, and these were color coded to distinguish VGAT puncta associated with gephyrin (blue) and VGAT puncta without gephyrin (red). B, C, Normalized density (B) and size (C) of VGAT puncta in control and 2-AG slices (MW test: p = 0. 0061 in B; p = 0.004 in C). D, E, Normalized density (D) and size (E) of gephyrin puncta in control and 2-AG slices (MW test: p = 0.54 in D; p = 0. 64 in E). F, G, Normalized density (F) and size (G) of VGAT/gephyrin colocalizations in control and 2-AG slices (MW test: p = 0.76 in F; p = 0.099 in G). H, I, Normalized density (F) and size (G) of VGAT puncta with and without gephyrin (two-way ANOVA Sidak test: p = 0.55 and p = 0.003 in H; p = 0.017 and p = 0.65 in I). Data from 13 image stacks in seven slices/group.
Acute activation of CB1 receptors affects nonpersistent boutons density only slightly
To get further insight into the role of CB1 receptors in the formation of inhibitory synapses, we performed two-photon live imaging in organotypic hippocampal slices to monitor GFP-labeled inhibitory bouton dynamics in response to short activation of CB1 receptors. Here we used short applications (5 min) of CB1 receptor agonists to mimic retrograde endocannabinoid signaling (Hu et al., 2019), but we wanted to avoid inducing synaptic weakening (Monday et al., 2020). We used the endogenous CB1 receptor ligand 2-AG as well as the chemically synthesized agonist WIN, which is widely used because of its high affinity and stability (Chevaleyre et al., 2007; Roland et al., 2014; Wang et al., 2018). We verified that brief WIN application only transiently and mildly suppressed inhibitory currents (data not shown). As previously reported (Frias et al., 2019), the majority of inhibitory boutons were present at all time points during the 140 min imaging period (persistent boutons), but a substantial fraction of inhibitory boutons appeared, disappeared, or reappeared during the imaging period (Fig. 3A; Schuemann et al., 2013; Frias et al., 2019). We will refer to the latter as NP boutons. Bath application of 100 μm 2-AG (5 min) did not affect overall bouton density (control, 30.8 ± 1.7 boutons/100 µm; 2-AG, 29.8 ± 1.7 boutons/100 μm; p = 0.81). The density of NP boutons appeared slightly increased after 2-AG compared with DMSO control (Fig. 3B,C), but this was mainly because of a large effect in a single axon. We calculated for each axon the average fraction of NP boutons that are present over time (NP presence). In control slices, there was a small decrease in NP presence over time, possibly reflecting a decrease in network activity level when the slices are transferred from the incubator to the microscope. After 2-AG application, NP presence appeared slightly more stable (Fig. 3D), but this difference did not reach statistical significance. We assessed whether this difference could be traced back to a more specific effect in a particular subgroup of NP boutons (see Materials and Methods; Frias et al., 2019), but we could not detect any differences in the densities of NP bouton subgroups in slices treated with control DMSO or 2-AG (Fig. 3E). There was also no difference in bouton duration (data not shown).
Figure 3.
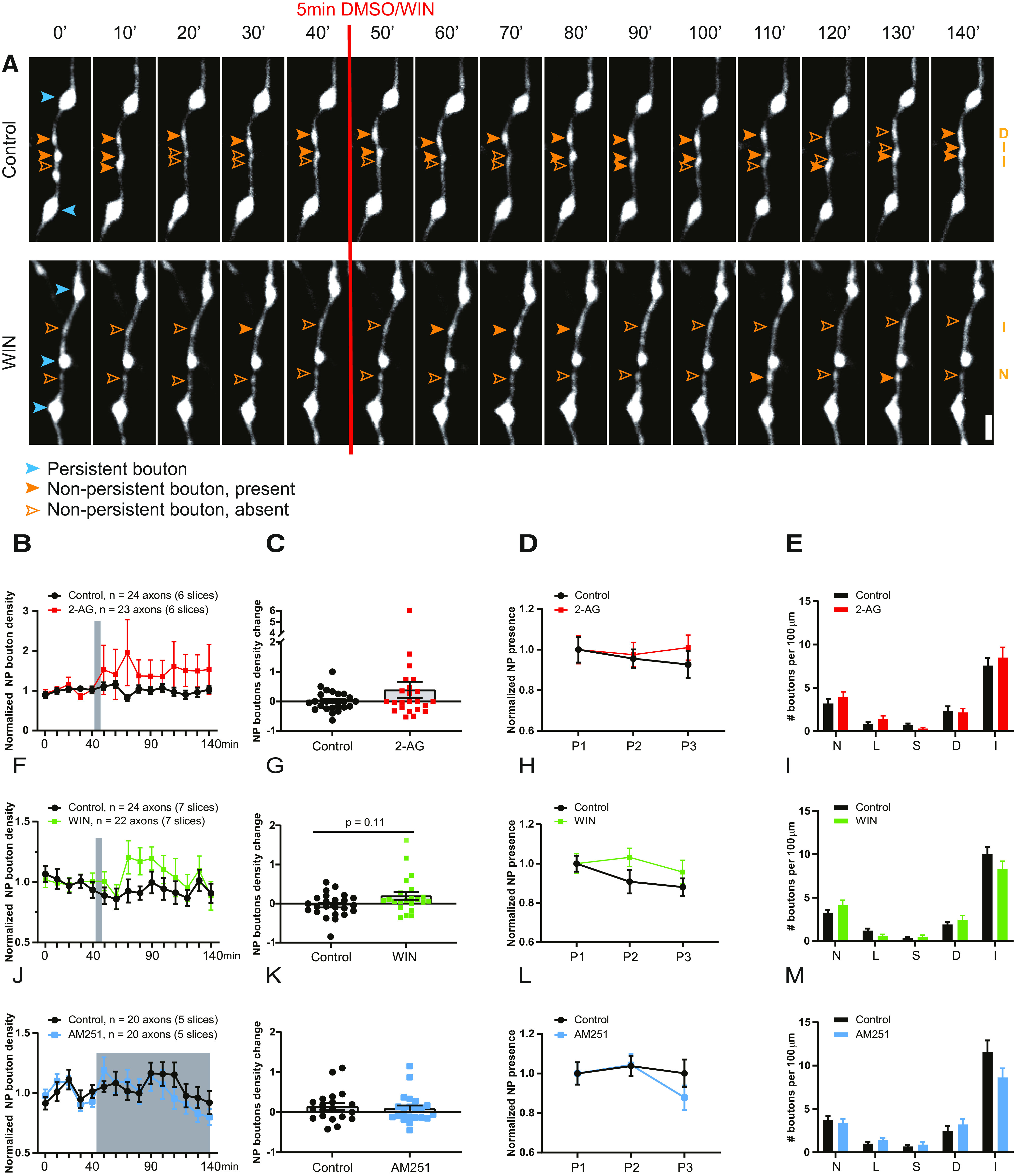
Brief activation of CB1 receptors slightly increases NP bouton density. A, Representative two-photon time-lapse images of GAD65-GFP-labeled inhibitory axons in the dendritic region of the hippocampal CA1 area (maximal projections of 17 z-sections). After a baseline of five time points (40 min), CB1 receptor agonist or DMSO was washed in for 5 min. Imaging was continued for another 10 time points (total imaging period is 140 min). P boutons (blue) and NP boutons (orange) are indicated by arrowheads. Empty arrowheads reflect a NP bouton that was absent at the time point. Subgroup of the NP boutons is indicated on the right in orange (abbreviations as in E). Scale bar, 2 µm. B, CB1 receptors were activated by bath application of 100 μm 2-AG for 5 min. Normalized NP bouton density over time in control (black) slices and after 2-AG (red) application (two-way ANOVA, p = 0.33). C, Maximum change in NP bouton density in control slices and after 2-AG application (MW test, p = 0.54). D, Normalized NP presence over time in control and 2-AG-treated slices. P1, Time points 1–5; P2, time points 6–10; P3, time points 11–15 in control and 2-AG-treated slices (two-way ANOVA, p = 0.61). E, Mean density of NP bouton subgroups in control slices and after 2-AG application. N, New boutons (MW test, p = 0.35); L, lost boutons (MW test, p = 0.44); S, stabilizing boutons (MW test, p = 0.21); D, destabilizing boutons (MW test, p = 0.91); I, intermittent boutons (MW test, p = 0.87). F, CB1 receptors were activated by bath application of 20 μm WIN for 5 min. Normalized NP bouton density over time in control (black) slices and after 2-AG (green) application (two-way ANOVA, p = 0.20). G, Maximum change in NP bouton density in control slices and after WIN application (MW test, p = 0.11). H, Normalized NP presence over time in control slices and after WIN application (two-way ANOVA, p = 0.20). I, Mean density of NP bouton subgroups in control slices and after WIN application [MW test: p = 0.40 (N); p = 0.06 (L); p = 0.79 (S); p = 0.70 (D); p = 0.10 (I)]. J, Slices were treated with the CB1 receptor antagonist AM251 (5 μm) after time point 5. Normalized NP bouton density over time in control (black) slices and during AM251 (blue) application (two-way ANOVA, p = 0.66). K, Maximum change in NP bouton density in control slices and during AM251 application (MW test, p = 0.6). L, Normalized NP presence over time in control slices and during AM251 application (two-way ANOVA, p = 0.56). M, Mean density of NP bouton subgroups in control slices and during AM251 application [MW test: p = 0.46 (N); p = 0.23 (L); p = 0.94 (S); p = 0.29 (D); p = 0.10 (I)]. Data in B–E from 24 axons in six control slices and 23 axons in six 2-AG slices. Data in F–I from 24 axons in seven control slices and 22 axons in seven WIN slices. Data in J–M from 20 axons in five control slices and 20 axons in five AM251 slices.
The endocannabinoid 2-AG is rather unstable in solution and gets rapidly degraded in biological tissue (Savinainen et al., 2012; Dócs et al., 2017). To exclude the possibility that 2-AG gets degraded before it can activate CB1 receptors, we repeated these experiments using 20 μm WIN. Short activation (5 min) of CB1 receptors by bath application of WIN slightly increased in NP bouton density (Fig. 3F,G). Although the increase appeared more robust compared with the 2-AG-induced effect, the effect was too small to reach statistical significance. Similar to 2-AG, the average NP presence appeared slightly increased (Fig. 3H), but we could not detect any changes in specific NP bouton subgroups (Fig. 3I). Together, these observations indicate that short CB1 receptor activation by 2-AG or WIN leads to only a small (if any) increase in NP bouton density in GFP-labeled inhibitory axons.
Endocannabinoids are produced on demand in postsynaptic neurons (Alger and Kim, 2011; Hashimotodani et al., 2013; Piomelli, 2014), but an ambient level of endocannabinoids is always present, even in slices (Szabó et al., 2014; Lee et al., 2015; Lenkey et al., 2015). Tonic CB1 receptor activation by endocannabinoids affects mostly perisomatic inhibitory synapses, while dendritic inhibitory synapses are reported to be less sensitive (Lee et al., 2010, 2015). To address whether tonic activation of CB1 receptors contributes to inhibitory bouton dynamics in our GFP-labeled axons (which mostly target dendrites; Wierenga et al., 2010), we applied 5 μm AM251, an antagonist of CB1 receptors. However, AM251 had no effect on NP bouton density (Fig. 3J,K), NP presence, or NP bouton subgroups (Fig. 3L,M).
Together, our experimental findings indicate that inhibitory bouton dynamics of the GFP-labeled axons are not under strong tonic endocannabinoid control and that short CB1 receptor activation by 2-AG or WIN only slightly increases NP inhibitory bouton density.
CB1 receptors regulate inhibitory bouton dynamics specifically in CB1R+ axons
The expression of CB1 receptors largely overlaps with the expression pattern of CCK in GABAergic interneurons (Katona et al., 1999, 2006). These interneurons are partially labeled in the GAD65-GFP mice that we used for the experiments (Wierenga et al., 2010). We previously estimated that ∼50% of the GFP-labeled inhibitory axons express CB1 receptors in our slices (Hu et al., 2019), and this may significantly dilute an effect of CB1 receptor activation on bouton dynamics (Fig. 3). We therefore used post hoc immunostaining immediately after two-photon live imaging to distinguish between axons with and without CB1 receptors (CB1R+ and CB1R– axons, respectively; Fig. 4A,B). In accordance with previous reports (Mikasova et al., 2008; Dudok et al., 2015), CB1 receptors covered the entire surface of CB1R+ inhibitory axons, and individual CB1R+ axons could be easily traced from the CB1 immunostainings (Fig. 4A,B). In addition, there was significant CB1 background staining, presumably reflecting CB1 receptors in pyramidal cells and glia cells (Bonilla-Del Río et al., 2021). CB1R– axons had a higher bouton density and higher bouton turnover compared with CB1R+ axons (Fig. 4C; see below), supporting the notion that CB1R+ and CB1R– GFP-labeled axons belong to separate subtypes of GABAergic cells.
Figure 4.
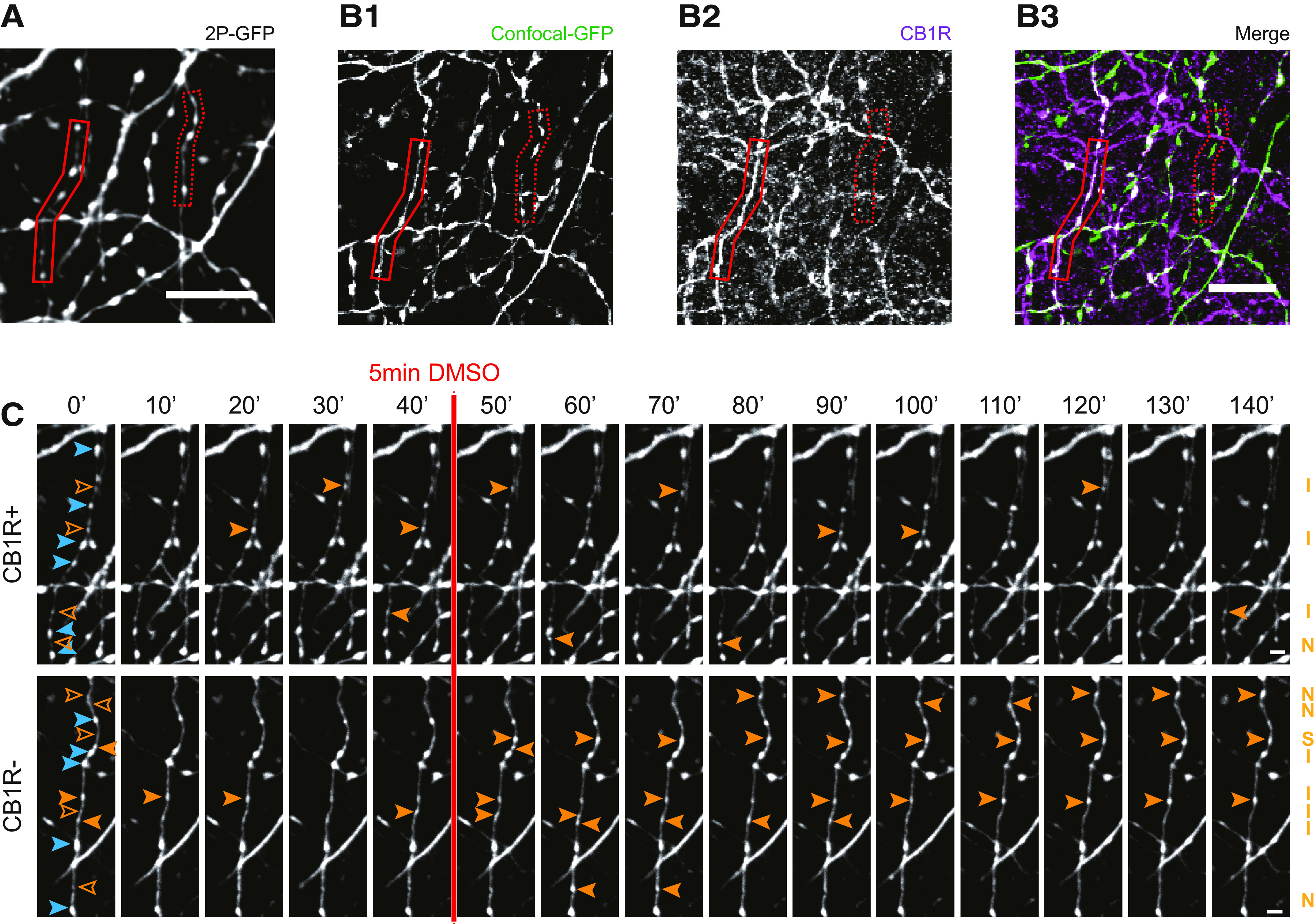
Distinction between CB1R+ and CB1R– axons using post hoc immunohistochemistry. A, z-Projection of representative two-photon image of GFP-labeled inhibitory axons. After two-photon live imaging, the slice was immediately fixated and further processed for immunohistochemistry to assess CB1R expression. B1, Confocal images of the same area after post hoc immunohistochemistry, showing the same GFP-labeled axons as in A (indicated with solid and dashed red boxes). B2, Immunostaining against CB1 receptors show a clear distinction between CB1R+ axons (solid red box), which express CB1 receptors and cover the entire axonal surface, and CB1R– axons (dashed red box), which do not express CB1 receptors. B3, merged image of B1 and B2. C, Two-photon time-lapse imaging of bouton dynamics in the CB1R+ and CB1R– axons indicated in A and B. Arrowheads indicate P (blue) and NP (orange) boutons, as in Figure 3A. Scale bars: A–B3, 10 µm; C, 2 μm.
We repeated the WIN application experiments, but now separately analyzed CB1R+ and CB1R– axons. In CB1R+ axons, the density of NP boutons significantly increased after WIN application (Fig. 5A,B). WIN also increased the average NP presence compared with control axons (Fig. 5C). When we analyzed the NP bouton subgroups, we found a specific increase in the density of new and stabilizing boutons (Fig. 5D,F), whereas other NP subgroups were unaffected (Fig. 5D–H). New boutons reflect immature synapses, which start to recruit presynaptic and postsynaptic proteins, while levels of VGAT and gephyrin at stabilizing boutons at the end of the imaging period are comparable to those of persistent boutons (Schuemann et al., 2013; Frias et al., 2019). In clear contrast, WIN had no effect on bouton density or dynamics in CB1R– axons in the same slices (Fig. 5I–P). These results indicate that axonal CB1 receptors are required for mediating the WIN-induced changes in bouton dynamics in inhibitory axons and exclude a role for CB1 receptors on other cells. Our results indicate that short activation of axonal CB1 receptors leads to an increase in NP bouton density by specifically promoting the formation and stabilization of inhibitory boutons.
Figure 5.
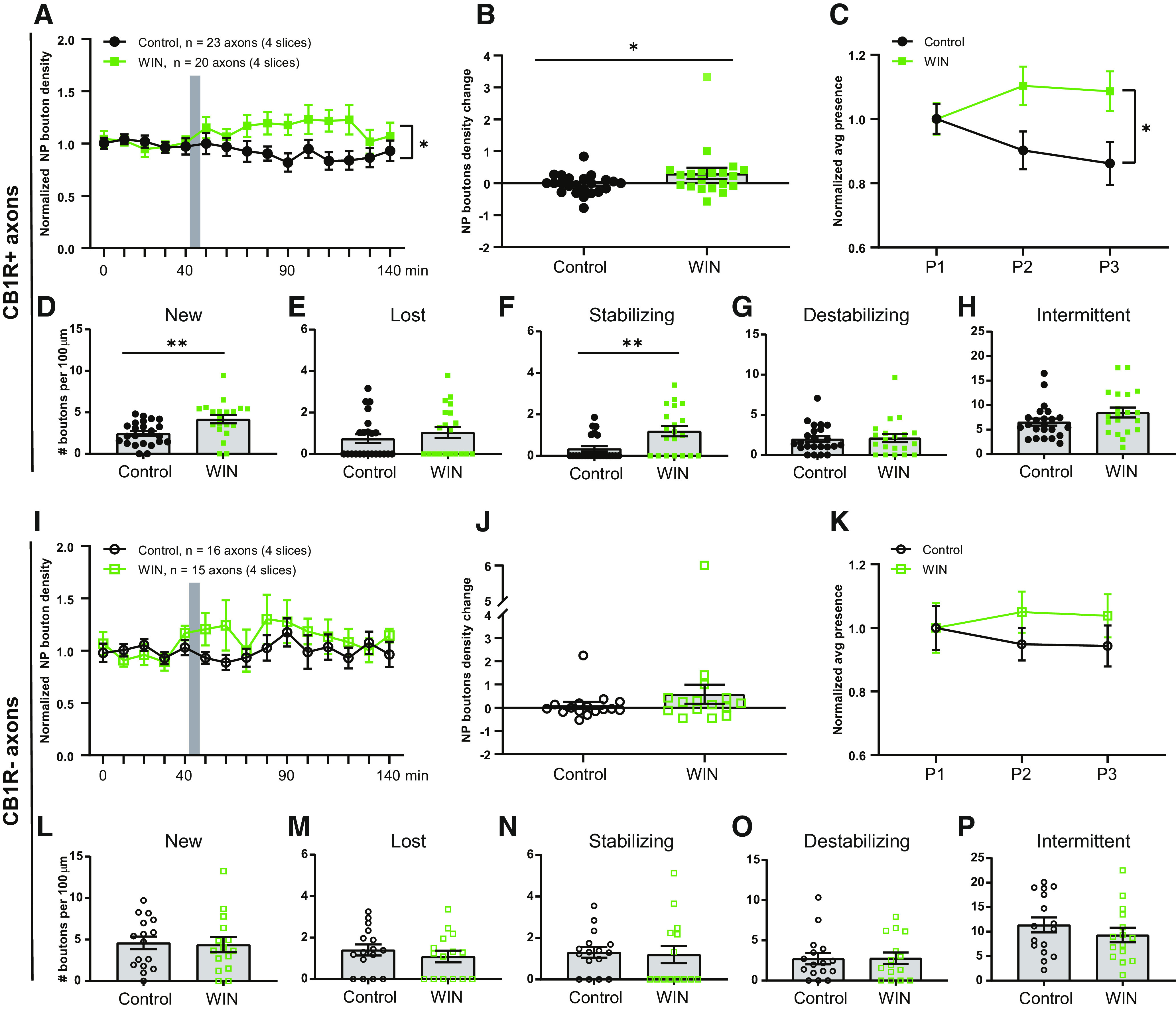
WIN promotes formation and stabilization of inhibitory boutons only in CB1R+ axons. A, Normalized NP bouton density in CB1R+ axons over time in control (black) slices and after WIN (green) application (two-way ANOVA, p = 0.018; interaction p = 0.026). B, Maximum change in NP bouton density in CB1R+ axons in control (black) slices and after WIN (green) application (MW test, p = 0.047). C, Normalized NP presence in CB1R+ axons over time in control slices and after WIN application (two-way ANOVA: p = 0.022; interaction, p = 0.045). D–H, Mean density of NP bouton subgroups in CB1R+ axons in control slices and after WIN application: new boutons (MW test, p = 0.002; D); lost boutons (MW test, p = 0.39; E); stabilizing boutons (MW test, p = 0.005; F); destabilizing boutons (MW test, p = 0.87; G); intermittent boutons (MW test, p = 0.16; H). I, Normalized NP bouton density in CB1R– axons over time in control (black) slices and after WIN (green) application (two-way ANOVA, p = 0.27). J, Maximum change in NP bouton density in CB1R– axons in control (black) slices and after WIN (green) application (MW test, p = 0.21). K, Normalized NP presence in CB1R– axons over time in control slices and after WIN application (two-way ANOVA, p = 0.37). L–P, Mean density of NP bouton subgroups in CB1R– axons in control slices and after WIN application: new boutons (MW test, p = 0.77; L); lost boutons (MW test, p = 0.46; M); stabilizing boutons (MW test, p = 0.50; N); destabilizing boutons (MW test, p = 0.99; O); and intermittent boutons (MW test, p = 0.34; P). Data from 23 CB1R+ and 16 CB1R– axons in four control slices and 20 CB1R+ and 15 CB1R– axons in four slices with WIN application.
WIN-induced bouton formation does not require Gi/o signaling and neuronal activity
CB1 receptors are G-protein-coupled receptors. Endocannabinoid signaling via CB1 receptors typically activates Gi/o-heterotrimeric proteins, resulting in a reduction of neurotransmitter release at presynaptic terminals (Lovinger, 2008; Castillo et al., 2012). We therefore tested whether WIN-induced bouton formation requires Gi/o signaling. We pretreated the slices with PTX (1 μg/ml) for 24 h to eliminate Gi/o signaling (Guo and Ikeda, 2004; Campbell and Smrcka, 2018) and then performed two-photon time-lapse live imaging as before. Axons with and without CB1R were distinguished using post hoc immunostaining (Fig. 6A–C). PTX pretreatment had no major effect on CB1 receptor expression patterns.
Figure 6.
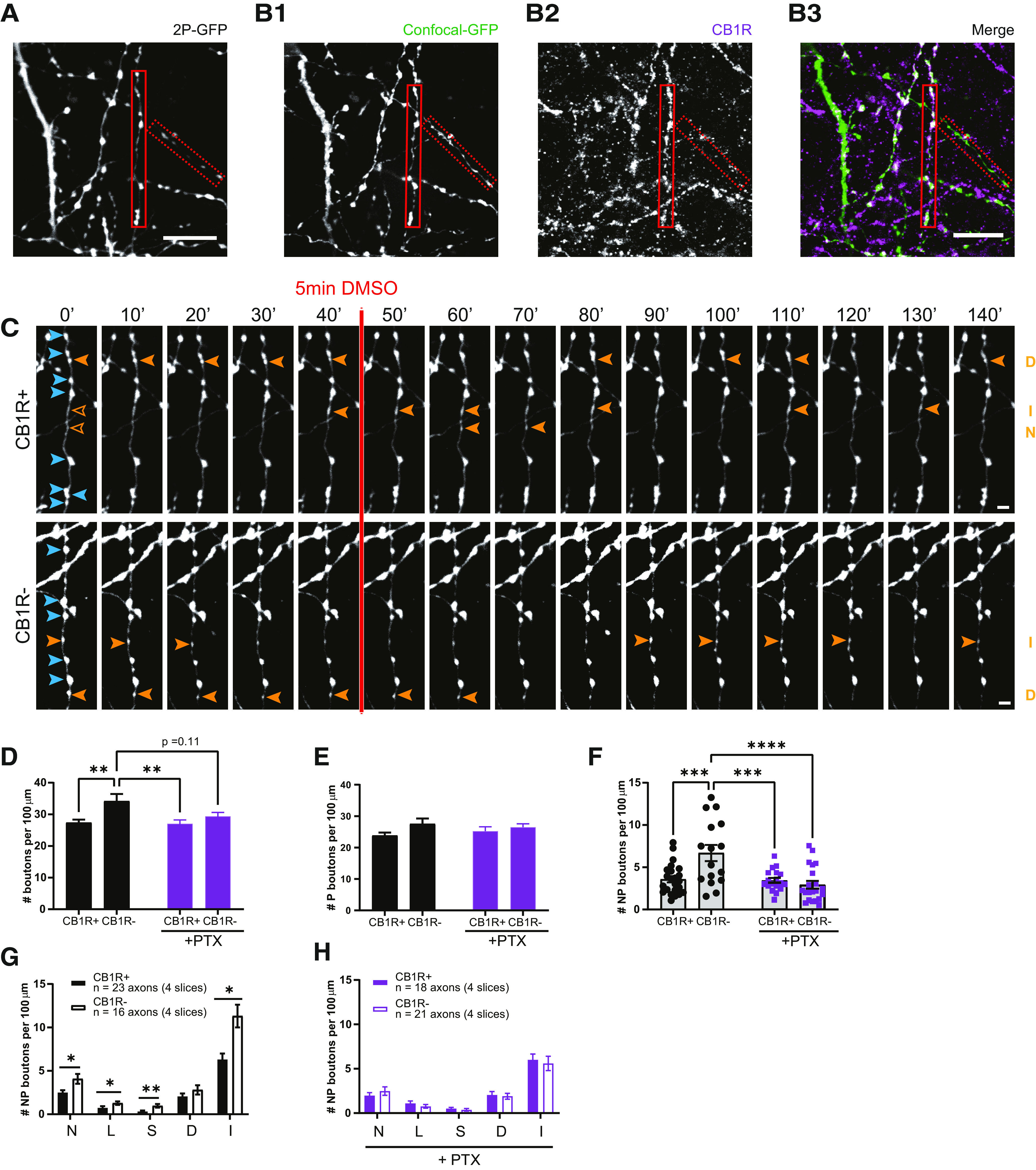
Gi/o signaling is an important regulator of inhibitory bouton dynamics. A, z-Projection of representative two-photon image of GFP-labeled inhibitory axons after PTX pretreatment. B1–B3, Confocal images of the same GFP-labeled axons (B1) as in A after post hoc immunohistochemistry for CB1 (B2). Solid and dashed red boxes indicate CB1R+ and CB1R− axons in the merged image (B3). C, Two-photon time-lapse imaging of bouton dynamics in the CB1R+ and CB1R– axons indicated in A and B1–B3 after PTX pretreatment. Arrowheads indicate P (blue) and NP (orange) boutons as in Figure 3A. D, Average bouton density during baseline in CB1R+ and CB1R– axons in control slices and after PTX pretreatment. Comparisons between CB1R+ and CB1R– axons: control, p = 0.0056; after PTX, p = 0.79; between control and PTX: CB1R– axons, p = 0.11; CB1R+ axons, p > 0.99; between CB1R+ (control) and CB1R– (PTX), p = 0.86; and between CB1R– (control) and CB1R+ (PTX), p = 0.0057 (two-way ANOVA Sidak test). E, Average density of P boutons during baseline in CB1R+ and CB1R– axons in control slices and after PTX pretreatment (p = 0.057 for axon type, two-way ANOVA Sidak test). F, Average density of NP boutons during baseline in CB1R+ and CB1R– axons in control slices and after PTX pretreatment. Comparisons between CB1R+ and CB1R– axons: control, p = 0.0007; after PTX, p = 0.99; between control and PTX: CB1R– axons, p < 0.0001; CB1R+ axons, p > 0.99; between CB1R+ (control) and CB1R– (PTX), p = 0.93; and between CB1R– (control) and CB1R+ (PTX), p = 0.0008 (two-way ANOVA Sidak test). G, Mean density of NP bouton subgroups in CB1R+ and CB1R– axons in control slices. N, New boutons (MW test, p = 0.035); L, lost boutons (MW test, p = 0.037); S, stabilizing boutons (MW test, p = 0.002); D, destabilizing boutons (MW test, p = 0.47); I, intermittent boutons (MW test, p = 0.010). H, Mean density of NP bouton subgroups in CB1R+ and CB1R– axons after PTX pretreatment (MW test, p = 0.45; N); p = 0.41; L); p = 0.36; S); p = 0.88; D); p = 0.40; I). Data from 23 CB1R+ and 16 CB1R– axons in four control slices, and 18 CB1R+ and 21 CB1R– axons in four PTX-pretreated slices. Scale bars: A, B, 10 µm; C, D, 2 μm.
Under control conditions, CB1R– axons had a higher bouton density compared with CB1R+ axons (Fig. 6D), which was mainly because of a higher density of NP boutons (Fig. 6E,F). The density for all NP bouton subgroups was almost twice as high in CB1R– axons compared with CB1R+ axons (Fig. 6G), showing that overall inhibitory bouton dynamics were more pronounced in CB1R– axons compared with CB1R+ axons. Unexpectedly, we observed that 24 h pretreatment with PTX affected bouton density. PTX pretreatment specifically downregulated bouton density in CB1R– axons, while bouton density in CB1R+ axons was largely unaffected (Fig. 6D). PTX specifically reduced the density of nonpersistent boutons in CB1R– axons (Fig. 6E,F). After PTX pretreatment, there was no longer a difference in NP bouton subgroups between CB1R+ and CB1R– inhibitory axons (Fig. 6H). This suggests that under normal conditions CB1R– axons have a higher Gi/o-protein activity compared with CB1R+ axons in these slices. These data imply that Gi/o signaling is an important regulator of inhibitory bouton dynamics.
We then tested whether acute activation of CB1 receptors via WIN can induce changes in inhibitory bouton dynamics in the absence of Gi/o signaling. We observed that short activation of CB1 receptors by WIN could still induce the formation of new inhibitory boutons in CB1R+ axons after pretreatment with PTX (Fig. 7A). This indicates that the formation of new inhibitory boutons by CB1 receptor activation is independent of Gi/o signaling. However, in the absence of Gi/o signaling, WIN application no longer promoted bouton stabilization (compare Figs. 7B, 5F), suggesting that bouton stabilization requires intact Gi/o signaling. As before, other NP bouton subgroups were not affected (Fig. 7C) and WIN application did not affect bouton formation (density of new boutons was 81 ± 23% of control; MW test, p = 0.51) or bouton dynamics (data not shown) in CB1R– axons. These data indicate that short activation of CB1 receptors on inhibitory axons by WIN promotes the formation of new boutons via a Gi/o-independent signaling pathway.
Figure 7.

CB1-mediated bouton formation does not require Gi/o signaling and is independent of activity. A, Mean density of new boutons in CB1R+ axons after control (black) and WIN (green) application in PTX-pretreated slices (MW test, p = 0.047). B, Mean density of stabilizing boutons in CB1R+ axons after control and WIN application in PTX-pretreated slices (MW test, p = 0.93). C, Mean density of other NP bouton subgroups in CB1R+ axons after control and WIN application in PTX-pretreated slices. L, Lost boutons (MW test, p = 0.82); D, destabilizing boutons (MW test, p = 0.37); I, intermittent boutons (MW test, p = 0.59). D, Mean density of new boutons in CB1R+ axons after control (black) and WIN (green) application in the presence of TTX (MW test, p = 0.013). E, Mean density of stabilizing boutons in CB1R+ axons after control and WIN application in the presence of TTX (MW test, p = 0.61). F, Mean density of other NP bouton subgroups in CB1R+ axons after control and WIN application in the presence of TTX. L, Lost boutons (MW test, p = 0.23); D, destabilizing boutons (MW test, p = 0.56); I, intermittent boutons (MW test, p = 0.16). Data in A–C from 18 axons in four slices with DMSO (control) application and 18 axons in four slices with WIN application. Data in D–F from 14 axons in four slices with DMSO (control) application and 15 axons in five slices with WIN application.
Gi/o-protein signaling can hyperpolarize neurons via activation of K+ channels (Bacci et al., 2004; Guo and Ikeda, 2004). Blocking ongoing Gi/o-protein activity with PTX may therefore enhance neuronal activity in our slices, which may by itself affect inhibitory bouton dynamics. However, as enhancing neuronal activity is expected to promote overall inhibitory bouton turnover (Schuemann et al., 2013; Frias et al., 2019), this does not appear to be in line with the observed decrease in inhibitory bouton dynamics in CB1R– axons after PTX. To address whether WIN-induced inhibitory bouton formation is affected by activity, we blocked network activity with TTX to reduce overall bouton dynamics (Schuemann et al., 2013; Frias et al., 2019). We observed that in the presence of TTX, brief activation of CB1 receptors with WIN still induced the specific increase in the density of new boutons (Fig. 7D). However, WIN no longer induced a change in the density of stabilizing boutons (Fig. 7E), consistent with our earlier finding that inhibitory bouton stabilization requires activity (Frias et al., 2019). Other NP bouton subgroups were not affected (Fig. 7F), and WIN application did not significantly affect bouton formation (179 ± 216% of control; MW test, p = 0.11) or other bouton dynamics (data not shown) in CB1R– axons. Together, these data demonstrate that CB1 receptor-mediated inhibitory bouton formation does not require Gi/o-protein signaling and is independent of neuronal activity.
Acute elevation of cAMP levels promotes inhibitory bouton formation via protein kinase A
In addition to the typical downstream signaling pathway via Gi/o-proteins, CB1R activation can trigger several other signaling pathways, including via G12/13-protein (Roland et al., 2014), Gq-protein (Lauckner et al., 2005), and Gs-protein (Glass and Felder, 1997; Finlay et al., 2017). Intriguingly, a novel form of CB1 receptor-mediated synaptic potentiation was recently reported, which was shown to depend on the activity of presynaptic protein kinase A (PKA; Cui et al., 2016; Wang et al., 2018). This raises the attention to CB1 receptor-mediated Gs signaling, as Gs-protein signaling enhances PKA activity via stimulation of cAMP production (Antoni, 2012; Taylor et al., 2013). We therefore tested whether inhibitory bouton dynamics were affected when we directly elevated cAMP levels via activation of adenylyl cyclase by 25 μm forskolin (5 min; Fig. 8A). We observed that brief application of forskolin induced the formation of new inhibitory boutons (Fig. 8B), while other NP subgroups were not affected (Fig. 8C,D). This suggests that the inhibitory bouton formation that we observed after CB1 receptor activation may be mediated by Gs signaling. The increase in inhibitory bouton formation after forskolin application appeared much stronger compared with WIN application (compare Figs. 8B, 3I), suggesting that most, if not all, GFP-labeled inhibitory axons responded to forskolin.
Figure 8.
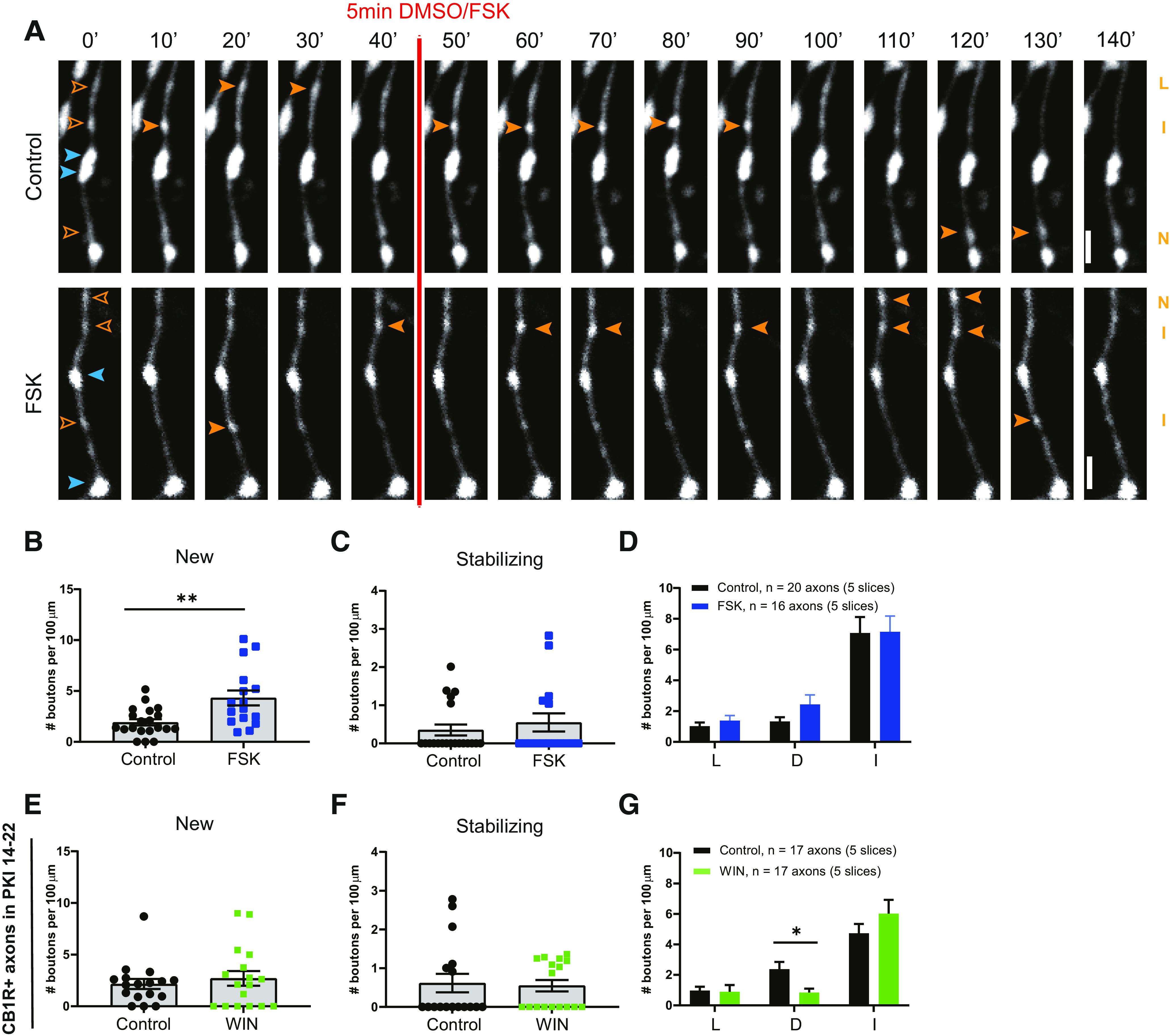
Inhibitory bouton formation is promoted by increasing intracellular cAMP levels with forskolin. A, Representative two-photon time-lapse images of bouton dynamics in GFP-labeled axons after control or forskolin application. Arrowheads indicate P (blue) and NP (orange) boutons as in Figure 3A. Scale bar, 2 µm. B, Mean density of new boutons in control (black) slices and after forskolin (blue) application (MW test, p = 0.007). C, Mean density of stabilizing boutons in control slices and after forskolin application (MW test, p = 0.67). D, Mean density of other subgroup of NP boutons in control slices and after forskolin application. L, Lost boutons (MW test, p = 0.46); D, destabilizing boutons (MW test, p = 0.37); I, intermittent boutons (MW test, p = 0.81). E, Mean density of new boutons in control (black) slices and after WIN (green) application in the presence of PKA blocker PKI 14-22 (MW test, p = 0.81). F, Mean density of stabilizing boutons in control slices and after WIN application in the presence of PKA blocker PKI 14-22 (MW test, p = 0.66). G, Mean density of other subgroup of NP boutons in control slices and after WIN application in the presence of PKA blocker PKI 14-22. L, lost boutons (MW test, p = 0.29); D, destabilizing boutons (MW test, p = 0.027); I, intermittent boutons (MW test, p = 0.45).
To directly test whether downstream PKA activity is required, we blocked PKA activity with PKI 14-22 (Chevaleyre et al., 2007). We observed that in the presence of PKI 14-22 WIN no longer triggered the formation or stabilization of inhibitory boutons (Fig. 8E,F). Interestingly, we observed a decrease in the density of destabilizing boutons after WIN treatment (Fig. 8G), suggesting that PKA activity levels may be important for the maintenance of inhibitory boutons. Together, these data show that the formation of new inhibitory boutons is promoted by increasing intracellular cAMP levels and PKA activity.
Gs signaling in inhibitory axons promotes inhibitory bouton formation
Bath application of forskolin strongly increases neuronal activity (Mitoma and Konishi, 1996; Gekel and Neher, 2008) and will raise cAMP levels in all cells in the slice. The observed specific increase in new inhibitory bouton formation after forskolin, without affecting overall bouton dynamics, is therefore quite remarkable. However, we cannot conclude that the observed increase in inhibitory bouton formation is a direct effect of elevated cAMP levels in the inhibitory axons. We made use of DREADDs (Urban and Roth, 2015; Roth, 2016) to achieve cell-specific manipulation of presynaptic cAMP levels. Gs-DREADDs allow the direct activation of the Gs-protein signaling pathway using the specific ligand CNO. To achieve sparse expression restricted to inhibitory neurons, we infected hippocampal slices from VGAT-Cre mice with Cre-dependent AAVs. We used two AAVs: one containing an HA-tagged Gs–DREADD construct and one containing GFP (Fig. 9A; for details, see Materials and Methods). Infections with these two AAVs resulted in sparse GFP-labeling of inhibitory cells and their axons, which partially overlapped with Gs-HA expression (Fig. 9B). Post hoc immunostaining allowed us to identify GFP-labeled axons with and without Gs-HA (HA+ and HA– axons) in the same slice (Fig. 9C,D). We performed two-photon microscopy to monitor bouton dynamics in GFP-expressing HA+ and HA– inhibitory axons (Fig. 9E). Bouton dynamics in VGAT-Cre slices were in line with previous data (Frias et al., 2019), indicating that the AAV infections did not alter overall bouton dynamics in inhibitory axons. After a 40 min baseline period, Gs-DREADDs were activated via bath application of CNO ligand. We found that CNO activation strongly increased the density of new boutons in Gs-HA+ axons compared with HA– axons (Fig. 9F). Other NP bouton subgroups were not affected, although the density of stabilizing boutons appeared to be somewhat increased (Fig. 9G,H). These data show that specific activation of Gs signaling in inhibitory axons mimics the WIN-induced inhibitory bouton formation.
Figure 9.
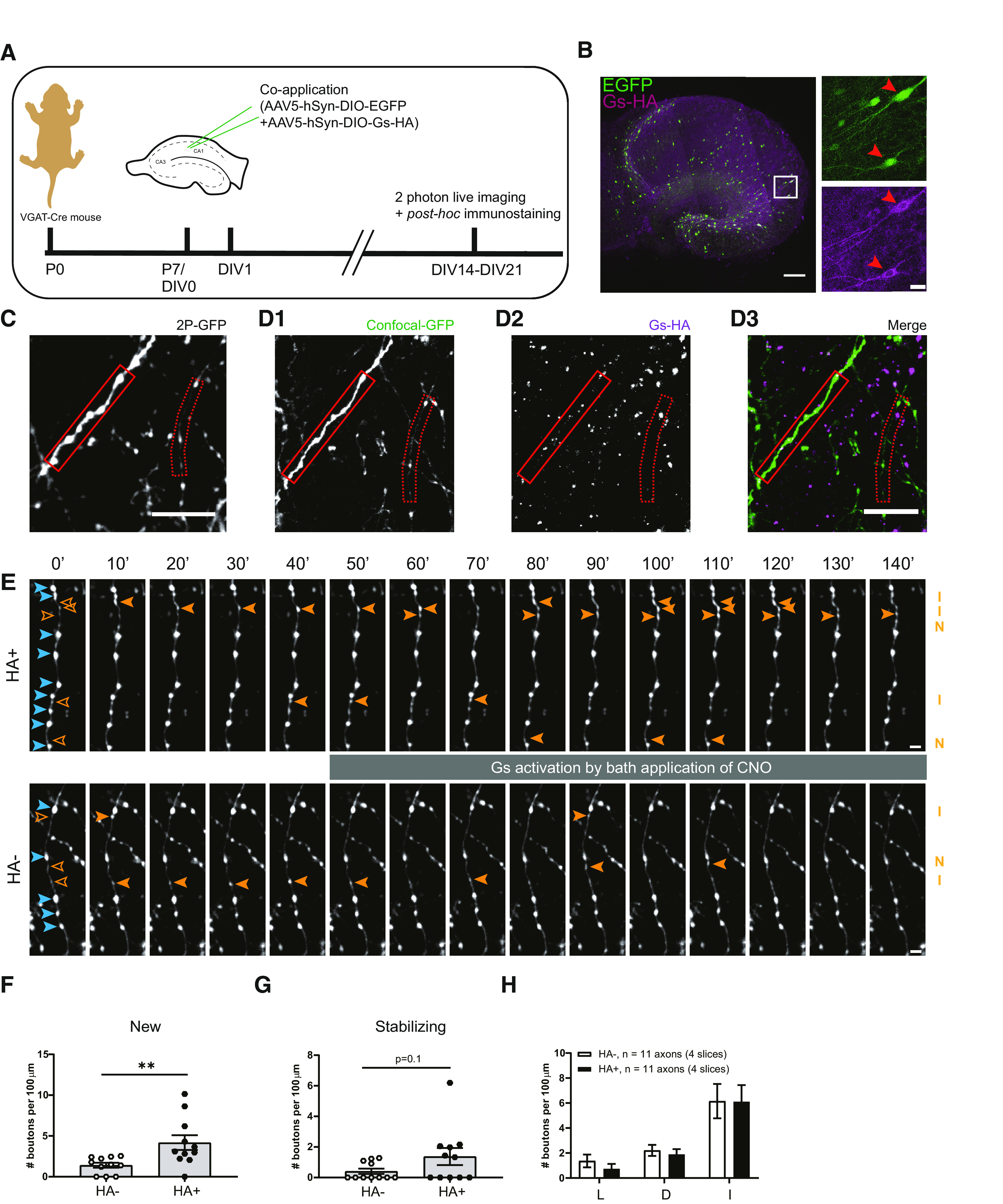
Specific activation of Gs at inhibitory axons induces new bouton formation. A, Experimental design. Hippocampal slice cultures are prepared from postnatal day 7 VGAT-Cre mouse pups. At DIV1, AAV5-hSyn-DIO-EGFP and AAV5-hSyn-DIO-Gs-HA viruses are applied to the VGAT-Cre slice cultures. After 2–3 weeks (DIV14–21) slices were used for two-photon live imaging and post hoc immunostaining to reveal Gs-HA expression. B, Representative example of VGAT-Cre slice culture at DIV20 showing sparse expression of GFP and Gs-HA in GABAergic cells. Right images (zoom from white box) show Gs-HA and EGFP coexpression in a subset of neurons (red arrowheads). C, The z-projection of representative two-photon image of GFP-labeled inhibitory axons in VGAT-Cre slice. D, Confocal images of the same area in C after post hoc immunohistochemistry against HA, showing the same GFP-labeled axons as in A (solid and dashed red boxes indicate HA+ and HA– axons, respectively). E, Two-photon time-lapse imaging of bouton dynamics in the HA+ and HA– axons indicated in C and D. Gs-DREADDs were activated by bath application of 10 μm CNO after the 40 min baseline period. Arrowheads indicate P (blue) and NP (orange) boutons, as in Figure 3A. F, Mean density of new boutons at HA+ and HA– axons in response to Gs-DREADD activation (MW test, p = 0.003). G, Mean density of stabilizing boutons at HA+ and HA– axons in response to Gs-DREADD activation (MW test, p = 0.10). H, Mean density of other subgroup of NP boutons at HA+ and HA– axons in response to Gs-DREADD activation. L, Lost boutons (MW test, p = 0.30); D, destabilizing boutons (MW test, p = 0.44); I, intermittent boutons (MW test, p = 0.85). Data from 11 HA+ and 11 HA– axons in four slices. Scale bars: B, overview, 200 μm; zoom, 20 μm; C, D, 10 μm; E, 2 μm.
Together, our results indicate that inhibitory bouton formation after brief CB1 receptor activation does not require Gi/o signaling, and that it is mimicked by the activation of Gs signaling in inhibitory axons. This suggests that CB1 receptors on inhibitory axons couple with Gs-proteins rather than with the conventional Gi/o effectors to trigger inhibitory bouton formation.
Discussion
Here we examined the signaling pathway underlying the CB1 receptor-mediated formation of new inhibitory synapses. We made several important observations. First of all, repeated CB1 activation led to an increase in mIPSC frequency and an increase in the density of presynaptic VGAT clusters, which were not associated with postsynaptic gephyrin. Inhibitory synapses that do not contain gephyrin are immature and show reduced transmission (Danglot et al., 2003; Yu et al., 2007; Patrizi et al., 2008; Niwa et al., 2012; Nguyen et al., 2016). Our observations are in line with the notion that presynaptic and postsynaptic signaling pathways during synapse formation are largely independent (Wierenga, 2017; Jiang et al., 2021) and suggest that CB1 receptors act purely presynaptically. Second, brief activation of CB1 receptors specifically triggered the formation of inhibitory synapses in CB1R+ axons. This indicates that the formation of inhibitory synapses is mediated by axonal CB1 receptors and that our data exclude a prominent role for CB1 receptors in astrocytes or postsynaptic neurons. Third, bouton turnover in inhibitory axons was strongly reduced when Gi/o-protein signaling was blocked by PTX pretreatment. This suggests that the modulation of axonal cAMP levels is an important regulator of bouton turnover in inhibitory axons. Fourth, CB1 receptor-mediated inhibitory bouton growth was independent of ongoing Gi/o signaling and activity, suggesting that signaling pathways downstream of axonal CB1 receptors differ from presynaptic CB1 receptors. Finally, inhibitory synapse formation was induced in response to an increase in cAMP after forskolin application and when Gs signaling was activated in inhibitory neurons via Gs-DREADDs. CB1 receptor-mediated inhibitory synapse formation required PKA activity. These findings revealed that an increase in cAMP is the key second messenger signal for inhibitory bouton formation and suggest that axonal CB1 receptors trigger inhibitory bouton formation by enhancing PKA activity.
Our present study has limitations that are important to mention here. First of all, we use transgenic mice in which several inhibitory neuron subtypes are labeled with GFP (Wierenga et al., 2010). This unspecific labeling diluted and hampered the detection of axon-specific effects (Fig. 2). However, we used it to our advantage by performing post hoc immunostaining to distinguish between different inhibitory axon types. This allowed comparison between CB1R+ and CB1R– or HA+ and HA– axons in the same slice and avoided comparison between slices from different GFP-labeled mouse lines. Another limitation of our study is that we have used bath application of CB1 agonist WIN to trigger inhibitory bouton formation to trigger inhibitory synapse formation in brain slices. It will be important to assess whether this also occurs in response to physiological relevant neural activity in vivo. Under physiological conditions, endocannabinoid signals are likely transient and highly localized (Hashimotodani et al., 2007; Monday and Castillo, 2017; Hu et al., 2019), providing spatial and temporal control over inhibitory synapse formation. We elevated cAMP levels to trigger inhibitory bouton formation by bath application of forskolin or by the activation of Gs-DREADDs in inhib`itory cells. While this allowed separation of the f`rmation and stabilization of inhibitory `boutons, it likely abolished spatial modulations. Axons contain several phosphodiesterases, which rapidly degrade cAMP and provide spatiotemporal compartmentalization of cAMP signaling (Baillie, 2009; Argyrousi et al., 2020). Pretreatment with PTX will disturb these cAMP modulations, and this strongly reduced inhibitory bouton dynamics and abolished the difference between CB1R+ and CB1R– axons (Fig. 6L). This indicates that CB1R– axons have higher Gi/o baseline activity compared with CB1R+ axons and suggests that cAMP modulation is an important factor regulating inhibitory bouton dynamics. Future research should further assess the relationship between cAMP signaling and inhibitory bouton turnover.
Synapse formation is a multistep process, with each step regulated by specific signaling pathways (Wierenga, 2017; Jiang et al., 2021). Our detailed two-photon analysis allows dissecting these steps and addressing the involved signaling pathways. Inhibitory synapse formation starts with the growth of a new bouton at an axonal location where the inhibitory axon is in close proximity to a dendrite (Wierenga et al., 2008; Dobie and Craig, 2011; Villa et al., 2016; Hu et al., 2019). Our data indicate that axonal CB1 receptors can trigger bouton formation, which does not require neuronal activity. We observed that CB1 receptor-mediated inhibitory bouton formation was not affected in the presence of TTX (Fig. 7D). In addition, we observed that forskolin, which strongly raises neuronal activity (data not shown), did not affect overall bouton turnover (Fig. 8D). This was unexpected given our previous observations that inhibitory bouton turnover is enhanced by neuronal activity (Schuemann et al., 2013; Frias et al., 2019). On the other hand, we observed that blocking Gi/o signaling strongly affected bouton turnover (Fig. 6F,H), which appeared independent of activity. These data suggest that axonal cAMP is the primary second messenger affecting inhibitory bouton formation, which is indirectly modulated by activity, possibly via changes in neuromodulatory signals.
Our data indicate that axonal CB1 receptors can directly trigger bouton formation via an increase in cAMP, while subsequent bouton stabilization and postsynaptic assembly require additional signaling. WIN-induced bouton stabilization was prevented when Gi/o signaling was blocked by PTX (Fig. 7B), and bouton stabilization was not altered by increasing cAMP levels with forskolin (Fig. 8C), although it may be facilitated with longer elevations (Fig. 9H). These data suggest that after the initial formation, CB1 receptors may also promote bouton stabilization via a more indirect pathway. We previously showed that bouton stabilization requires neuronal activity and involves local actin remodeling via a reduction in rho kinase (ROCK) activity (Frias et al., 2019). Interactions between CB1 receptor signaling and ROCK activity (Berghuis et al., 2007) and actin remodeling (Njoo et al., 2015; Zhou et al., 2019) have been reported, but future research should further clarify the precise nature of these interactions.
CB1 receptors are highly versatile and are involved in many neuronal processes via multiple downstream pathways, including axon guidance and synaptic plasticity (Berghuis et al., 2007; Roland et al., 2014; Njoo et al., 2015; Araque et al., 2017; Monday and Castillo, 2017). There are multiple factors, including interacting proteins (Guggenhuber et al., 2016), that determine which downstream signaling pathway is activated after CB1 receptor activation (Flores-Otero et al., 2014; Nogueras-Ortiz and Yudowski, 2016), and this functional selectivity of CB1 receptors may have important clinical relevance (Ibsen et al., 2017; Laprairie et al., 2017; Sholler et al., 2020). It was recently reported that the duration of CB1 receptor activation determines the direction of plasticity at corticostriatal synapses with brief activation inducing LTP, while prolonged activation induces LTD (Cui et al., 2015, 2016). Our data suggest that brief activation of axonal CB1 receptors promotes the formation of new inhibitory boutons via Gs-mediated elevation of cAMP levels, but we have not extensively tested longer activations or different ligand concentrations. An intriguing possibility is that the subcellular location of CB1 receptors affects downstream signaling pathway or that different CB1 receptor isoforms are localized to different subcellular locations (Marti-Solano et al., 2020). CB1 receptors at presynaptic terminals couple to Gi/o to affect GABA release (Guo and Ikeda, 2004; Lee et al., 2015), while CB1 receptors in the axonal shaft of the same inhibitory axons may couple to Gs-proteins. Although CB1 receptors prefer coupling to Gi-proteins, they can switch to Gs-proteins when Gi-proteins are not available or are already occupied (Glass and Felder, 1997; Caballero-Florán et al., 2016; Eldeeb et al., 2016; Finlay et al., 2017). This may suggest that Gi-proteins are only available at presynaptic terminals, while Gs-protein coupling could be dominant in axons.
Our experiments indicate that PKA is an important effector downstream of cAMP to trigger inhibitory bouton formation. Presynaptic PKA activity is also involved in CB1-mediated synaptic plasticity (Chevaleyre et al., 2007; Cui et al., 2016) and may act by altering local clustering and interbouton exchange of synaptic vesicles (Patzke et al., 2019; Chenouard et al., 2020). PKA resides close to the plasma membrane and preferably phosphorylates membrane proteins in its close proximity (Tillo et al., 2017). Potential PKA targets to mediate inhibitory bouton formation remain yet to be identified. In addition, PKA-independent pathways downstream of cAMP signaling may also play a role, for instance via Epac2 (Kawasaki et al., 1998). Epac2 activity can strongly increase synaptic transmission (Gekel and Neher, 2008; Fernandes et al., 2015), yet a role in synapse formation has not been reported. Interestingly, Epac2 was recently found to be downstream of Gs-coupled β-adrenergic receptors to mediate presynaptic LTP at parallel fiber synapses to Purkinje cells (Martín et al., 2020). cAMP signaling via PKA, Epac2, or Rho GTPases may affect the axonal cytoskeleton. Actin is important in the formation, stabilization, and maintenance of presynaptic terminals (Bednarek and Caroni, 2011; Pielage et al., 2011; Chia et al., 2013, 2014; Frias et al., 2019; Chenouard et al., 2020), and cAMP fluctuations may drive local modifications in the actin cytoskeleton (Bernier et al., 2019) underlying structural presynaptic changes.
Our findings suggest that axonal CB1 receptors serve an important role in local, on-demand synapse formation. Our observation that inhibitory bouton formation was more prominent after cAMP elevation than after WIN application (compare Figs. 8B, 9F, Fig. 3I) suggests that axonal cAMP signaling is an important second messenger signal mediating bouton formation not only in CB1R+, but perhaps in all, inhibitory axons. Intriguingly, our observations are reminiscent of cAMP-mediated bouton formation in zebrafish (Yoshida and Mishina, 2005), Aplysia (Nazif et al., 1991; Bailey and Kandel, 1993; Upreti et al., 2019), and Drosophila axons (Zhong et al., 1992; Koon et al., 2011; Maiellaro et al., 2016). This raises the possibility that axonal cAMP signaling is a universal second messenger system for regulating structural plasticity in axons. Activation of CB1 receptors via dendritic endocannabinoid signaling (Hu et al., 2019) then represents one specific way to trigger cAMP-mediated bouton formation in CB1R+ axons in response to strong excitatory synaptic activity. Other axons may use different axonal receptors to mediate bouton formation. Indeed, GABAergic interneurons express many different G-proteins (Cox et al., 2008; Helboe et al., 2015; Puighermanal et al., 2017), which often provide neuromodulatory context signals from other brain areas (Hattori et al., 2017). Our findings raise the intriguing possibility that neuromodulatory receptors on the axonal surface provide the opportunity to build a new inhibitory bouton on demand, triggered by axon-specific and context-dependent signaling.
Footnotes
This work was supported by a CSC scholarship (J.L.), by Alzheimer Nederland (Grant WE.03-2018-11, to M.R.) and by the Netherlands Organization for Scientific Research (Grant OCENW.KLEIN.150, to M.S.G.), and as part of the research program of the Foundation for Fundamental Research on Matter (Grants #15PR3178 and #16NEPH05, to D.L.H.K.). We thank Lotte Herstel for helping with experiments shown in Figure 1, E and F, and René van Dorland for excellent technical support.
The authors declare no competing financial interests.
References
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68:247–286. 10.1016/s0301-0082(02)00080-1 [DOI] [PubMed] [Google Scholar]
- Alger BE, Kim J (2011) Supply and demand for endocannabinoids. Trends Neurosci 34:304–315. 10.1016/j.tins.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni FA (2012) New paradigms in cAMP signalling. Mol Cell Endocrinol 353:3–9. 10.1016/j.mce.2011.10.034 [DOI] [PubMed] [Google Scholar]
- Araque A, Castillo PE, Manzoni OJ, Tonini R (2017) Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 124:13–24. 10.1016/j.neuropharm.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw A, Duff G, Zabouri N, Cécyre B, Chainé N, Cherif H, Tea N, Lutz B, Ptito M, Bouchard JF (2011) Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J Neurosci 31:1489–1499. 10.1523/JNEUROSCI.4134-09.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyrousi EK, Heckman PRA, Prickaerts J (2020) Role of cyclic nucleotides and their downstream signaling cascades in memory function: being at the right time at the right spot. Neurosci Biobehav Rev 113:12–38. 10.1016/j.neubiorev.2020.02.004 [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA (2004) Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431:312–315. 10.1038/nature02913 [DOI] [PubMed] [Google Scholar]
- Bailey CH, Chen M (1989) Time course of structural changes at identified sensory neuron synapses during long-term sensitization in Aplysia. J Neurosci 9:1774–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER (1993) Structural changes accompanying memory storage. Annu Rev Physiol 55:397–426. 10.1146/annurev.ph.55.030193.002145 [DOI] [PubMed] [Google Scholar]
- Baillie GS (2009) Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J 276:1790–1799. 10.1111/j.1742-4658.2009.06926.x [DOI] [PubMed] [Google Scholar]
- Bednarek E, Caroni P (2011) Β-adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron 69:1132–1146. 10.1016/j.neuron.2011.02.034 [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD (1999) Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol 516:227–248. 10.1111/j.1469-7793.1999.227aa.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T (2007) Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316:1212–1216. 10.1126/science.1137406 [DOI] [PubMed] [Google Scholar]
- Bernier L-P, Bohlen CJ, York EM, Choi HB, Kamyabi A, Dissing-Olesen L, Hefendehl JK, Collins HY, Stevens B, Barres BA, MacVicar BA (2019) Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Rep 27:2895–2908. [DOI] [PubMed] [Google Scholar]
- Bonilla-Del Río I, Puente N, Mimenza A, Ramos A, Serrano M, Lekunberri L, Gerrikagoitia I, Christie BR, Nahirney PC, Grandes P (2021) Acute Δ9-tetrahydrocannabinol prompts rapid changes in cannabinoid CB 1 receptor immunolabeling and subcellular structure in CA1 hippocampus of young adult male mice. J Comp Neurol 529:2332–2346. 10.1002/cne.25098 [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM (2011) Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus 21:354–373. 10.1002/hipo.20768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero-Florán RN, Conde-Rojas I, Oviedo Chávez A, Cortes-Calleja H, Lopez-Santiago LF, Isom LL, Aceves J, Erlij D, Florán B (2016) Cannabinoid-induced depression of synaptic transmission is switched to stimulation when dopaminergic tone is increased in the globus pallidus of the rodent. Neuropharmacology 110:407–418. 10.1016/j.neuropharm.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Campbell AP, Smrcka AV (2018) Targeting G protein-coupled receptor signalling by blocking G proteins. Nat Rev Drug Discov 17:789–803. 10.1038/nrd.2018.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Donato F, Muller D (2012) Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci 13:478–490. 10.1038/nrn3258 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Kim AN, Peters AJ, Komiyama T (2015) Subtype-specific plasticity of inhibitory circuits in motor cortex during motor learning. Nat Neurosci 18:1109–1115. 10.1038/nn.4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenouard N, Xuan F, Tsien RW (2020) Synaptic vesicle traffic is supported by transient actin filaments and regulated by PKA and NO. Nat Commun 11:5318. 10.1038/s41467-020-19120-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. 10.1016/s0896-6273(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE (2007) Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1α. Neuron 54:801–812. 10.1016/j.neuron.2007.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PH, Li P, Shen K (2013) Cellular and molecular mechanisms underlying presynapse formation. J Cell Biol 203:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PH, Chen B, Li P, Rosen MK, Shen K (2014) Local F-actin network links synapse formation and axon branching. Cell 156:208–220. 10.1016/j.cell.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Barberis A, Higley MJ (2019) Preserving the balance: diverse forms of long-term GABAergic synaptic plasticity. Nat Rev Neurosci 20:272–281. 10.1038/s41583-019-0141-5 [DOI] [PubMed] [Google Scholar]
- Cox DJ, Racca C, LeBeau FEN (2008) Β-adrenergic receptors are differentially expressed in distinct interneuron subtypes in the rat hippocampus. J Comp Neurol 509:551–565. 10.1002/cne.21758 [DOI] [PubMed] [Google Scholar]
- Cui Y, Paille V, Xu H, Genet S, Delord B, Fino E, Berry H, Venance L (2015) Endocannabinoids mediate bidirectional striatal spike-timing-dependent plasticity. J Physiol 593:2833–2849. 10.1113/JP270324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Prokin I, Xu H, Delord B, Genet S, Venance L, Berry H (2016) Endocannabinoid dynamics gate spike-timing dependent depression and potentiation. Elife 5:e13185. 10.7554/eLife.13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danglot L, Triller A, Bessis A (2003) Association of gephyrin with synaptic and extrasynaptic GABAA receptors varies during development in cultured hippocampal neurons. Mol Cell Neurosci 23:264–278. 10.1016/s1044-7431(03)00069-1 [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA (2003) Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol 550:135–147. 10.1113/jphysiol.2003.039099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie FA, Craig AM (2011) Inhibitory synapse dynamics: coordinated presynaptic and postsynaptic mobility and the major contribution of recycled vesicles to new synapse formation. J Neurosci 31:10481–10493. 10.1523/JNEUROSCI.6023-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dócs K, Mészár Z, Gonda S, Kiss-Szikszai A, Holló K, Antal M, Hegyi Z (2017) The ratio of 2-AG to its isomer 1-AG as an intrinsic fine tuning mechanism of CB1 receptor activation. Front Cell Neurosci 11:39. 10.3389/fncel.2017.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P (2013) Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504:272–276. 10.1038/nature12866 [DOI] [PubMed] [Google Scholar]
- Donato F, Chowdhury A, Lahr M, Caroni P (2015) Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning. Neuron 85:770–786. 10.1016/j.neuron.2015.01.011 [DOI] [PubMed] [Google Scholar]
- Dudok B, Barna L, Ledri M, Szabó SI, Szabadits E, Pintér B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, Varga C, Lee S-H, Matolcsi M, Cervenak J, Kacskovics I, Watanabe M, Sagheddu C, Melis M, Pistis M, Soltesz I, et al. (2015) Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat Neurosci 18:75–86. 10.1038/nn.3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldeeb K, Leone-kabler S, Howlett AC (2016) CB1 cannabinoid receptor-mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J Basic Clin Physiol Pharmacol 27:311–322. 10.1515/jbcpp-2015-0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, Contractor A (2015) Epac2 mediates cAMP-dependent potentiation of neurotransmission in the hippocampus. J Neurosci 35:6544–6553. 10.1523/JNEUROSCI.0314-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay DB, Cawston EE, Grimsey NL, Hunter MR, Korde A, Vemuri VK, Makriyannis A, Glass M (2017) Gαs signalling of the CB1 receptor and the influence of receptor number. Br J Pharmacol 174:2545–2562. 10.1111/bph.13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CE, Méndez P (2014) Shaping inhibition: activity dependent structural plasticity of GABAergic synapses. Front Cell Neurosci 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Otero J, Ahn KH, Delgado-Peraza F, Mackie K, Kendall DA, Yudowski GA (2014) Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun 5:4589. 10.1038/ncomms5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias CP, Liang J, Bresser T, Scheefhals L, van Kesteren M, van Dorland R., Hu HY, Bodzeta A, Van Bergen en Henegouwen PMP, Hoogenraad CC, Wierenga CJ (2019) Semaphorin4D induces inhibitory synapse formation by rapid stabilization of presynaptic boutons via MET co-activation. J Neurosci 39:4221–4237. 10.1523/JNEUROSCI.0215-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekel I, Neher E (2008) Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci 28:7991–8002. 10.1523/JNEUROSCI.0268-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333. 10.1523/JNEUROSCI.17-14-05327.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Miyamae T, Pafundo DE, Yoshino H, Rotaru DC, Hoftman G, Datta D, Zhang Y, Hammond M, Sampson AR, Fish KN, Ermentrout GB, Lewis DA (2015) Functional maturation of GABA synapses during postnatal development of the monkey dorsolateral prefrontal cortex. Cereb Cortex 25:4076–4093. 10.1093/cercor/bhu122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenhuber S, Alpar A, Chen R, Schmitz N, Wickert M, Mattheus T, Harasta AE, Purrio M, Kaiser N, Elphick MR, Monory K, Kilb W, Luhmann HJ, Harkany T, Lutz B, Klugmann M (2016) Cannabinoid receptor-interacting protein Crip1a modulates CB1 receptor signaling in mouse hippocampus. Brain Struct Funct 221:2061–2074. 10.1007/s00429-015-1027-6 [DOI] [PubMed] [Google Scholar]
- Guo J, Ikeda SR (2004) Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol 65:665–674. 10.1124/mol.65.3.665 [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-shosaku T, Kano M (2007) Ca2+-assisted receptor-driven endocannabinoid release: mechanisms that associate presynaptic and postsynaptic activities. Curr Opin Neurobiol 17:360–365. 10.1016/j.conb.2007.03.012 [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tanimura A, Kita Y, Sano Y, Shimizu T, Di Marzo V, Kano M (2013) Acute inhibition of diacylglycerol lipase blocks endocannabinoid-mediated retrograde signalling: evidence for on-demand biosynthesis of 2-arachidonoylglycerol. J Physiol 591:4765–4776. 10.1113/jphysiol.2013.254474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori R, Kuchibhotla KV, Froemke RC, Komiyama T (2017) Functions and dysfunctions of neocortical inhibitory neuron subtypes. Nat Neurosci 20:1199–1208. 10.1038/nn.4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Pagano Zottola AC, Delamarre A, Cannich A, Vincent P, Varilh M, Robin LM, Terral G, García-Fernández MD, Colavita M, Mazier W, Drago F, Puente N, Reguero L, Elezgarai I, et al. (2016) A cannabinoid link between mitochondria and memory. Nature 539:555–559. 10.1038/nature20127 [DOI] [PubMed] [Google Scholar]
- Helboe L, Egebjerg J, de Jong IEM (2015) Distribution of serotonin receptor 5-HT6 mRNA in rat neuronal subpopulations: a double in situ hybridization study. Neuroscience 310:442–454. 10.1016/j.neuroscience.2015.09.064 [DOI] [PubMed] [Google Scholar]
- Herstel LJ, Wierenga CJ (2021) Network control through coordinated inhibition. Curr Opin Neurobiol 67:34–41. 10.1016/j.conb.2020.08.001 [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M (2009) Experience leaves a lasting structural trace in cortical circuits. Nature 457:313–317. 10.1038/nature07487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HY, Kruijssen DL, Frias CP, Rózsa B, Hoogenraad CC, Wierenga CJ (2019) Endocannabinoid signaling mediates local dendritic coordination between excitatory and inhibitory synapses. Cell Rep 27:666–675. 10.1016/j.celrep.2019.03.078 [DOI] [PubMed] [Google Scholar]
- Ibsen MS, Connor M, Glass M (2017) Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis Cannabinoid Res 2:48–60. 10.1089/can.2016.0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Sando R, Südhof TC (2021) Multiple signaling pathways are essential for synapse formation induced by synaptic adhesion molecules. Proc Natl Acad Sci U S A 118: 10.1073/pnas.2000173118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullié D, Stoeber M, Sibarita J, Zieger HL, Bartol TM, Arttamangkul S, Sejnowski TJ, Hosy E, von Zastrow M (2020) A discrete presynaptic vesicle cycle for neuromodulator receptors. Neuron 105:663–677.e8. 10.1016/j.neuron.2019.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380. 10.1152/physrev.00019.2008 [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35:529–558. 10.1146/annurev-neuro-062111-150420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF (1999) Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. 10.1523/JNEUROSCI.19-11-04544.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urbán GM, Wallace M, Ledent C, Jung K-M, Piomelli D, Mackie K, Freund TF (2006) Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci 26:5628–5637. 10.1523/JNEUROSCI.0309-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282:2275–2279. 10.1126/science.282.5397.2275 [DOI] [PubMed] [Google Scholar]
- Keck T, Scheuss V, Jacobsen RI, Wierenga CJ, Eysel UT, Bonhoeffer T, Hübener M (2011) Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 71:869–882. 10.1016/j.neuron.2011.06.034 [DOI] [PubMed] [Google Scholar]
- Kirchner JH, Gjorgjieva J (2021) Emergence of local and global synaptic organization on cortical dendrites. Nat Commun 12:4005. 10.1038/s41467-021-23557-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindienst T, Winnubst J, Roth-alpermann C, Bonhoeffer T, Lohmann C (2011) Activity-dependent clustering of functional synaptic inputs on developing hippocampal dendrites. Neuron 72:1012–1024. 10.1016/j.neuron.2011.10.015 [DOI] [PubMed] [Google Scholar]
- Knott GW, Quairiaux C, Genoud C, Welker E (2002) Formation of dendritic spines with GABAergic synapses induced by whisker stimulation in adult mice. Neuron 34:265–273. 10.1016/S0896-6273(02)00663-3 [DOI] [PubMed] [Google Scholar]
- Koon AC, Ashley J, Barria R, DasGupta S, Brain R, Waddell S, Alkema MJ, Budnik V (2011) Autoregulatory and paracrine control of synaptic and behavioral plasticity by octopaminergic signaling. Nat Neurosci 14:190–199. 10.1038/nn.2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL (2012) Recurrent network activity drives striatal synaptogenesis. Nature 485:646–650. 10.1038/nature11052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Denovan-Wright EM (2017) Cannabinoid receptor ligand bias: implications in the central nervous system. Curr Opin Pharmacol 32:32–43. 10.1016/j.coph.2016.10.005 [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K (2005) The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A 102:19144–19149. 10.1073/pnas.0509588102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus MS, Josh Huang Z (2011) Distinct maturation profiles of perisomatic and dendritic targeting GABAergic interneurons in the mouse primary visual cortex during the critical period of ocular dominance plasticity. J Neurophysiol 106:775–787. 10.1152/jn.00729.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Földy C, Soltesz I (2010) Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J Neurosci 30:7993–8000. 10.1523/JNEUROSCI.6238-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Ledri M, Tóth B, Marchionni I, Henstridge CM, Dudok B, Kenesei K, Barna L, Szabó SI, Renkecz T, Oberoi M, Watanabe M, Limoli CL, Horvai G, Soltesz I, Katona I (2015) Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA release. J Neurosci 35:10039–10057. 10.1523/JNEUROSCI.4112-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkey N, Kirizs T, Holderith N, Máté Z, Szabó G, Vizi ES, Hájos N, Nusser Z (2015) Tonic endocannabinoid-mediated modulation of GABA release is independent of the CB1 content of axon terminals. Nat Commun 6:6557. 10.1038/ncomms7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW (2005) Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324. 10.1038/nrn1648 [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Sturgess K, Erdélyi F, Szabó G, Molnár Z, Paulsen O (2004) Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb Cortex 14:1122–1133. 10.1093/cercor/bhh072 [DOI] [PubMed] [Google Scholar]
- Lovinger DM (2008) Presynaptic modulation by endocannabinoids. Handbook of Experimental Pharmacology, Vol 184, pp 435–477. Berlin, Heidelberg:Springer. 10.1007/978-3-540-74805-2_14 [DOI] [PubMed] [Google Scholar]
- Maffei A, Charrier C, Caiati MD, Barberis A, Mahadevan V, Woodin MA, Tyagarajan SK (2017) Emerging mechanisms underlying dynamics of GABAergic synapses. J Neurosci 37:10792–10799. 10.1523/JNEUROSCI.1824-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiellaro I, Lohse MJ, Kittel RJ, Calebiro D (2016) cAMP signals in drosophila motor neurons are confined to single synaptic boutons. Cell Rep 17:1238–1246. 10.1016/j.celrep.2016.09.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Szabo GG, Kim HK, Alexander A, Bui AD, Lee SH, Lutz B, Soltesz I (2016) Cannabinoid control of learning and memory through HCN channels. Neuron 89:1059–1073. 10.1016/j.neuron.2016.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, García-Font N, Suárez-Pinilla AS, Bartolomé-Martín D, Ferrero JJ, Luján R, Torres M, Sánchez-Prieto J (2020) β-adrenergic receptors/epac signaling increases the size of the readily releasable pool of synaptic vesicles required for parallel fiber LTP. J Neurosci 40:8604–8617. 10.1523/JNEUROSCI.0716-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Solano M, Crilly SE, Malinverni D, Munk C, Harris M, Pearce A, Quon T, Mackenzie AE, Wang X, Peng J, Tobin AB, Ladds G, Milligan G, Gloriam DE, Puthenveedu MA, Babu MM (2020) Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature 587:650–656. 10.1038/s41586-020-2888-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikasova L, Groc L, Choquet D, Manzoni OJ (2008) Altered surface trafficking of presynaptic cannabinoid type 1 receptor in and out synaptic terminals parallels receptor desensitization. Proc Natl Acad Sci U S A 105:18596–18601. 10.1073/pnas.0805959105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitoma H, Konishi S (1996) Long-lasting facilitation of inhibitory transmission by monoaminergic and cAMP-dependent mechanism in rat cerebellar GABAergic synapses. Neurosci Lett 217:141–144. [PubMed] [Google Scholar]
- Monday HR, Castillo PE (2017) Closing the gap: long-term presynaptic plasticity in brain function and disease. Curr Opin Neurobiol 45:106–112. 10.1016/j.conb.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monday HR, Bourdenx M, Jordan B, Castillo P (2020) CB 1 receptor-mediated inhibitory LTD triggers presynaptic remodeling via protein synthesis and ubiquitination. Elife 9:e54812. 10.7554/eLife.54812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins C, Fishell G, Tsien RW (2016) Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89:1131–1156. 10.1016/j.neuron.2016.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Díez A, Araque A (2014) Astrocytes in endocannabinoid signalling. Philos Trans R Soc B Biol Sci 369:20130599. 10.1098/rstb.2013.0599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazif FA, Byrne JH, Cleary LJ (1991) cAMP induces long-term morphological changes in sensory neurons of Aplysia. Brain Res 539:324–327. 10.1016/0006-8993(91)91638-h [DOI] [PubMed] [Google Scholar]
- Nguyen QA, Horn ME, Nicoll RA (2016) Distinct roles for extracellular and intracellular domains in neuroligin function at inhibitory synapses. Elife 5:e19236. 10.7554/eLife.19236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu D, Michaelsen-Preusse K, Güner Ü, van Dorland R, Wierenga CJ, Lohmann C (2018) A BDNF-mediated push-pull plasticity mechanism for synaptic clustering. Cell Rep 24:2063–2074. 10.1016/j.celrep.2018.07.073 [DOI] [PubMed] [Google Scholar]
- Nishiyama J, Yasuda R (2015) Biochemical Computation for Spine Structural Plasticity. Neuron 87:63–75. 10.1016/j.neuron.2015.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa F, Bannai H, Arizono M, Fukatsu K, Triller A, Mikoshiba K (2012) Gephyrin-independent GABA(A)R mobility and clustering during plasticity. PLoS One 7:e36148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njoo C, Agarwal N, Lutz B, Kuner R (2015) The Cannabinoid receptor CB1 Interacts with the WAVE1 complex and plays a role in actin dynamics and structural plasticity in neurons. PLoS Biol 13:e1002286. 10.1371/journal.pbio.1002286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueras-Ortiz C, Yudowski GA (2016) The multiple waves of cannabinoid 1 receptor signaling. Mol Pharmacol 90:620–626. 10.1124/mol.116.104539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh WC, Lutzu S, Castillo PE, Kwon H (2016) De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353:1037–1040. 10.1126/science.aaf5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo GVE, Lucion AB, Calcagnotto ME (2018) Postnatal development of inhibitory synaptic transmission in the anterior piriform cortex. Int J Dev Neurosci 71:1–9. 10.1016/j.ijdevneu.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Patrizi A, Scelfo B, Viltono L, Briatore F, Fukaya M, Watanabe M, Strata P, Varoqueaux F, Brose N, Fritschy J, Sassoè-Pognetto M, Sassoe M (2008) Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc Natl Acad Sci U S A 105:13151–13156. 10.1073/pnas.0802390105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke C, Brockmann MM, Dai J, Gan KJ, Grauel MK, Fenske P, Liu Y, Acuna C, Rosenmund C, Südhof TC (2019) Neuromodulator signaling bidirectionally controls vesicle numbers in human synapses. Cell 179:498–513. 10.1016/j.cell.2019.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielage J, Bulat V, Zuchero JB, Fetter RD, Davis GW (2011) Hts/adducin controls synaptic elaboration and elimination. Neuron 69:1114–1131. 10.1016/j.neuron.2011.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D (2014) More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology 76:228–234. 10.1016/j.neuropharm.2013.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puighermanal E, Cutando L, Boubaker-Vitre J, Honoré E, Longueville S, Hervé D, Valjent E (2017) Anatomical and molecular characterization of dopamine D1 receptor-expressing neurons of the mouse CA1 dorsal hippocampus. Brain Struct Funct 222:1897–1911. 10.1007/s00429-016-1314-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W (1967) Distinguishing theoretical synaptic potentials computed for different soma-dendritic distributions of synaptic input. J Neurophysiol 30:1138–1168. 10.1152/jn.1967.30.5.1138 [DOI] [PubMed] [Google Scholar]
- Roland AB, Ricobaraza A, Carrel D, Jordan BM, Rico F, Simon A, Humbert-Claude M, Ferrier J, McFadden MH, Scheuring S, Lenkei Z (2014) Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife 3:e03159. 10.7554/eLife.03159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL (2016) DREADDs for Neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger S, Vittori C, Bednarek E, Genoud C, Strata P, Sacchetti B, Caroni P (2011) Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature 473:514–518. 10.1038/nature09946 [DOI] [PubMed] [Google Scholar]
- Ruiter M, Herstel LJ, Wierenga CJ (2020) Reduction of dendritic inhibition in CA1 pyramidal neurons in amyloidosis models of early Alzheimer's disease. J Alzheimers Dis 78:951–964. 10.3233/JAD-200527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter M, Lützkendorf C, Liang J, Wierenga CJ (2021) Amyloid-β oligomers induce only mild changes to inhibitory bouton dynamics. J Alzheimers Dis Rep 5:153–160. 10.3233/ADR-200291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinainen JR, Saario SM, Laitinen JT (2012) The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 204:267–276. 10.1111/j.1748-1716.2011.02280.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl B, Thomas CI, Ryan MA, Kamasawa N, Fitzpatrick D (2021) Cortical neuron response selectivity derives from strength in numbers of synapses. Nature 590:111–114. 10.1038/s41586-020-03044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuemann A, Klawiter A, Bonhoeffer T, Wierenga CJ (2013) Structural plasticity of GABAergic axons is regulated by network activity and GABAA receptor activation. Front Neural Circuits 7:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholler DJ, Huestis MA, Amendolara B, Vandrey R, Cooper ZD (2020) Therapeutic potential and safety considerations for the clinical use of synthetic cannabinoids. Pharmacol Biochem Behav 199:173059. 10.1016/j.pbb.2020.173059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó GG, Lenkey N, Holderith N, Andrási T, Nusser Z, Hájos N (2014) Presynaptic calcium channel inhibition underlies CB1 cannabinoid receptor-mediated suppression of GABA release. J Neurosci 34:7958–7963. 10.1523/JNEUROSCI.0247-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Jaenisch R, Sur M (2021) The role of GABAergic signalling in neurodevelopmental disorders. Nat Rev Neurosci 22:290–307. 10.1038/s41583-021-00443-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP (2013) PKA: lessons learned after twenty years. Biochim Biophys Acta 1834:1271–1278. 10.1016/j.bbapap.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo SE, Xiong WH, Takahashi M, Miao S, Andrade AL, Fortin DA, Yang G, Qin M, Smoody BF, Stork PJS, Zhong H (2017) Liberated PKA catalytic subunits associate with the membrane via myristoylation to preferentially phosphorylate membrane substrates. Cell Rep 19:617–629. 10.1016/j.celrep.2017.03.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upreti C, Konstantinov E, Kassabov SR, Bailey CH, Kandel ER (2019) Serotonin induces structural plasticity of both extrinsic modulating and intrinsic mediating circuits in vitro in Aplysia Californica. Cell Rep 28:2955–2965.e3. 10.1016/j.celrep.2019.08.016 [DOI] [PubMed] [Google Scholar]
- Urban DJ, Roth BL (2015) DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 55:399–417. 10.1146/annurev-pharmtox-010814-124803 [DOI] [PubMed] [Google Scholar]
- Villa KL, Berry KP, Subramanian J, Cha JW, Oh WC, Kwon H-B, Kubota Y, So PTC, Nedivi E (2016) Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89:756–769. 10.1016/j.neuron.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Jia Y, Pham DT, Palmer LC, Jung K-M, Cox CD, Rumbaugh G, Piomelli D, Gall CM, Lynch G (2018) Atypical endocannabinoid signaling initiates a new form of memory-related plasticity at a cortical input to hippocampus. Cereb Cortex 28:2253–2266. 10.1093/cercor/bhx126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ (2017) Live imaging of inhibitory axons: synapse formation as a dynamic trial-and-error process. Brain Res Bull 129:43–49. 10.1016/j.brainresbull.2016.09.018 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Wadman WJ (1999) Miniature inhibitory postsynaptic currents in CA1 pyramidal neurons after kindling epileptogenesis. J Neurophysiol 82:1352–1362. 10.1152/jn.1999.82.3.1352 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Becker N, Bonhoeffer T (2008) GABAergic synapses are formed without the involvement of dendritic protrusions. Nat Neurosci 11:1044–1052. 10.1038/nn.2180 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Müllner FE, Rinke I, Keck T, Stein V, Bonhoeffer T (2010) Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PLoS One 5:e15915. 10.1371/journal.pone.0015915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Mishina M (2005) Distinct roles of calcineurin-nuclear factor of activated T cells and protein kinase A-cAMP response element-binding protein signaling in presynaptic differentiation. J Neurosci 25:3067–3079. 10.1523/JNEUROSCI.3738-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Jiang M, Miralles CP, Li R-W, Chen G, de Blas AL (2007) Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci 36:484–500. 10.1016/j.mcn.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu CF (1992) Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci 12:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Han B, Xia C, Zhuang X (2019) Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 365:929–934. 10.1126/science.aaw5937 [DOI] [PMC free article] [PubMed] [Google Scholar]