

SUMMARY
Brain function is compromised in myotonic dystrophy type 1 (DM1), but the underlying mechanisms are not fully understood. To gain insight into the cellular and molecular pathways primarily affected, we studied a mouse model of DM1 and brains of adult patients. We found pronounced RNA toxicity in the Bergmann glia of the cerebellum, in association with abnormal Purkinje cell firing and fine motor incoordination in DM1 mice. A global proteomics approach revealed downregulation of the GLT1 glutamate transporter in DM1 mice and human patients, which we found to be the result of MBNL1 inactivation. GLT1 downregulation in DM1 astrocytes increases glutamate neurotoxicity and is detrimental to neurons. Finally, we demonstrated that the upregulation of GLT1 corrected Purkinje cell firing and motor incoordination in DM1 mice. Our findings show that glial defects are critical in DM1 brain pathophysiology and open promising therapeutic perspectives through the modulation of glutamate levels.
Graphical Abstract
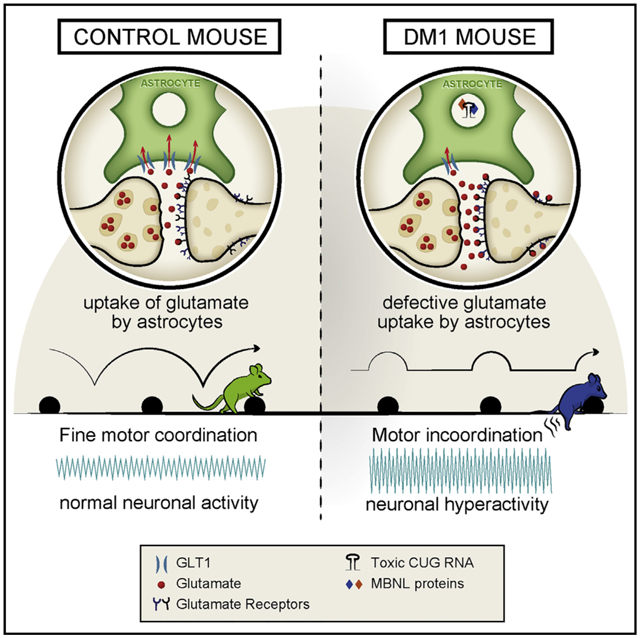
In Brief
Neural dysfunction in myotonic dystrophy is not fully understood. Using a transgenic mouse model of the disease, Sicot et al. find electrophysiological and motor evidence for cerebellar dysfunction in association with pronounced signs of RNA toxicity in Bergmann glia. Upregulation of a defective glial-specific glutamate transporter corrects cerebellum phenotypes.
INTRODUCTION
Repeat-containing RNA can cause neurological diseases through a trans-dominant gain of function (Mohan et al., 2014; Sicot and Gomes-Pereira, 2013). RNA toxicity is best described in myotonic dystrophy type 1 (DM1), but it operates in an increasing number of conditions (Sicot et al., 2011). DM1 is the most common muscular dystrophy in adults, with a variable prevalence ranging from 0.5 to 18 cases in 100,000 individuals (Theadom et al., 2014). DM1 is a multisystemic disorder that affects the skeletal muscle, heart, and the CNS, among other tissues (Udd and Krahe, 2012). Five main clinical forms of DM1 can be distinguished based on age at onset: congenital, childhood, juvenile, adult, and mild or late onset (Dogan et al., 2016). CNS impairment is more pronounced in the early-onset cases. Among these, the congenital patients exhibit moderate to severe intellectual disability. The childhood- and juvenile-onset cases can also show reduced IQ, low cognitive processing speed, and visuospatial impairment, as well as attention and executive deficits (Angeard et al., 2007, 2011). The main CNS manifestations in the adult form include dysexecutive behavior (such as apathy, lack of motivation, and inflexibility), reduced attention and visuospatial construction ability, daytime sleepiness, and impaired social cognition (Meola and Sansone, 2007; Serra et al., 2016; Sistiaga et al., 2010). Overall, the quality of life of DM1 patients is significantly impaired by their cognitive deficits (Antonini et al., 2006; Dogan et al., 2016). Brain disease is further supported by histopathological changes, such as the aggregation of hyperphosphorylated Tau protein isoforms, particularly in the amygdala, hippocampus, and entorhinal and temporal cortex (Caillet-Boudin et al., 2014). White matter lesions, gray matter changes, metabolic deficits, and changes in functional connectivity were reported in multiple brain areas (Caliandro et al., 2013; Minnerop et al., 2011; Serra et al., 2014, 2015; Weber et al., 2010; Wozniak et al., 2014), implicating the dysregulation of complex brain networks and various cell types (Schneider-Gold et al., 2015; Serra et al., 2016). However, the link between the distribution of DM1 pathology across brain territories and cell types and the neurological symptoms of the disease must be further elucidated.
DM1 is caused by the expansion of an unstable CTG repeat in the 3′ untranslated region (3′ UTR) of the DM protein kinase (DMPK) gene (Brook et al., 1992). Repeat number correlates with disease severity and inversely with age of onset. Expanded DMPK transcripts accumulate in nuclear RNA foci and perturb the activity of multiple RNA-binding proteins. Among these, the sequestration of muscleblind-like (MBNL) proteins and the upregulation of the CUGBP/Elav-like family (CELF) affect primarily alternative splicing but also RNA transcription, localization and polyadenylation, miRNA processing, protein translation, and phosphorylation of downstream targets (Batra et al., 2014; Goodwin et al., 2015; Hernández-Hernández et al., 2013a, 2013b; Sicot et al., 2011; Wang et al., 2012). Today we do not know the extent or distribution of these events in the CNS, their cellular specificity, or how they contribute to neuropathogenesis.
To investigate DM1 brain disease, we have been using DMSXL mice, which carry a human DMPK transgene containing >1,000 CTG repeats (Gomes-Pereira et al., 2007; Seznec et al., 2000). DMSXL homozygotes express enough toxic transcripts to perturb muscular, cardiac, and respiratory function (Algalarrondo et al., 2015; Huguet et al., 2012; Panaite et al., 2013), in association with RNA foci and missplicing (Hernández-Hernández et al., 2013a; Huguet et al., 2012). The expression of expanded CUG RNA in the CNS affects behavior and synaptic function of DMSXL mice (Hernández-Hernández et al., 2013a). In contrast, DM20 mice, overexpressing short DMPK transcripts, do not show RNA foci accumulation, obvious phenotypes, or synaptic protein dysfunction (Hernández-Hernández et al., 2013a; Seznec et al., 2001). The differences between mouse lines corroborate the toxicity of expanded CUG RNA repeats in the CNS of DMSXL mice. However, the underlying molecular and cellular mechanisms leading to brain impairment are not entirely known, which delays the development of efficient therapeutic strategies in the CNS.
To overcome this limitation, we have combined molecular, electrophysiological, and behavioral approaches to gain insight into the susceptible cell populations, dysfunctional connections, and affected molecular pathways in the CNS of DM1. It was our aim to better understand brain disease pathogenesis and find promising therapeutic targets. We found evidence of cerebellar glial dysfunction, which is caused by the downregulation of a glutamate transporter that affects neuronal physiology.
RESULTS
Bergmann Glia Show Abundant RNA Accumulation in DMSXL Cerebellum
To investigate the impact of the DM1 expansion on different cell populations and networks in the CNS, we investigated the distribution of the canonical molecular signs of the disease (the toxic CUG RNA foci) in different areas of the DMSXL mouse brain. We were particularly intrigued by the distinctive and peculiar distribution of RNA foci in the cerebellum. The cerebellum is a well-organized brain region with a highly specific and uniform laminar arrangement of cells into distinct, easily identified anatomical layers (Voogd and Glickstein, 1998). Fluorescence in situ hybridization (FISH) revealed that CUG RNA foci were rarely found in Purkinje cells, but in contrast, they were abundant in a population of neighboring cells, extending toward the molecular layer. MBNL1 and MBNL2 co-localized with RNA in these foci-rich cells in DMSXL mice (Figure 1A), in contrast to wild-type animals (Figure S1A). MBNL proteins remained distributed throughout the nucleus and cytoplasm of DMSXL Purkinje cells, even in those rare neurons showing RNA foci accumulation, without pronounced sequestration (Figure 1A).
Figure 1. RNA Foci Accumulate in Cerebellar Bergmann Astrocytes and Deregulate Alternative Splicing.
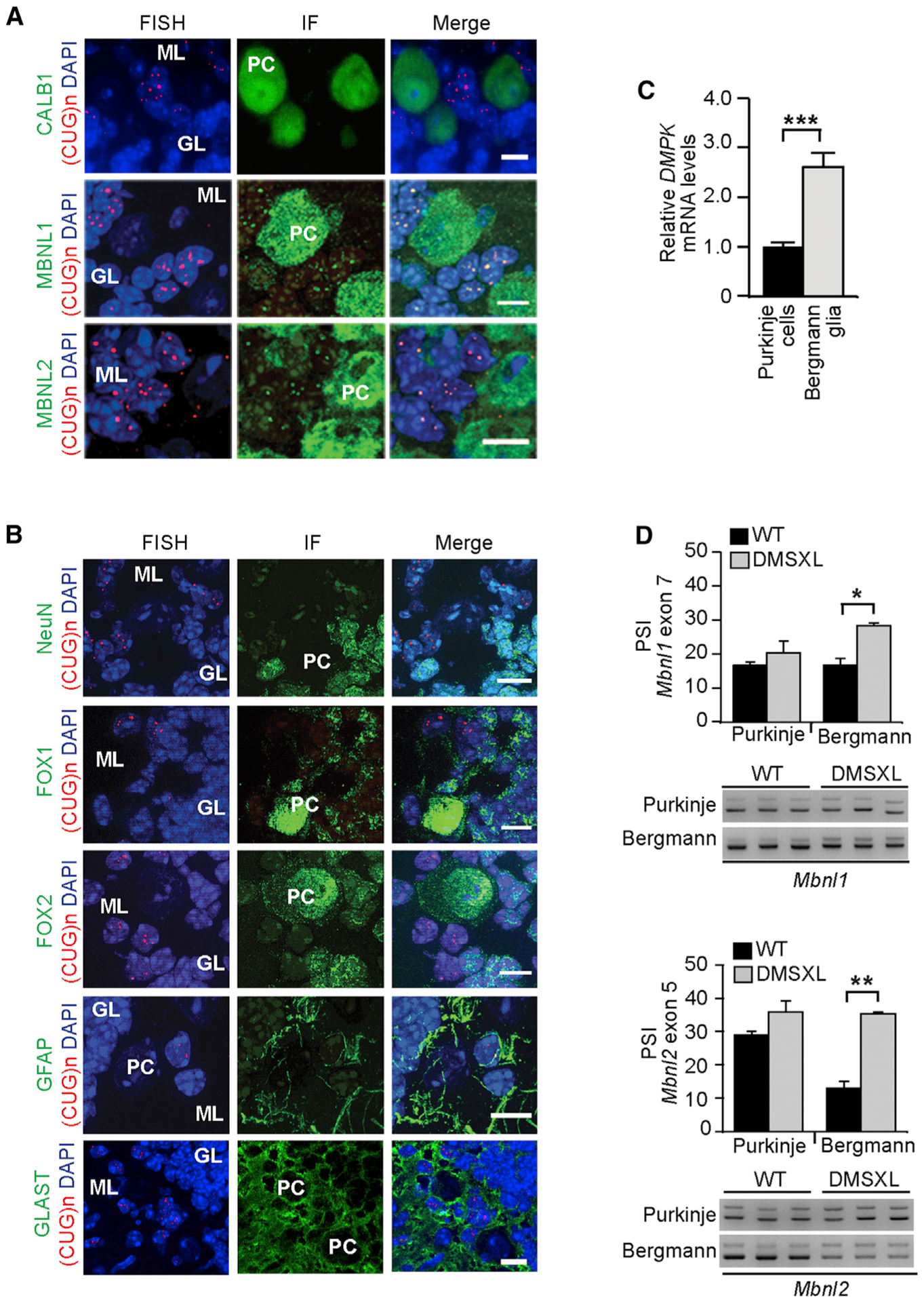
(A) FISH detection of RNA foci (red) and immunofluorescence of calbindin 1 (CALB1) and MBNL proteins (green) in mouse cerebellum. An example of a rare foci-positive Purkinje cell is shown in the bottom panel.
(B) Identification of cell types showing abundant nuclear RNA foci (red) in DMSXL cerebellum through immunodetection of NeuN, Fox1, Fox2, GFAP, or GLAST (green). DAPI was used for nuclear staining.
(C) Nested qRT-PCR of DMPK transgene expression (±SEM) in Purkinje cells and Bergmann astrocytes microdissected from DMSXL cerebellum (n = 3).
(D) Percent of spliced in (PSI) of alternative exon 7 of Mbnl1 and exon 5 of Mbnl2 transcripts (±SEM). Purkinje cells and neighboring Bergmann glia were collected by laser microdissection from DMSXL and wild-type (WT) mice (n = 3, each genotype). Three independent technical replicates were studied for each mouse. Representative analyses of alternative splicing by nested RT-PCR are shown.
PC, Purkinje cells; GL, granular layer; ML, molecular layer. The scale bar represents 10 μm.
*p < 0.05, **p < 0.01, ***p < 0.001; Mann-Whitney U test. See also Figure S1.
Given the intriguing distribution of RNA foci in DMSXL cerebellum, we sought to identify the nature of foci-rich cells through immunodetection of cell-specific markers. NeuN stains almost exclusively mature granular neurons, whereas Fox1 and Fox2 stain Purkinje and Golgi cells; Fox2 stains also the granular neurons (Kim et al., 2011). We found that foci accumulated preferentially in non-neuronal cells of the molecular layer, which did not express NeuN, Fox1, or Fox2. The non-neuronal nature of these cells was confirmed by GFAP staining (Figure 1B). The distinctive localization around the Purkinje cells into the molecular layer of the cerebellum, the expression of GFAP, and the lack of neuronal markers strongly suggested that the foci-rich cells were Bergmann astrocytes. We have confirmed their nature by the immunodetection of GLAST/SLC1A3, a glial glutamate transporter, which in adult mouse cerebellum is primarily expressed in Bergmann glia (Regan et al., 2007): immunofluorescence combined with FISH revealed greater GLAST expression near Purkinje cells, usually in foci-rich cells (Figure 1B).
To elucidate the reasons behind the preferential accumulation of CUG RNA foci in Bergmann glia, we quantified the levels of expanded DMPK transcripts in Bergmann astrocytes and Purkinje cells collected from DMSXL cerebellum by laser capture microdissection. The purity of the collected cells was controlled by RT-PCR amplification of cell-specific transcripts (Figure S1B). qRT-PCR revealed levels of toxic CUG RNA nearly three times higher in Bergmann glia than in adjacent Purkinje cells (Figure 1C). We conclude that higher transgene expression in cerebellar Bergmann glia contributes to the higher foci abundance in this cell type.
RNA Spliceopathy Is More Pronounced in the Bergmann Glia of the Cerebellum
Higher levels of CUG RNA and foci in Bergmann astrocytes predict pronounced spliceopathy in this cell type. Thus, we studied splicing defects in microdissected DMSXL Bergmann and Purkinje cells. We have previously shown that Mbnl1 and Mbnl2 transcripts show consistent missplicing in DMSXL mouse brains (Hernández-Hernández et al., 2013a) and serve as robust markers of spliceopathy in our mouse model. In line with our hypothesis, Mbnl1 and Mbnl2 splicing was significantly dysregulated in DMSXL Bergmann glia while remaining unaltered in Purkinje cells (Figure 1D). Overall, the missplicing of these transcripts was mild in the analysis of whole DMSXL cerebellum (Figure S1C), suggesting that splicing abnormalities are pronounced in Bergmann astrocytes but diluted in whole-tissue samples. Finally, we assessed the contributing role of CELF proteins to cerebellum pathology. Western blot analysis in whole DMSXL cerebellum revealed mild upregulation of CELF2 but no significant changes in CELF1 levels (Figure S1D).
Electrophysiological Abnormalities of Purkinje Cells in DMSXL Cerebellum
We next investigated whether Bergmann RNA toxicity in DMSXL mice was sufficient to affect cerebellar function. The functional output of the cerebellar cortex is determined by the Purkinje cell firing, which can be electrophysiologically identified by two types of firing patterns: complex spikes and simple spikes (Cheron et al., 2013). We performed electrophysiological recordings in the Purkinje cell layer of alert DMSXL mice and found significantly higher simple spike firing rates (86.8 ± 7.6 versus 50.5 ± 4.2 Hz) and rhythmicity index (0.13 ± 0.02 versus 0.07 ± 0.01) in DMSXL mice relative to the wild-type controls, indicative of neuronal hyperactivity of Purkinje cells (Figure 2A). In addition, spontaneous fast local field potential (LFP) oscillations were found throughout the cerebellum in all DMSXL mice but were absent in wild-type controls. DMSXL fast LFP oscillations appeared as spindle-shaped episodes of oscillation with a frequency of 200 ± 27 Hz and maximal amplitude of 0.48 ± 0.26 mV (Figure 2B). We quantified calcium buffering proteins and studied DMSXL cerebellum histology, but we did not find obvious changes in steady-state protein levels or overt histopathology that could contribute to the defective Purkinje neuronal activity and cerebellum dysfunction (Figures S2A and S2B).
Figure 2. Cerebellum Dysfunction in DMSXL Mice Revealed by Behavioral and Electrophysiological Phenotyping.
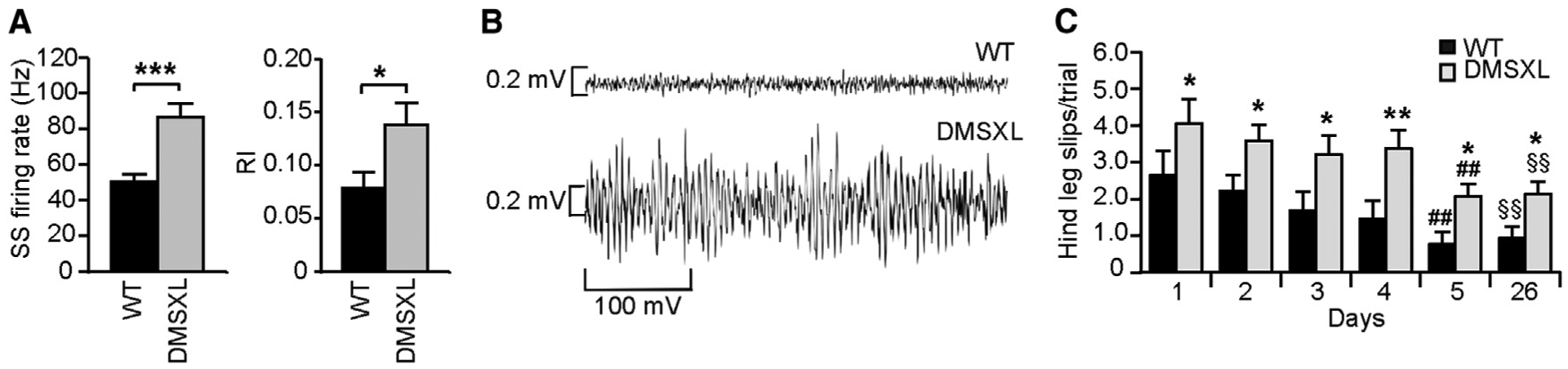
(A) In vivo electrophysiological recordings of single spike (SS) frequency and rhythmicity index (RI) in 2-month-old alert DMSXL (n = 4) and WT mice (n = 3). Error bars represent the SEM. *p < 0.05, ***p < 0.001; Mann-Whitney U test.
(B) Representative fast LFP oscillation in the cerebellum of one DMSXL compared to WT control.
(C) Assessment of cerebellum-dependent motor-coordination of DMSXL (n = 20) and WT controls (n = 21), at 3–4 months in the runway test over 5 consecutive days (days 1–5). The graph represents the average number of hind leg slips per trial (±SEM) (*p < 0.05, **p < 0.01; two-way ANOVA). Learning was assessed by the number of slips at day 5 relative to day 1 (##p < 0.01; two-way ANOVA). Retention of the task was assessed at day 26, following a period of 3 weeks without testing, relative to day 1 (§§p < 0.01; two-way ANOVA).
See also Figure S2.
Cerebellum-Dependent Motor Incoordination in DMSXL Mice
To confirm cerebellar dysfunction in DMSXL mice, we assessed a cerebellum-dependent behavior phenotype. In the runway test, mice run along an elevated platform and must surmount low obstacles intended to impede their progress. The test assesses cerebellum-dependent motor coordination, and in contrast with rotarod, it is minimally influenced by muscle performance (Bearzatto et al., 2005). The number of slips of the right hind leg is a direct indication of motor incoordination.
Both wild-type and DMSXL showed a progressive and significant decrease in number of hind leg slips from day 1 to day 5 (Figure 2C), indicating the capacity to learn new cerebellum-dependent tasks. However, DMSXL mice showed significantly higher numbers of slips from day 1, pointing to deficits in the fine-tuning of movements and cerebellar dysfunction. After 3 weeks, the test still revealed significantly lower number of slips relative to day 1 in both wild-type and DMSXL mice, demonstrating efficient task retention by both genotypes (Figure 2C). In summary, although capable of acquiring and retaining new cerebellum-dependent motor tasks, DMSXL mice showed signs of motor incoordination.
In conclusion, both electrophysiological and behavioral assessment demonstrated that the expression and accumulation of toxic RNA foci in DMSXL cerebellum (particularly in Bergmann glia) are associated with cerebellum pathology, which is characterized by abnormal Purkinje cell firing and fine motor incoordination.
The GLT1 Glutamate Transporter Is Downregulated in DMSXL Cerebellum
To decipher the mechanisms of abnormal Purkinje cell activity and cerebellar dysfunction, we used global proteomics to identify expression changes and dysregulated pathways in DMSXL cerebellum. We first studied whole-cell lysates and found that the expression of 241 proteins was altered in DMSXL cerebellum. A Gene Ontology analysis on this 241-protein set revealed that many altered proteins were membrane-bound, but we did not find a biological process predominantly dysregulated in DMSXL cerebellum (Figure 3A). To refine our search, we specifically investigated the membrane-bound proteome of mouse cerebellum and found 60 proteins with altered expression in DMSXL cerebellum. This protein set showed enrichment for ion transport, synaptic transmission, and glutamate signaling (Figure 3B) and included the glial high-affinity glutamate transporter (GLT1) (excitatory amino acid transporter 2 [EAAT2] or solute carrier family 1 member 2 [SLC1A2]). GLT1 is a membrane transporter that in the cerebellum is mainly expressed by the Bergmann glia to clear glutamate released during synaptic transmission from the extracellular space, avoiding excessive stimulation of postsynaptic neurons (Kanai and Hediger, 2004). Therefore, we tested the hypothesis that abnormal GLT1 expression in DMSXL cerebellum results in defective neuroglial communication and abnormal DMSXL Purkinje cell firing.
Figure 3. GLT1 Downregulation in DMSXL Cerebellum.
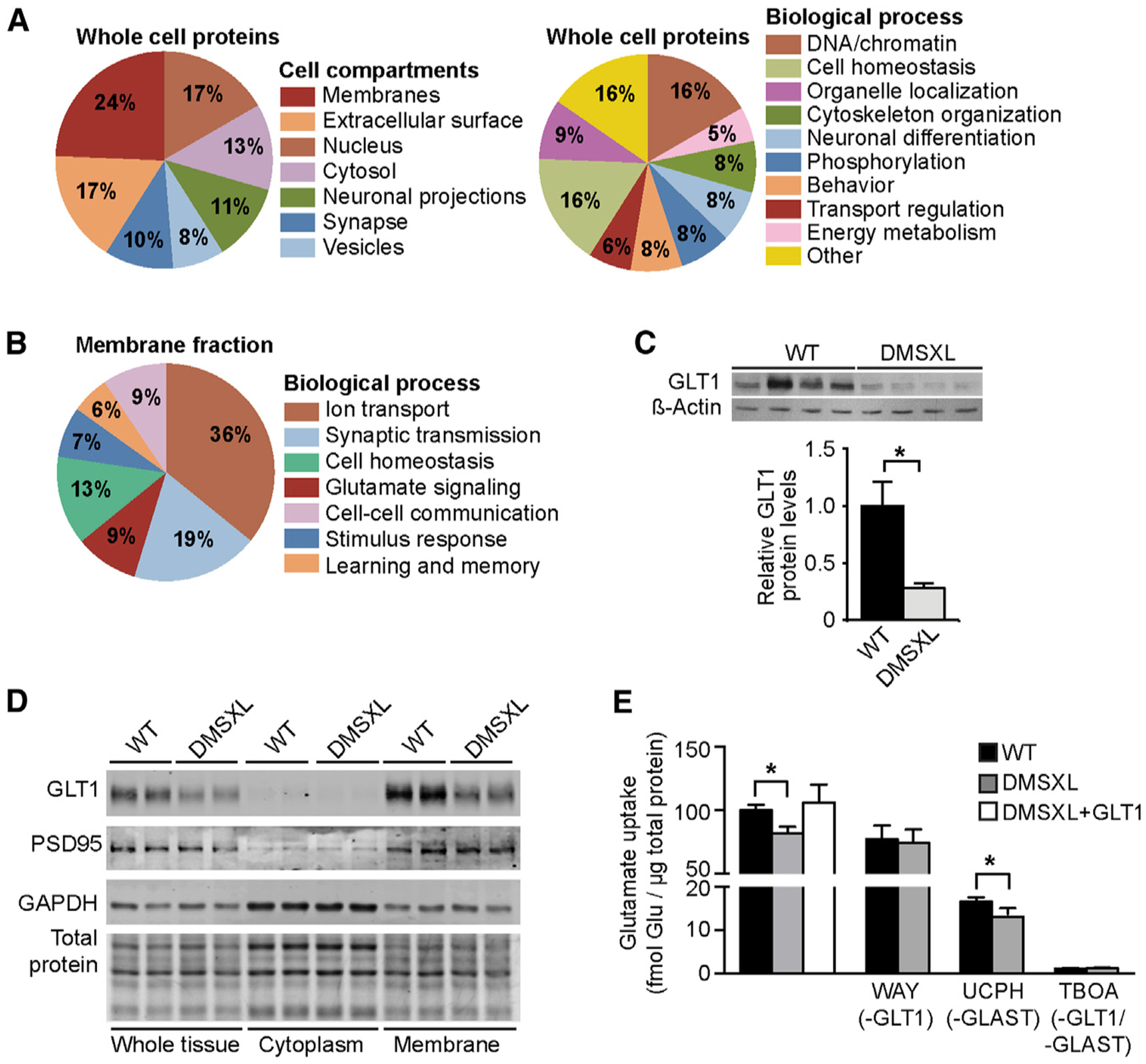
(A) Isobaric tag for relative and absolute quantification (iTRAQ) analysis of whole cell protein fractions collected from the cerebellum of 2-month-old DMSXL mice and WT littermates (n = 4, each genotype). Pie charts indicate the percentage of altered proteins in the cellular components and biological processes showing the highest significant enrichment in the proteomics expression data.
(B) Quantitative proteomics analysis of the membrane-bound proteins from the cerebellum of 2-month-old DMSXL mice and WT littermates (n = 2, each genotype). Pie charts indicate the percentage of altered membrane proteins in the biological processes showing the highest significant enrichment.
(C) Western blot quantification of GLT1 protein levels in the cerebellum of DMSXL and WT mice (n = 4, each genotype) at 2 months of age. β-actin was used as loading control.
(D) Western blot detection of GLT1 in cytosolic and membrane protein fractions of DMSXL and WT cerebellum at 2 months of age (n = 2, each genotype). GAPDH and PSD95 confirmed cytosolic and membrane protein enrichment, respectively. Total protein was visualized by stain-free protocols and used as loading control.
(E) Uptake of radioactive glutamate by WT and DMSXL astrocytes: under control conditions, following DMSXL transfection with GLT1-expressing plasmids, or upon selective inhibition of GLT1 (WAY213623), GLAST (UCPH 101), or both (TBOA) (n = 3, each group).
Errors bars represent the SEM. *p < 0.05; Mann-Whitney U test. See also Figure S3.
We first confirmed GLT1 downregulation by western blot in DMSXL cerebellum (Figure 3C), as well as in other mouse brain regions (Figure S3A), but not in control DM20 mice (Figure S3B). Semiquantitative analysis of GLT1 immunofluorescence intensity by confocal microscopy showed a significant reduction in the DMSXL molecular layer, close to the Purkinje cells (Figure S3C). In contrast to GLT1, the levels of GLAST remained unchanged in DMSXL brains (Figure S3D), demonstrating that the impact of expanded DMPK transcripts is specific to the GLT1 glutamate transporter. Fractioning of DMSXL cerebellar tissue revealed downregulation of GLT1 in the membrane-bound protein faction (Figure 3D), in line with defective glutamate transport across the membrane. To investigate the functional impact of reduced GLT1 levels, we measured the uptake of radioactive glutamate by DMSXL astrocytes in the presence of WAY213623 (GLT1-specific inhibitor), UCPH (GLAST-specific inhibitor), or TBOA (pan-glutamate transporter inhibitor). We found a significant reduction in total and in GLT1-mediated glutamate uptake when compared to wild-type controls (Figure 3E), consistent with GLT1 downregulation in DMSXL astrocytes (Figure S3E). In contrast, GLAST-mediated transport was unaltered, while TBOA nearly abolished glutamate uptake in both cultures. Transfection of DMSXL astrocytes with GLT1-expressing plasmids corrected defective glutamate transport (Figure 3E). Altogether, these results demonstrate the causative role of GLT1 downregulation in defective glutamate transport by DMSXL astrocytes. GLT1 downregulation is not explained by Bergmann cell loss, as revealed by the quantification of Bergmann-specific transcripts in DMSXL cerebellum, which showed no reduced expression relative to wild-type controls (Figure S3F). Overall, the reduction of GLT1 within the molecular layer is consistent with Bergmann dysfunction.
Human DM1 Cerebellum Shows Bergmann-Specific RNA Foci Accumulation and GLT1 Downregulation
We then assessed the implications of the mouse findings to the human condition through the analysis of post-mortem DM1 brains. In human DM1 cerebellum, although small foci were rarely detected in Purkinje cells, large and more abundant foci accumulated predominantly in calbindin 1 (CALB1)-negative cells, co-localizing with MBNL1 and MBNL2 (Figure 4A). Like in DMSXL mice, the preferential accumulation of RNA foci was concentrated in the Bergmann glia, which expressed GFAP and GLAST in the absence of NeuN, Fox1, or Fox2 neuronal markers (Figure 4B). The analysis of selected MBNL1 and MBNL2 candidate splicing events revealed that human DM1 cerebellar tissue showed mild spliceopathy (Figure S4A), in association with CELF2 upregulation (Figure S4B).
Figure 4. Analysis of RNA Foci Accumulation and GLT1 Downregulation in Human DM1 Cerebellum.
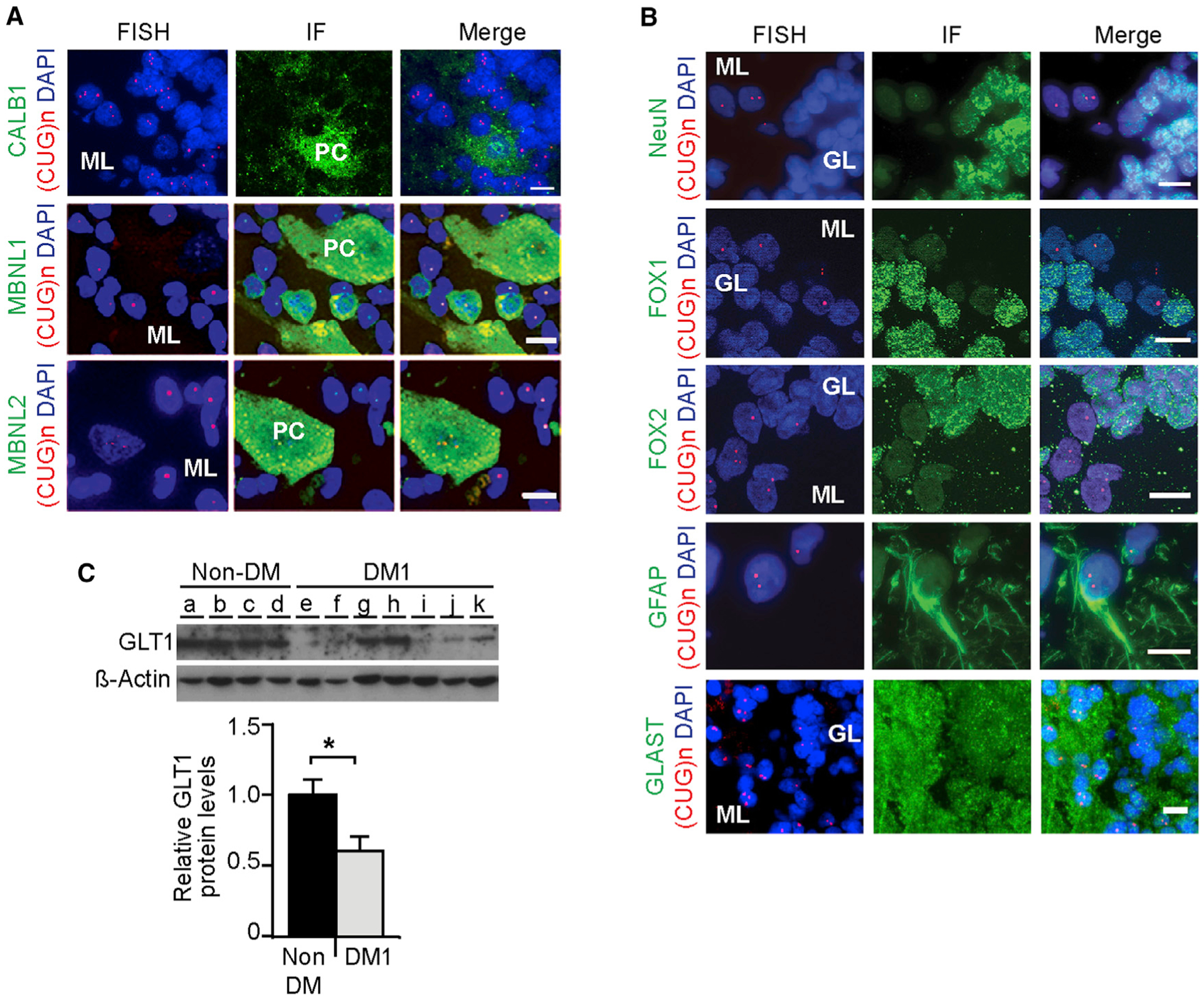
(A) FISH of RNA foci (red) combined with immunofluorescence detection of CALB1 and MBNL proteins (green) in post-mortem brains from adult DM1 patients. Some rare and small RNA foci were detected in human Purkinje cells but were not associated with pronounced sequestration of MBNL proteins.
(B) FISH of RNA foci (red) and protein markers (green) of different cerebellar cell populations near Purkinje cells.
(C) Western blot quantification of GLT1 in DM1 cerebellum (n = 7) relative to non-DM controls (n = 4). β-actin was used as loading control.
The scale bar represents 10 μm. Error bars represent the SEM. *p < 0.05; Mann-Whitney U test. See also Figure S4.
Finally, we studied GLT1 protein expression in human DM1 cerebellum and found a dramatic reduction in five of seven adult DM1 patients: overall, GLT1 was reduced by ~50% relative to non-DM controls (Figure 4C). GLT1 was also decreased in DM1 frontal cortex and brainstem (Figure S4C), indicating wider dysregulation of this glutamate transporter throughout the CNS.
GLT1 Downregulation Is Mediated by MBNL1 Inactivation
To gain insight into the mechanisms of GLT1 downregulation, we quantified transcript levels and found significantly lower levels of GLT1 mRNA in the cerebellum and frontal cortex of the DM1 patients with pronounced protein downregulation (Figure 5A). In amyotrophic lateral sclerosis (ALS), missplicing of GLT1 results in RNA degradation and loss of protein (Lin et al., 1998). To test whether similar mechanisms operate in DM1, we studied ALS-associated exon missplicing and abnormal intron retention. We did not find obvious splicing abnormalities in DM1 patients or in DMSXL mice (Figures 5SA and 5SB).
Figure 5. Transcriptional GLT1 Downregulation Is Mediated by MBNL1 Inactivation.
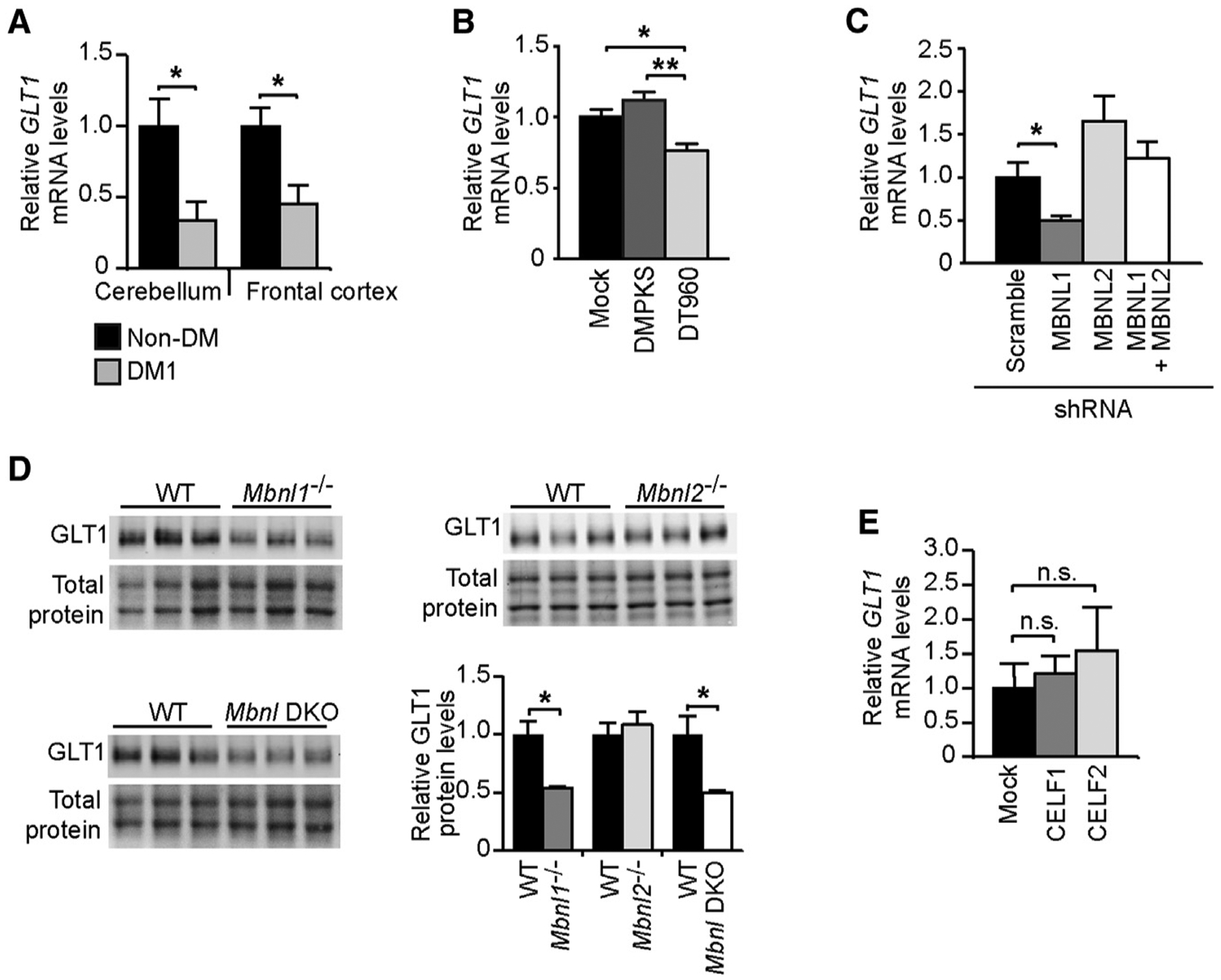
(A) Quantification of GLT1 transcripts (±SEM) in the cerebellum and frontal cortex of DM1 patients showing the most pronounced protein decrease (n = 5) relative to non-DM controls (n = 4).
(B) Quantification of GLT1 transcripts in human T98G glial cells transfected with expanded DMPK constructs containing 960 interrupted CTG repeats (DT960) relative to no-repeat (DMPKS) and mock transfected controls.
(C) Quantification of GLT1 mRNA following MBNL1 and/or MBNL2 knockdown in T98G cells.
(D) Western blot quantification of GLT1 protein levels in 2- to 4-month-old Mbnl1 KO, Mbnl2 KO, in Mbnl1/Mbnl2 double-KO mice (DKO) relative to WT littermates (n = 3, each genotype). Representative western blot membranes of at least three technical replicates. Total protein was visualized by stain-free protocols and used as loading control.
(E) Quantification of GLT1 mRNA in T98G cells overexpressing CELF1 or CELF2.
Data are represented as the mean (±SEM) of three independent experiments in (B), (C), and (E). *p < 0.05, **p < 0.01; Mann-Whitney U test in (A) and (D), one-way ANOVA in (B) and (C); n.s., not statistically significant. See also Figure S5.
We then tested whether GLT1 downregulation was the direct result of the expression of CUG-containing RNA or a secondary consequence associated with DM1 brain disease progression. To this end, we transfected human T98G glioblastoma cells with expanded DMPK constructs and found that CUG RNA expansions reduced GLT1 transcript levels relative to no-repeat control constructs (Figure 5B).
MBNL proteins regulate various aspects of RNA metabolism. Hence, we tested whether MBNL1 or MBNL2 inactivation was sufficient to lower GLT1 levels. We used short hairpin RNA (shRNA) to knockdown MBNL1 and/or MBNL2 in T98G cells (Figure S5C). qRT-PCR revealed that MBNL1 downregulation alone was sufficient to decrease GLT1 mRNA levels, while MBNL2 inactivation left GLT1 transcripts unchanged (Figure 5C). The simultaneous treatment with MBNL1 and MBNL2 shRNA resulted in a modest downregulation of both MBNL proteins (Figure S5C), which was insufficient to affect GLT1 mRNA levels. We confirmed the determinant role of MBNL1 in vivo through the analysis of Mbnl1 and Mbnl2 knockout (KO) mice (Charizanis et al., 2012; Kanadia et al., 2003): only the cerebellum of Mbnl1 −/− mice showed significant downregulation of GLT1. In contrast to double-shRNA-transfected T98G cells, the inactivation of MBNL1 and MBNL2 proteins in Mbnl1/Mbnl2 double-KO mice (Goodwin et al., 2015) significantly reduced GLT1 levels (Figures 5D and S5D).
In an attempt to provide insight into the prevalent role of MBNL1 over MBNL2 in the regulation of the glial-specific GLT1 glutamate transporter, we quantified the expression of MBNL proteins in mouse primary neurons and astrocytes. The analysis revealed that the relative expression of MBNL1 is 2-fold higher in mouse primary astrocytes relative to neurons (Figure S5E), suggesting a more important role of MBNL1 in glial cells.
Because CELF2 is upregulated in DM1 brains, we also studied whether CELF proteins could regulate GLT1 expression and contribute to abnormal GLT1 levels. Transient transfection of CELF1 and CELF2 in T98G cells (Figure S5F) did not result in lower GLT1 mRNA levels (Figure 5E).
In summary, our results demonstrate that GLT1 protein down-regulation in DM1 is associated with lower transcript levels without evidence of missplicing and is mediated by partial inactivation of MBNL1 in glial cells, independently of CELF proteins.
GLT1 Downregulation in DMSXL Astrocytes Is Associated with Increased Glutamate Neurotoxicity
Glutamate transporters guard against prolonged elevation of extracellular glutamate concentration and protect neurons from excitotoxicity (Kanai and Hediger, 2004). To investigate the impact of GLT1 downregulation on neuronal physiology in DM1, we tested whether primary DMSXL astrocytes expressing significantly lower levels of GLT1 (Figure S3E) failed to protect neurons against glutamate neurotoxicity in culture. To this end, we co-cultured neurons and astrocytes of mixed genotypes and allowed neurites to extend for 8 days. Then, we added 50 μM of glutamate to the medium and monitored neuronal damage by measuring neurite collapse by fluorescence live-cell videomicroscopy (Figure 6A). Neurite collapse was significantly more pronounced in wild-type and DMSXL neurons co-cultured with DMSXL astrocytes than in neurons grown with wild-type astrocytes (Figure 6B). Antagonists of NMDA and AMPA receptors reduced glutamate-induced neurite collapse and eliminated differences between genotypes (Figure 6B), demonstrating the mediating role of glutamate receptors in the neurotoxicity detected in neuroglial cultures. The increased glutamate neurotoxicity in the presence of DMSXL astrocytes was not accounted for by significant changes in the expression of NMDA and AMPA receptors: western blot quantification GRIN1 (NMDA receptor subunit type 1) and GRIA2 (AMPA receptor subunit type 2) did not show significant changes in DMSXL cell cultures (Figure 6C) or in brain tissue (Figures S6A and S6B). To determine whether the deleterious effect of astrocytes on neurons was directly mediated by GLT1 downregulation, we transfected DMSXL astrocytes with GLT1 before glutamate neurotoxicity assessment. GLT1 transfection of DMSXL astrocytes rescued the neurite collapse (of both wild-type and DMSXL neurons) to levels that were indistinguishable from those measured in the presence of wild-type astrocytes (Figure 6D).
Figure 6. Neuronal Glutamate Toxicity in Mixed Cultures of Neurons and Astrocytes.
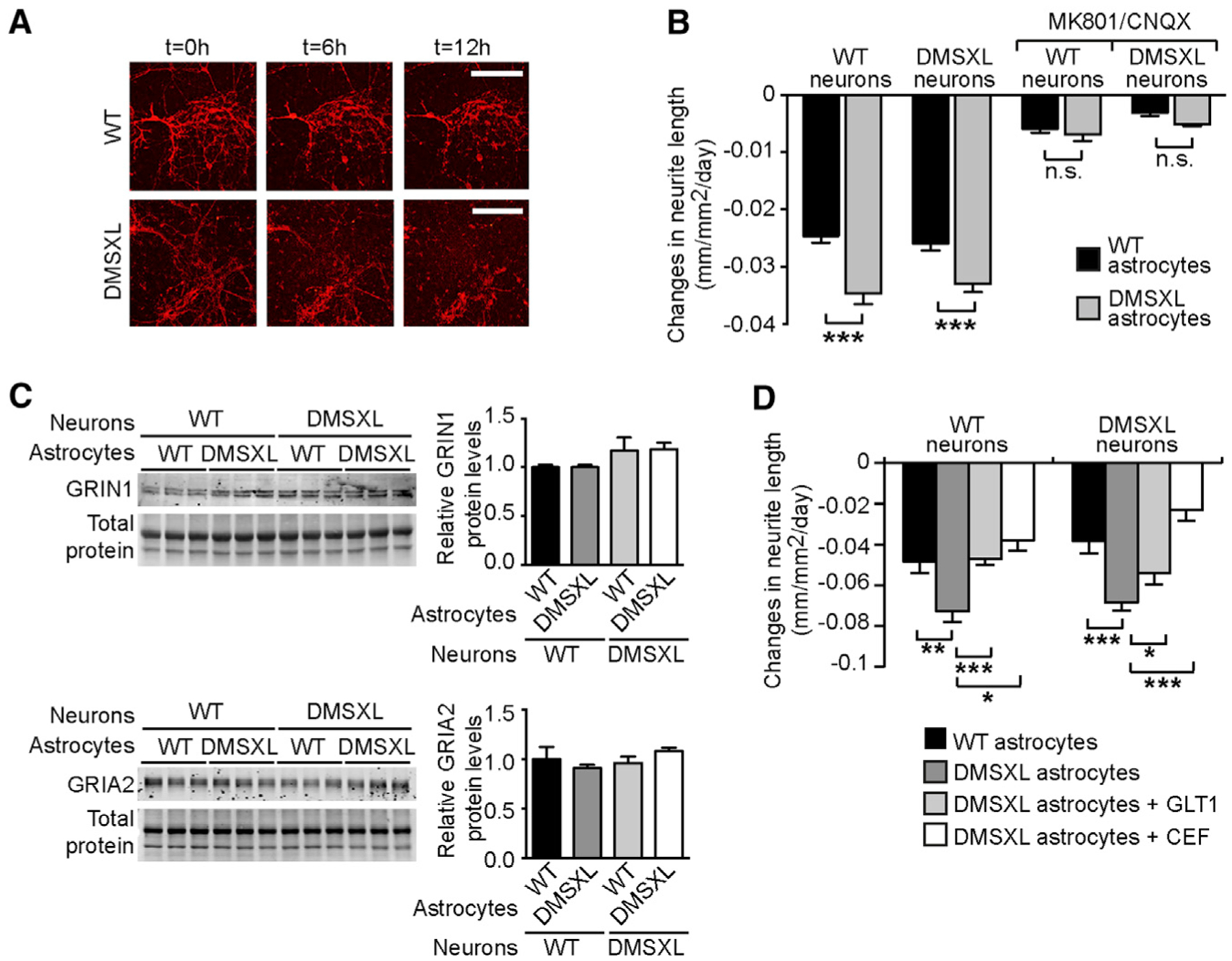
(A) Representative images of WT neurite collapse co-cultured with WT and DMSXL astrocytes. Glutamate (50 μM) was added to the medium at t = 0 hr, and neurite length was monitored by the expression of fluorescent red mKate2 protein under the control of the neuron-specific synapsin-1 promoter. The scale bar represents 200 μm.
(B) Rate of glutamate-induced neurite collapse of WT and DMSXL primary neurons, cultured with WT or DMSXL astrocytes. (+)-MK 801 (NMDA receptor antagonist) and CNQX (AMPA receptor antagonist) were added to block glutamate receptors (n = 4 independent co-cultures, each mixed genotype combination). The graph shows the quantification of three independent experiments (±SEM).
(C) Representative western blot of GRIN1 (NMDA receptor subunit) and GRIA2 (AMPA receptor subunit) in mixed neuroglial cultures (n = 3 independent mixed cultures). Data are represented as the mean (±SEM). Total protein was visualized by stain-free protocols and used as loading control.
(D) Rate of glutamate-induced neurite collapse of WT and DMSXL neurons. Mouse primary neurons were cultured with WT astrocytes, control DMSXL astrocytes, DMSXL astrocytes transfected with GLT1, or DMSXL astrocytes treated with 10 μM ceftriaxone (CEF) (n = 4 independent co-cultures, each combination).
Error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001; one way-ANOVA; n.s., not statistically significant. See also Figure S6.
Altogether, these results demonstrate that the downregulation of GLT1 in DMSXL astrocytes perturbs the neuroglial interplay and has a negative impact on neuronal physiology, failing to protect against glutamate excitotoxicity.
GLT1 Upregulation by Ceftriaxone Corrects the Cerebellum Phenotype of DMSXL Mice
To explore the role of GLT1 downregulation in DMSXL cerebellar dysfunction, we first used LFP oscillations in the Purkinje cell layer of the cerebellar vermis to compare the extracellular electrical activity of DMSXL and Glt1-deficient mice, which show a ~60% reduction in GLT1 (Tanaka et al., 1997). Both DMSXL and heterozygous Glt1+/− mice exhibited a frequency peak of oscillations around 200 Hz, similar to wild-type controls (Figures 7A and 7B), but the amplitude of the power peak of LFP oscillations was significantly higher in Glt1+/− and in DMSXL mice (Figures 7C and 7D). In other words, the abnormal neuronal activity of DMSXL Purkinje cells is recreated by the partial inactivation of Glt1, in agreement with a mediating role of GLT1 downregulation in the onset of DMSXL cerebellar phenotypes.
Figure 7. Rescuing of DMSXL Cerebellum Phenotype following GLT1 Upregulation by Ceftriaxone.
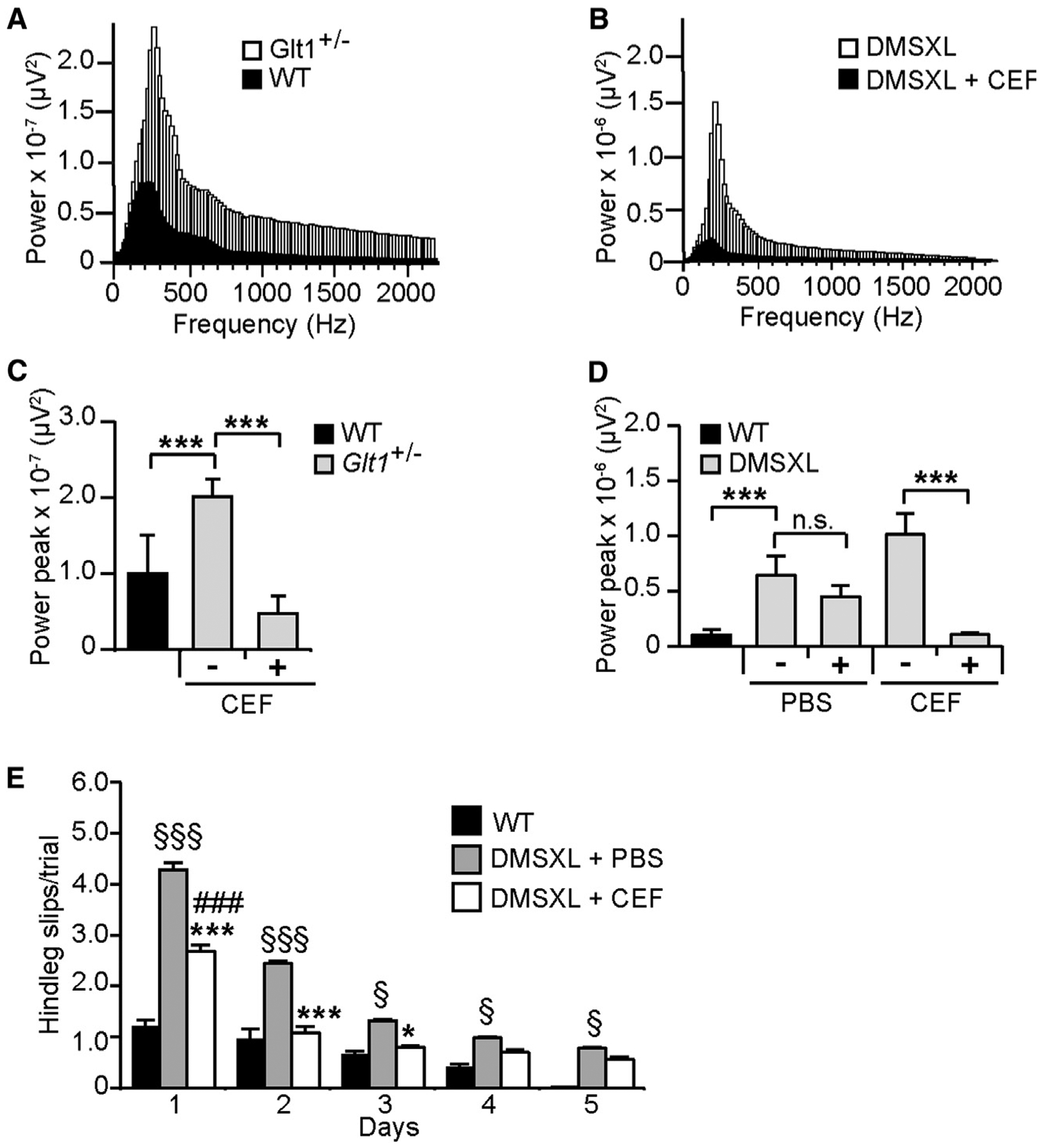
(A) Representative profiles of LFP oscillations recorded over 690 min in the Purkinje cell layer of a Glt1+/− mouse and a WT control.
(B) Representative profiles of LFP oscillations recorded over 690 min in DMSXL mice before and following ceftriaxone injection.
(C) The power peak of the LFP oscillation (±SEM) was calculated by fast Fourier transform (FFT) analysis in Glt1+/− mice (n = 4) and WT controls (n = 3). Glt1+/− LFP oscillations were assessed before and following ceftriaxone injection. ***p < 0.001; one-way ANOVA.
(D) Effect of ceftriaxone on the power peak of the LFP oscillation (±SEM) in DMSXL mice (n = 5). Control DMSXL mice were injected with PBS (n = 4). Non-injected WT mice are shown as controls (n = 3). ***p < 0.001; one-way ANOVA; n.s., not statistically significant.
(E) Runway assessment of motor coordination of DMSXL mice injected with ceftriaxone relative to control DMSXL animals injected with PBS (n = 9, each group; *p < 0.001, ***p < 0.001; two-way ANOVA). The graphs represent the mean number of hind leg slips per trial (±SEM). Ceftriaxone-treated animals were compared with non-treated WT controls (n = 9; ###p < 0.001 at day 1; two-way ANOVA). DMSXL mice injected with PBS performed consistently worse than WT controls throughout the entire test (§p < 0.05, §§§p < 0.001; two-way ANOVA). Mice were studied at 2 months of age.
See also Figure S7.
To further demonstrate the implications of GLT1 in DMSXL cerebellar dysfunction, we injected DMSXL animals with ceftriaxone for 5 consecutive days. Ceftriaxone is a β-lactam antibiotic that activates GLT1 expression (Rothstein et al., 2005). Ceftriaxone corrected GLT1 protein levels in DMSXL cerebellum (Figure S7A) and glutamate neurotoxicity in DMSXL neuroglial co-cultures (Figure 6D). LFP oscillations were recorded in the same animal before and following ceftriaxone treatment. Ceftriaxone did not change the frequency peak of LFP oscillations in DMSXL mice (Figure 7B) but resulted in a remarkable reduction in the amplitude of the power peak of Glt1+/− and DMSXL Purkinje LFP oscillations down to wild-type values (Figures 7C and 7D). In contrast, Purkinje LFP oscillations did not change significantly in sham-treated DMSXL mice.
Finally, we assessed whether ceftriaxone-induced GLT1 upregulation (Figure S7A) ameliorated DMSXL motor coordination. A 5-day regimen of ceftriaxone significantly reduced the average number of hind leg slips from day 1 relative to PBS-injected DMSXL controls (Figure 7E). From day 2, the number of slips of ceftriaxone-treated DMSXL mice was indistinguishable from wild-type controls. To investigate whether improved Purkinje cell firing and mouse motor performance could be mediated by an effect of ceftriaxone on transgene expression, we measured DMPK transcripts following treatment, but we did not find differences relative to PBS-injected DMSXL controls (Figure S7B). Overall, these results demonstrate that reduced GLT1 is a critical contributing factor to DMSXL cerebellar pathophysiology, which can be rescued by ceftriaxone-induced upregulation of this glutamate transporter.
DISCUSSION
We found prevalent signs of RNA toxicity in the Bergmann glia of a mouse model of DM1, in association with cerebellar abnormalities, such as network-mediated Purkinje cell excitability and motor incoordination. We demonstrated that defective expression of GLT1 glutamate transporter in astrocytes plays a determinant role in mediating these phenotypes.
Bergmann glia consist of a population of astrocytes whose cell bodies are embedded in the Purkinje layer. Their cellular processes create a microenvironment essential for the good functioning of the Purkinje synapses (Bellamy, 2006). Purkinje cells are the sole output of the cerebellar cortex. Their firing is an integrated response to their intrinsic excitability and to the excitatory and inhibitory inputs from the cerebellar network (Cheron et al., 2013). Thus, the electrophysiological recordings of spontaneous firing rate and rhythmicity in vivo provide an efficient way of assessing the functional states of the cerebellar neuronal network, independently of confounding factors, such as the muscle pathology and the reduced body weight of DMSXL mice (Gomes-Pereira et al., 2007; Huguet et al., 2012). Hence, the fast LFP oscillations registered in DMSXL mice revealed pathological changes in integrated Purkinje cell activity (Cheron et al., 2008), which may result from the synchronization of high-frequency rhythmic firing caused by intrinsic Purkinje cell excitability (Cheron et al., 2004), granular cell hyperexcitability (Bearzatto et al., 2006; Cheron et al., 2004), or altered synaptic plasticity (Servais et al., 2007). Abnormal spontaneous fast LFP oscillations were previously reported in ataxic mice showing cerebellum-dependent motor incoordination (Bearzatto et al., 2006; Cheron et al., 2004, 2005; Servais et al., 2007), supporting their contribution to the motor phenotype of DMSXL mice in the runway test. In contrast, other patterns of abnormal firing, such as slow firing (Servais and Cheron, 2005) or bursting (Cheron et al., 2009), were associated with milder or more severe incoordination, respectively.
Despite their functional abnormalities, Purkinje cells did not display abundant RNA foci or pronounced missplicing in DMSXL mice, hinting that neuronal hyperactivity is not mediated by an autonomous trans-dominant effect of CUG repeats operating in Purkinje cells alone. Our data suggest that DMPK transcript levels in Purkinje cells are insufficient to trigger RNA foci accumulation and toxicity. We propose that Purkinje cell hyperexcitation is mediated by abnormalities in the neighboring Bergmann glia, which show high DMPK expression and abundant RNA foci, in association with GLT1 downregulation.
GLT1 is a glial-specific glutamate transporter that recaptures excitatory glutamate from the synaptic cleft and protects from neurotoxicity due to excessive glutamate stimulation (Bellamy, 2006). The most important role of the glial glutamate transporters in the cerebellum is to avoid neurotransmitter spillover and activation of extra-synaptic receptors, thereby maintaining synapse independence. In the cerebellum, Bergmann astrocytes closely appose Purkinje cells, dictating a robust effect of GLT1 on synaptic transmission through proximity: even small changes in the density of Bergmann GLT1 transporters have a significant impact on Purkinje cell function (Tzingounis and Wadiche, 2007). Therefore, GLT1 downregulation can lead to increased synaptic glutamate, chronic Purkinje cell hyperexcitation, and emergence of fast oscillations. The critical role of GLT1 downregulation in the cerebellar dysfunction of DMSXL mice is corroborated by two observations. First, heterozygous Glt1+/− mice show similar electrophysiological LFP abnormalities. Second, ceftriaxone-mediated GLT1 upregulation corrects the spontaneous hyperactivity of DMSXL Purkinje cells and motor incoordination. The similar benefits of ceftriaxone treatment and GLT1 transfection on neuronal physiology in co-cultures strongly point to a rescuing mechanism of ceftriaxone mediated by GLT1 upregulation. Moreover, ceftriaxone was capable of correcting the Purkinje cell activity in Glt1+/− mice, in further support of a specific effect on this target. In conclusion, the benefits of GLT1 upregulation in DMSXL neuroglial co-cultures and in mice demonstrate the role of defective neuroglial interactions in DM1 brain disease.
Inactivation of Glt1 causes lethal spontaneous seizures in KO mice, in association with selective hippocampal neurodegeneration, but no morphological changes in the cerebellum or signs of ataxia (Tanaka et al., 1997). Heterozygous Glt1+/− mice show a ~60% reduction in GLT1 protein levels, and like DMSXL mice, they exhibit abnormally high cerebellar fast oscillations, together with mild behavioral phenotypes in the absence of neurodegeneration (Kiryk et al., 2008). Altogether, our results suggest that partial inactivation of GLT1 in DM1 is more likely associated with neuronal dysfunction than cell death. Broader neuronal hyperexcitability and dysfunction in DM1 beyond cerebellum is supported by the observation of GLT1 downregulation in multiple brain areas and by the increased susceptibility of DMSXL mice to pentylenetetrazol (PTZ)-induced seizures (Charizanis et al., 2012).
GLT1 is a highly regulated transporter, modulated by changes in RNA transcription, splicing and stability, post-translational modifications, and protein activity (Kim et al., 2011). MBNL1 inactivation alone decreased GLT1 transcripts and protein. In contrast, MBNL2 inactivation alone did not affect GLT1 levels, maybe because of the compensating increase of MBNL1 protein levels (Batra et al., 2014; Goodwin et al., 2015; Mohan et al., 2014). It is conceivable that MBNL1, but not MBNL2, specifically regulates GLT1 expression in glial cells. In line with this view, the higher expression of MBNL1 in mouse primary astrocytes, when compared to mouse primary neurons, hints at a predominant role of MBNL1 in the regulation of glia-specific transcripts. Poly(A)-RNA sequencing revealed changes in the alternative polyadenylation of the GLT1 transcripts in the brain of DM1 and myotonic dystrophy type 2 (DM2) patients and in Mbnl double-KO mice (Goodwin et al., 2015). These data suggest that MBNL loss of function perturbs GLT1 polyadenylation, leading to altered levels of this glutamate transporter.
Deficits in GLT1 were reported in several neurological diseases, including Alzheimer’s disease, Huntington’s disease, ALS, and fragile X syndrome (Kim et al., 2011), but the underlying mechanisms and contribution to disease manifestations have not been fully resolved. As in our DM1 mouse model, GLT1 downregulation in mouse models of fragile X syndrome is associated with enhanced neuronal excitability (Higashimori et al., 2013). In DM1, the downregulation of GLT1 and altered glutamate levels in adult patients (Takado et al., 2015) suggest an impairment of the glutamatergic system. Regulation of GLT1 activity and extracellular glutamate may improve the homeostasis and neurotransmission in DM1 brains. Ceftriaxone, in particular, is well tolerated, permeable to the blood-brain barrier, and augments GLT1 promoter activity and glutamate uptake, but other small-molecule GLT1 activators have been described (Kong et al., 2014).
The cerebellum controls motor coordination, skilled voluntary movements, posture, and gait. The implication of the cerebellum in DM1 neuropathology has not been sufficiently studied. However, imaging studies suggest cerebellar abnormalities: brain voxel-based morphometry revealed white matter decrease (Minnerop et al., 2011), while fMRI showed altered connectivity in cerebellar regions implicated in planning of movements and motor coordination (Serra et al., 2016). The frequency of stumbles and falls in DM1 is 10-fold higher than in healthy controls (Wiles et al., 2006). Several aspects of DM1 disease biology could lead to gait difficulties, among these the weakness of the leg muscles (Hammarén et al., 2014). However, because in these studies muscular impairment was often an inclusion criterion, the results are only generalizable to muscularly impaired DM1 individuals, excluding those that show gait affection without muscle weakness. A study demonstrated limited contribution of muscle weakness to gait abnormalities in DM1 and suggested a role for sensory deficits (Bachasson et al., 2016). In line with this view, altered brain connectivity has been associated with patients’ motor deficits (Toth et al., 2015). Adaptive cognitive strategies usually mitigate the risk of falls caused by muscle impairment, but they might be compromised in DM1 due to brain dysfunction. There is a need for clinical assessment of cerebellum deficits and their contribution to impaired balance and frequent stumbles and falls in DM1. The cerebellum may also participate in DM1 through non-motor functions. Cerebellar lesions can result in executive dysfunction, blunting or flattening of affect, constrictions in social interaction, and impaired spatial cognition (Schmahmann and Sherman, 1998). Defective Bergmann or Purkinje cell communication could mediate, at least partly, similar cognitive and behavioral deficits previously reported in DM1, but further studies are required.
In summary, our data provide insight into DM1 brain mechanisms and demonstrate how glial molecular abnormalities affect neuronal activity through neuroglial miscommunication. They open the route to the clinic, providing exciting therapeutic perspectives through the modulation of GLT1 levels and glutamate signaling. Therapies aiming to restore GLT1 protein and glutamate neurotransmission could have applicability in DM1.
EXPERIMENTAL PROCEDURES
Transgenic Mice
All animal experiments were conducted according to the ARRIVE guidelines (Animal Research: Reporting In Vivo Experiments). This project has been conducted with the authorization for animal experimentation No. 75 003 in the animal facility with the approval No. B 91 228 107, both delivered by Prefecture de Police and the French Veterinary Department.
Human Tissue Samples
Mouse cerebellum tissues were microdissected at different ages and stored at −80°C. Human cerebellum samples were collected from different laboratories: Dr. Yasuhiro Suzuki (Asahikawa Medical Center) and Dr. Tohru Matsuura (Okayama University). All experiments using human samples were approved by the ethics committees of the host institutions. Written informed-consent specimen use for research was obtained from all patients. Information relative to patients was previously described (Hernández-Hernández et al., 2013a) and is summarized in Tables S1 and S2.
Statistical Analysis
Statistical analyses were performed with Prism (GraphPad), SPSS (v.14.0, SPSS), Statistica (v.6.0, StatSoft), and/or Excel software. When two groups were compared, we first performed a normality test. Parametric data were compared using a two-tailed Student’s t test (with equal or unequal variance, as appropriate). Non-parametric data were compared using a two-tailed Mann-Whitney U test. For one-way ANOVA, if statistical significance was achieved, we performed post-test analysis to account for multiple comparisons. Statistical significance was set at p < 0.05. The data are presented as mean ± SEM.
Supplementary Material
Highlights.
Bergmann glia show marked RNA toxicity in the cerebellum of DM1 mice and patients
DM1 mice show reduced motor coordination associated with Purkinje cell hyperexcitability
GLT1 is downregulated in astrocytes, causing glutamate neurotoxicity
GLT1 upregulation rescues excitotoxicity, Purkinje firing, and motor coordination
ACKNOWLEDGMENTS
We thank Dr. Thomas Cooper for providing the DMPKS, DT960, CELF1, and CELF2 plasmids; Dr. Nicolas Reyes for the GLT1-EGFP-expressing plasmid; Dr. Rob Willemsen for FXTAS mouse brain slices; and Dr. Jeffrey Rothstein for anti-GLT1 antibody. We are grateful to the personnel of CERFE (Centre d’Exploration et de Recherche Fonctionelle Expérimentale, Genopole, Evry, France) and LEAT (Laboratoire d’Experimentation Animale, Imagine Institute, Paris, France) for attentively caring for the mice. We thank Léonard Bertrand and Elodie Dandelot for help with the graphical abstract. This study was supported by grants from AFM-Téléthon (France, project grant 16161 to M.G.-P.), INSERM (France), Université Paris Descartes (France), and Fondation ARC (France); as well as PhD fellowships from Ministére Français de la Recherche et Technologie (France, to G.S. and D.M.D.), AFM-Téléthon (France, to G.S.) and Imagine Foundation (France, to S.O.B.). This program received a state subsidy managed by the National Research Agency under the “Investments for the Future” program bearing the reference ANR-10-IAHU-01 and under the program ANR-10BLAN-1121-01. L.S., G.C., G.G., and M.G.-P. have a patent on GLT1 upregulation in DM1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.06.006.
REFERENCES
- Algalarrondo V, Wahbi K, Sebag F, Gourdon G, Beldjord C, Azibi K, Balse E, Coulombe A, Fischmeister R, Eymard B, et al. (2015). Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul. Disord 25, 308–320. [DOI] [PubMed] [Google Scholar]
- Angeard N, Gargiulo M, Jacquette A, Radvanyi H, Eymard B, and Héron D (2007). Cognitive profile in childhood myotonic dystrophy type 1: is there a global impairment? Neuromuscul. Disord 17, 451–458. [DOI] [PubMed] [Google Scholar]
- Angeard N, Jacquette A, Gargiulo M, Radvanyi H, Moutier S, Eymard B, and Héron D (2011). A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul. Disord 21, 468–476. [DOI] [PubMed] [Google Scholar]
- Antonini G, Soscia F, Giubilei F, De Carolis A, Gragnani F, Morino S, Ruberto A, and Tatarelli R (2006). Health-related quality of life in myotonic dystrophy type 1 and its relationship with cognitive and emotional functioning. J. Rehabil. Med 38, 181–185. [DOI] [PubMed] [Google Scholar]
- Bachasson D, Moraux A, Ollivier G, Decostre V, Ledoux I, Gidaro T, Servais L, Behin A, Stojkovic T, Hébert LJ, et al. (2016). Relationship between muscle impairments, postural stability, and gait parameters assessed with lower-trunk accelerometry in myotonic dystrophy type 1. Neuromuscul. Disord 26, 428–435. [DOI] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, and Swanson MS (2014). Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 56, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearzatto B, Servais L, Cheron G, and Schiffmann SN (2005). Age dependence of strain determinant on mice motor coordination. Brain Res. 1039, 37–42. [DOI] [PubMed] [Google Scholar]
- Bearzatto B, Servais L, Roussel C, Gall D, Baba-Aïssa F, Schurmans S, de Kerchove d’Exaerde A, Cheron G, and Schiffmann SN (2006). Targeted calretinin expression in granule cells of calretinin-null mice restores normal cerebellar functions. FASEB J. 20, 380–382. [DOI] [PubMed] [Google Scholar]
- Bellamy TC (2006). Interactions between Purkinje neurones and Bergmann glia. Cerebellum 5, 116–126. [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. [DOI] [PubMed] [Google Scholar]
- Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, and Sergeant N (2014). Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front. Mol. Neurosci 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliandro P, Silvestri G, Padua L, Bianchi ML, Simbolotti C, Russo G, Masciullo M, and Rossini PM (2013). fNIRS evaluation during a phonemic verbal task reveals prefrontal hypometabolism in patients affected by myotonic dystrophy type 1. Clin. Neurophysiol 124, 2269–2276. [DOI] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. (2012). Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheron G, Gall D, Servais L, Dan B, Maex R, and Schiffmann SN (2004). Inactivation of calcium-binding protein genes induces 160 Hz oscillations in the cerebellar cortex of alert mice. J. Neurosci 24, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheron G, Servais L, Wagstaff J, and Dan B (2005). Fast cerebellar oscillation associated with ataxia in a mouse model of Angelman syndrome. Neuroscience 130, 631–637. [DOI] [PubMed] [Google Scholar]
- Cheron G, Servais L, and Dan B (2008). Cerebellar network plasticity: from genes to fast oscillation. Neuroscience 153, 1–19. [DOI] [PubMed] [Google Scholar]
- Cheron G, Sausbier M, Sausbier U, Neuhuber W, Ruth P, Dan B, and Servais L (2009). BK channels control cerebellar Purkinje and Golgi cell rhythmicity in vivo. PLoS ONE 4, e7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheron G, Dan B, and Márquez-Ruiz J (2013). Translational approach to behavioral learning: lessons from cerebellar plasticity. Neural Plast. 2013, 853654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan C, De Antonio M, Hamroun D, Varet H, Fabbro M, Rougier F, Amarof K, Arne Bes MC, Bedat-Millet AL, Behin A, et al. (2016). Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a nationwide multiple databases cross-sectional observational study. PLoS ONE 11, e0148264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Pereira M, Foiry L, Nicole A, Huguet A, Junien C, Munnich A, and Gourdon G (2007). CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet. 3, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernández Gómez FJ, Eddarkaoui S, Sergeant N, Buée L, Kimura T, et al. (2015). MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. 12, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarén E, Kjellby-Wendt G, Kowalski J, and Lindberg C (2014). Factors of importance for dynamic balance impairment and frequency of falls in individuals with myotonic dystrophy type 1—a cross-sectional study—including reference values of Timed Up & Go, 10m walk and step test. Neuromuscul. Disord 24, 207–215. [DOI] [PubMed] [Google Scholar]
- Hernández-Hernández O, Guiraud-Dogan C, Sicot G, Huguet A, Luilier S, Steidl E, Saenger S, Marciniak E, Obriot H, Chevarin C, et al. (2013a). Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 136, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Hernández O, Sicot G, Dinca DM, Huguet A, Nicole A, Buée L, Munnich A, Sergeant N, Gourdon G, and Gomes-Pereira M (2013b). Synaptic protein dysregulation in myotonic dystrophy type 1: disease neuropathogenesis beyond missplicing. Rare Dis. 1, e25553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimori H, Morel L, Huth J, Lindemann L, Dulla C, Taylor A, Freeman M, and Yang Y (2013). Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet 22, 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet A, Medja F, Nicole A, Vignaud A, Guiraud-Dogan C, Ferry A, Decostre V, Hogrel JY, Metzger F, Hoeflich A, et al. (2012). Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8, e1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, and Swanson MS (2003). A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- Kanai Y, and Hediger MA (2004). The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 447, 469–479. [DOI] [PubMed] [Google Scholar]
- Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, et al. (2011). Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J. Cell. Physiol 226, 2484–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, Knapska E, Filipkowski RK, Kaczmarek L, and Danysz W (2008). Behavioral characterization of GLT1 (+/−) mice as a model of mild glutamatergic hyper-function. Neurotox. Res 13, 19–30. [DOI] [PubMed] [Google Scholar]
- Kong Q, Chang LC, Takahashi K, Liu Q, Schulte DA, Lai L, Ibabao B, Lin Y, Stouffer N, Das Mukhopadhyay C, et al. (2014). Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Invest 124, 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, and Rothstein JD (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602. [DOI] [PubMed] [Google Scholar]
- Meola G, and Sansone V (2007). Cerebral involvement in myotonic dystrophies. Muscle Nerve 36, 294–306. [DOI] [PubMed] [Google Scholar]
- Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, Schneider-Gold C, Betz RC, Helmstaedter C, Tittgemeyer M, et al. (2011). The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 134, 3530–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A, Goodwin M, and Swanson MS (2014). RNA-protein interactions in unstable microsatellite diseases. Brain Res. 1584, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaite PA, Kuntzer T, Gourdon G, Lobrinus JA, and Barakat-Walter I (2013). Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis. Model. Mech 6, 622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, and Rothstein JD (2007). Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J. Neurosci 27, 6607–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, et al. (2005). Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, and Sherman JC (1998). The cerebellar cognitive affective syndrome. Brain 121, 561–579. [DOI] [PubMed] [Google Scholar]
- Schneider-Gold C, Bellenberg B, Prehn C, Krogias C, Schneider R, Klein J, Gold R, and Lukas C (2015). Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS ONE 10, e0130352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra L, Silvestri G, Petrucci A, Basile B, Masciullo M, Makovac E, Torso M, Spanò B, Mastropasqua C, Harrison NA, et al. (2014). Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol. 71, 603–611. [DOI] [PubMed] [Google Scholar]
- Serra L, Petrucci A, Spanò B, Torso M, Olivito G, Lispi L, Costanzi-Porrini S, Giulietti G, Koch G, Giacanelli M, et al. (2015). How genetics affects the brain to produce higher-level dysfunctions in myotonic dystrophy type 1. Funct. Neurol 30, 21–31. [PMC free article] [PubMed] [Google Scholar]
- Serra L, Cercignani M, Bruschini M, Cipolotti L, Mancini M, Silvestri G, Petrucci A, Bucci E, Antonini G, Licchelli L, et al. (2016). “I know that you know that I know”: neural substrates associated with social cognition deficits in DM1 patients. PLoS ONE 11, e0156901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais L, and Cheron G (2005). Purkinje cell rhythmicity and synchronicity during modulation of fast cerebellar oscillation. Neuroscience 134, 1247–1259. [DOI] [PubMed] [Google Scholar]
- Servais L, Hourez R, Bearzatto B, Gall D, Schiffmann SN, and Cheron G (2007). Purkinje cell dysfunction and alteration of long-term synaptic plasticity in fetal alcohol syndrome. Proc. Natl. Acad. Sci. USA 104, 9858–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H, Junien C, and Gourdon G (2000). Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet 9, 1185–1194. [DOI] [PubMed] [Google Scholar]
- Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, et al. (2001). Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet 10, 2717–2726. [DOI] [PubMed] [Google Scholar]
- Sicot G, and Gomes-Pereira M (2013). RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 1832, 1390–1409. [DOI] [PubMed] [Google Scholar]
- Sicot G, Gourdon G, and Gomes-Pereira M (2011). Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum. Mol. Genet 20 (R2), R116–R123. [DOI] [PubMed] [Google Scholar]
- Sistiaga A, Urreta I, Jodar M, Cobo AM, Emparanza J, Otaegui D, Poza JJ, Merino JJ, Imaz H, Martí-Massó JF, and López de Munain A (2010). Cognitive/personality pattern and triplet expansion size in adult myotonic dystrophy type 1 (DM1): CTG repeats, cognition and personality in DM1. Psychol. Med 40, 487–495. [DOI] [PubMed] [Google Scholar]
- Takado Y, Terajima K, Ohkubo M, Okamoto K, Shimohata T, Nishizawa M, Igarashi H, and Nakada T (2015). Diffuse brain abnormalities in myotonic dystrophy type 1 detected by 3.0 T proton magnetic resonance spectroscopy. Eur. Neurol 73, 247–256. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. [DOI] [PubMed] [Google Scholar]
- Theadom A, Rodrigues M, Roxburgh R, Balalla S, Higgins C, Bhattacharjee R, Jones K, Krishnamurthi R, and Feigin V (2014). Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology 43, 259–268. [DOI] [PubMed] [Google Scholar]
- Toth A, Lovadi E, Komoly S, Schwarcz A, Orsi G, Perlaki G, Bogner P, Sebok A, Kovacs N, Pal E, and Janszky J (2015). Cortical involvement during myotonia in myotonic dystrophy: an fMRI study. Acta Neurol. Scand 132, 65–72. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, and Wadiche JI (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci 8, 935–947. [DOI] [PubMed] [Google Scholar]
- Udd B, and Krahe R (2012). The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 11, 891–905. [DOI] [PubMed] [Google Scholar]
- Voogd J, and Glickstein M (1998). The anatomy of the cerebellum. Trends Cogn. Sci 2, 307–313. [DOI] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. (2012). Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber YG, Roebling R, Kassubek J, Hoffmann S, Rosenbohm A, Wolf M, Steinbach P, Jurkat-Rott K, Walter H, Reske SN, et al. (2010). Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 74, 1108–1117. [DOI] [PubMed] [Google Scholar]
- Wiles CM, Busse ME, Sampson CM, Rogers MT, Fenton-May J, and van Deursen R (2006). Falls and stumbles in myotonic dystrophy. J. Neurol. Neurosurg. Psychiatry 77, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Lim KO, Hemmy LS, and Day JW (2014). Tractography reveals diffuse white matter abnormalities in myotonic dystrophy type 1. J. Neurol. Sci 341, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.