
Figure 1. Mutation spectra of natural isolates of S. Cerevisiae.
Principal component analysis of segregating mutation spectrum variation from a subset of the 1011 yeast strains. (A). Mutation spectrum PCA of all natural variants under 50% derived allele frequency. Each strain’s mutation spectrum histogram is projected as a single point, colored to indicate its population of origin (Peter et al., 2018). The inset summarizes the loadings of the first and second principal component vectors. (B). Mutation spectrum PCA of rare variants (derived allele count 2–4). Singleton variants are excluded to minimize the impact of sequencing error. Strains appearing more than 1.8 standard deviations from the origin along both PC1 and PC2 are labeled with their strain names.

Figure 1—figure supplement 1. Mutation spectrum PCA after subsampling to avoid overlap between lineages.

To eliminate any clustering of strains as a result of shared variation, we randomly assigned each mutation of frequency k/n to one of the k haplotypes carrying the derived allele, then computed each strain’s spectrum based on these resampled mutation counts. The results still show similar clustering of outlier lineages to what is seen in Figure 1A without this subsampling, although with slightly more noise and dispersion.
Figure 1—figure supplement 2. PCA of synonymous variant mutation spectra.

To avoid the confounding effects of selection on nonsynonymous variants, we compute the spectrum of synonymous mutations present in each strain and normalize it by the spectrum of mutational opportunities for synonymous variants to arise. The resulting spectra display a similar PCA structure to what is seen in Figure 1A.
Figure 1—figure supplement 3. Mutation PCA using all variants stratified by triplet context.

This PCA shows substructure among the 96-dimensional mutation spectra obtained by classifying all variants by the left and right adjacent base pairs as well as the ancestral and derived alleles. Clustering is qualitatively similar to what is observed in Figure 1A using six-dimensional mutation spectra, classifying each variant by its ancestral and derived allele but no surrounding sequence context.
Figure 1—figure supplement 4. PCA of singleton mutation spectra.

This PCA clusters mutation spectra that were computed using only singletons: variants present in the focal strain and no other strains. It shows a similar structure to Figure 1B, where spectra were computed from nonsingleton rare variants.
Figure 1—figure supplement 5. Mutation spectrum comparison of natural variants versus de novo mutations from a previous mutation accumulation (MA) study.
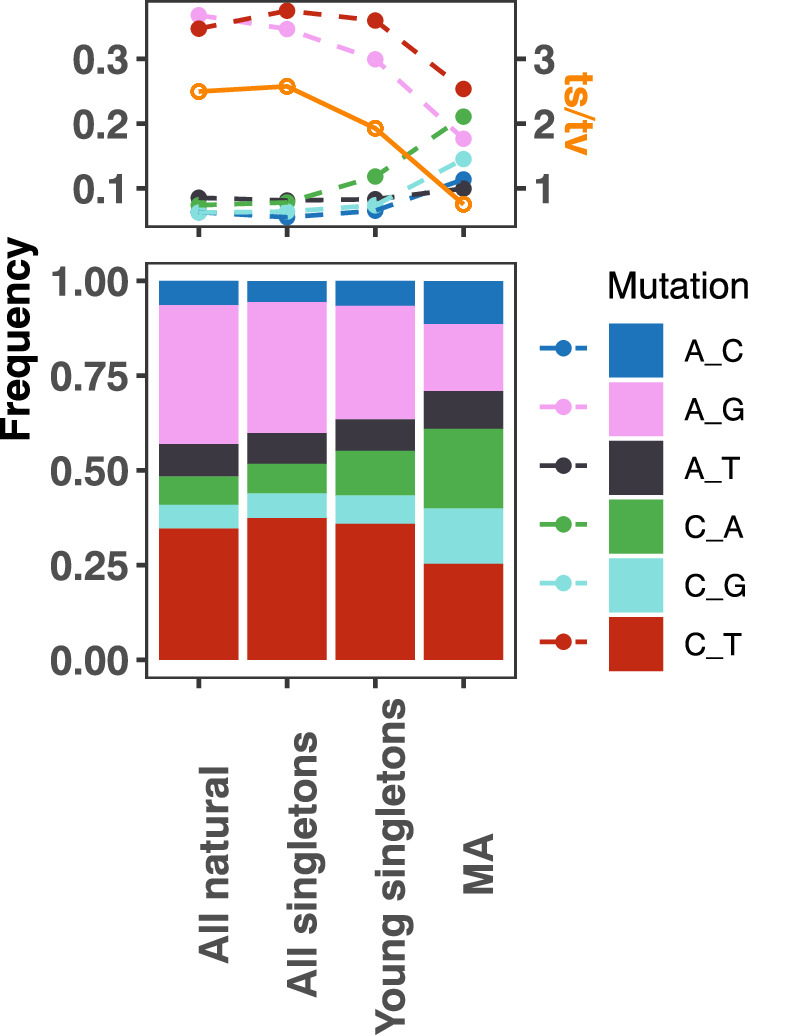
From left to right are mutation spectra from (1) all natural variants, (2) all singletons, (3) young singletons (see Materials and methods), and (4) MA experiments (Sharp et al., 2018, from haploids in RDH54+ backgrounds). These datasets are ordered such that mutations are expected to get younger from left to right. The top panel represents the frequency of each mutation type as a solid dot and the ts/tv ratio with an orange dot. The bottom panel shows the stacked frequency of each mutation type. Young singletons are defined similarly to the convention of Zhu et al., 2017 as having a density of less than 0.0087 count/kb.