

Abstract
Triple negative breast cancer (TNBC) is considered an early onset subtype of breast cancer that carries with it a poorer prognosis in young rather than older women for reasons that remain poorly understood. Hematopoiesis in the bone marrow becomes altered with age, and may therefore affect the composition of tumor-infiltrating hematopoietic cells and subsequent tumor progression. In this study, we investigated how age- and tumor-dependent changes to bone marrow-derived hematopoietic cells impact TNBC progression. Using multiple mouse models of TNBC tumorigenesis and metastasis, we found that a specific population of bone marrow cells upregulated CSF-1R and secreted the growth factor granulin (GRN) to support stromal activation and robust tumor growth in young mice. However, the same cell population in old mice expressed low levels of CSF1R and GRN, and failed to promote tumor outgrowth, suggesting that age influences the tumorigenic capacity of bone marrow cells in response to tumor-associated signals. Importantly, bone marrow cells from young mice were sufficient to activate a tumor-supportive microenvironment and induce tumor progression in old mice. These results indicate that hematopoietic age is an important determinant of TNBC aggressiveness and provide rationale for investigating age-stratified therapies designed to prevent the pro-tumorigenic effects of activated bone marrow cells.
Keywords: triple-negative breast cancer, age, metastasis, tumor microenvironment, hematopoietic cells
INTRODUCTION
Aging is associated with increased breast cancer risk (1), while young age at diagnosis is associated with higher recurrence rates (2). Triple-negative breast cancer (TNBC) accounts for ~15% of breast cancer cases and carries with it poor prognosis due to lack of targeted therapies and limited success of chemotherapy (3). Older women with TNBC have better outcome than younger women for unknown reasons (4). Nevertheless, older patients tend to forego treatment due to co-morbidities, resulting in a poor outcome for these patients (5). Complicating our understanding of TNBC progression with age is the fact that although women over the age of 60 represent nearly half of new breast cancer cases (6), only 10% of patients enrolled in clinical trials are over the age of 65 (7). Treatment of TNBC patients therefore presents age-specific challenges that might be overcome if more were understood about the disease.
The prevailing view that accumulation of DNA damage over time translates into age-associated increased cancer risk (8) does not explain the fact that TNBC incidence is relatively unchanged with age (4). It is likely that age-related changes to the non-neoplastic cells within the tumor microenvironment (TME) also influence disease progression. Indeed, aging has multiple effects on tissue homeostasis that have either pro- or anti-tumorigenic consequences (9–13). Nevertheless, very little is known about the aging TNBC microenvironment.
The bone marrow is an important source of hematopoietic cells that comprise the TME (14) and one of the major organs affected by systemic signaling in cancer (15). It is now well established that hematopoiesis is altered with age (16,17), leading to altered effector cell production and changes in lineage potential (18). Given the importance of bone marrow cells (BMCs) to tumor progression (19), surprisingly little is known about how aging affects tumor-infiltrating BMCs.
Here, we investigated how age affects the composition and function of pro-tumorigenic BMCs and their impact on TNBC progression. Consistent with clinical observations, we found that TNBC was more aggressive in young mice than in old mice. We identified age- and tumor-dependent molecular and functional changes to hematopoietic cells that impacted TNBC progression. Importantly, BMCs from young mice with TNBC were sufficient to activate a supportive TME and stimulate tumor progression in old mice. The results presented here indicate that hematopoietic age is an important determinant of TNBC progression.
MATERIALS AND METHODS
Cell lines
HMLER-HR and BPLER transformed human mammary epithelial tumor cells were generated and maintained as described (20,21). MCF7Ras cells (22) and GFP+ immortalized human mammary fibroblasts (hMFs) were a generous gift from Dr. Robert Weinberg (Whitehead Institute, Cambridge, MA, USA) and maintained in DMEM with 10% fetal calf serum (Hyclone), and penicillin-streptomycin (Sigma). All cell lines were validated as mycoplasma-negative and all appropriate human cell lines authenticated at the Molecular Diagnostics Laboratory at Dana Farber Cancer Center.
Animals and Tumor Xenografts
Female NCR-NU (nude) were purchased from Taconic and FVB/NJ mice were purchased from Taconic and Jackson Labs (stock no. 001800). All experiments were conducted in accordance with regulations of the Children’s Hospital Institutional Animal Care and Use Committee (protocol 12-11-2308R) and MIT Committee on Animal Care protocol (1005-076-08). Old mice were defined as >10 months and young mice were defined as < 26 weeks at the start of any given experiment. Tumor cells were prepared in 20% Matrigel (BD Biosciences) and injected subcutaneously into mice (see Supplemental Information for cell numbers). Tumors were measured using calipers with volume calculated as 4/3*π*(radius3).
Generation and expression profiling of CAFs
GRN-dependent CAFs and GRN-independent CAFs were generated as published (23,24), respectively. For gene expression analyses, total RNA was isolated from CAFs using standard procedures (Qiagen) and hybridized to Affymetrix HG-U133A plus 2 arrays (samples run in triplicate). See Supplemental Information for details. Full data sets were deposited online: GEO GSE75333.
Flow cytometric analysis and sorting
BMCs were isolated as reported (25) and suspended in sterile PBS containing 2% FCS and 0.01% NaN3. FcγR II/III receptors were blocked using anti-CD16/32, and cells labeled with 7-AAD (BioLegend) and appropriate antibodies for 30 min at 4°C. Sca1+/cKit- cells were analyzed and/or isolated from total bone marrow using Canto II or FACSAria IIu/FACSDiva (BD Biosciences). Analyses were performed using FlowJo (TreeStar). Antibody details are listed in Table S3.
Immunohistochemistry and microscopy
Dissected tissues were fixed in 4% paraformaldehyde for 24 hours, stored in 70% ethanol for 24 hours, then embedded in paraffin and sectioned onto ProbeOn Plus microscope slides (Fisher Scientific). Sectioning and hematoxylin and eosin (H&E) stains were performed by the DF/HCC Research Pathology Core at Brigham and Women’s Hospital. Immunohistochemistry was performed as previously described (25). Images were captured with identical exposure and gain using a Nikon Eclipse Ni microscope and quantified using CellProfiler as previously described (23). Antibody details and dilutions are listed in Table S3.
Real-time quantitative reverse transcription PCR
Total RNA was isolated from sorted BMCs with Trizol reagent (Invitrogen), purified using RNeasy Mini Kit (Qiagen) and 100 ng mRNA reverse transcribed (ProtoScript AMV First Strand cDNA Synthesis Kit; New England Biolabs). Taqman Master Mix (Applied Biosystems) was used to develop real-time PCR for GRN with an ABI 7900HT quantitative-PCR system (Applied Biosystems), according to the manufacturers’ instructions. Samples were analyzed in triplicate for a minimum of 3 animals per group. Values were normalized to GAPDH expression and analyses performed using the ΔΔCt method. Primer details are listed in Table S4.
Human and mouse osteopontin ELISA
Whole blood was collected in EDTA coated tubes (VWR) and centrifuged at 1.5g for 8 minutes to isolate ~200 μl of plasma per mouse. Human or mouse plasma osteopontin concentrations were determined by ELISA according to manufacturer’s instructions (R&D) and analyzed using an ABI 7900HT plate reader (Applied Biosciences).
Bone marrow cell gene expression analysis
RNA was obtained using RNeasy Plus Micro Kit (Qiagen); 500 pg mRNA was used to generate biotinylated cRNA according to the Nugen Ovation Pico WTA system. Following fragmentation, 5 μg of cRNA were hybridized to the Affymetrix GeneChip Mouse Gene 2.0 ST microarrays and chips scanned using the GeneChip Scanner 3000 7G. See Supplemental Information and GEO GSE74120 for microarray analyses.
Statistical analyses
Data are represented as mean ± SEM and analyzed by one-way ANOVA and/or Student’s t-test, with Kaplan Meier curves for survival analysis using GraphPad Prism 6.0, unless otherwise stated. P < 0.05 was considered statistically significant.
RESULTS
Breast cancer growth in young and old mice
In order to investigate the effects of aging on host factors that support cancer progression, we established models that mimic TNBC progression with age. We used human TNBC cells, termed BPLER, previously shown to form aggressively growing mammary tumors in young mice (21,26). When injected into young (8–10 weeks) and old (>10 months) nude mice, BPLER tumors showed delayed onset (~16 days) and slower growth kinetics in the old cohort (Fig. 1A). At the experimental end point (45d), tumors from both cohorts formed with 100% incidence; however, the tumors from old mice were ~4-fold smaller (Fig. 1A).
Figure 1. Model of age-dependent triple-negative and luminal breast cancer.
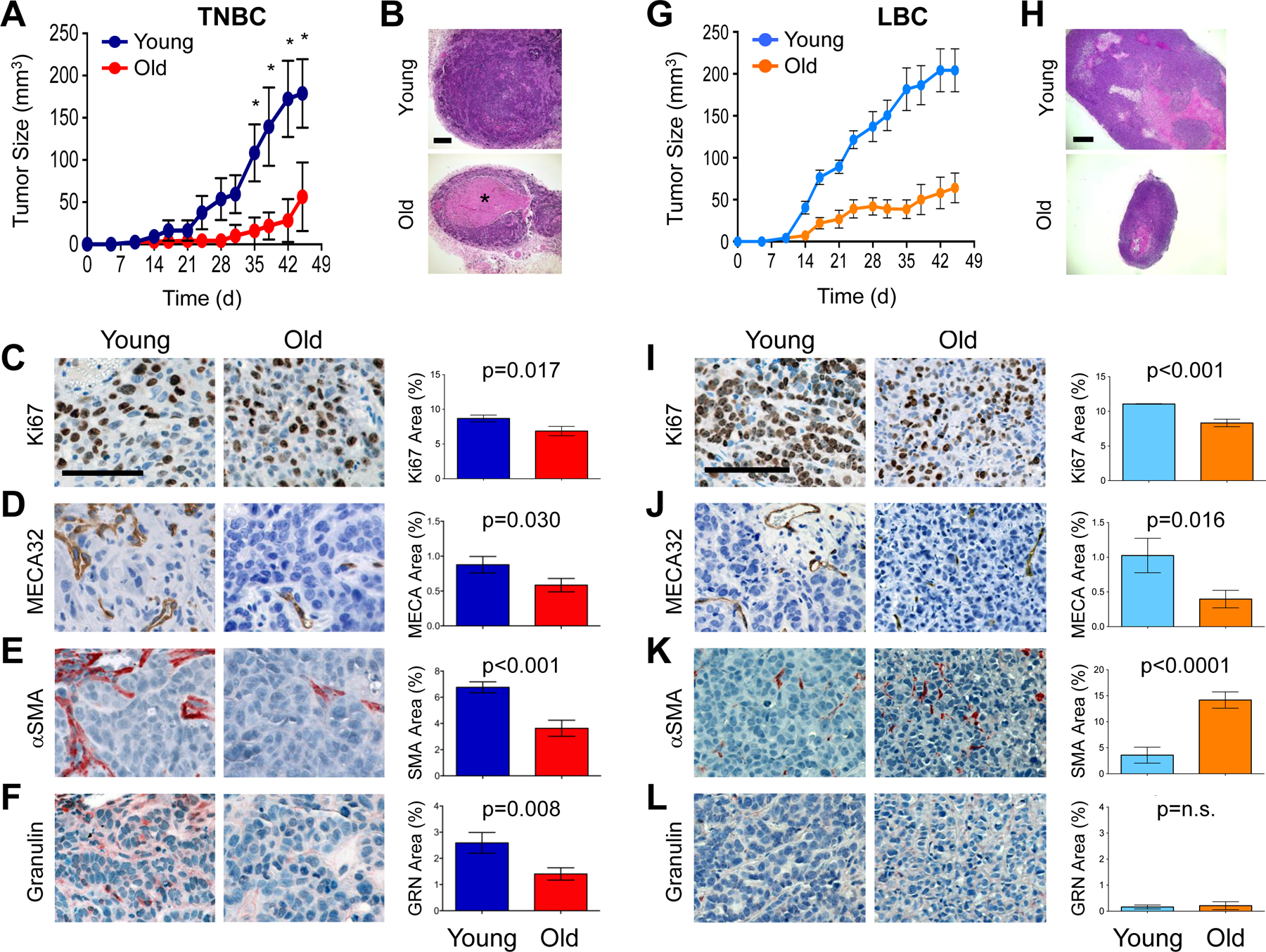
(A) Human BPLER breast tumor volume and growth rate in young (blue) and old (red) nude mice (n=5 per cohort). (B) Representative H&E images of BPLER tumor sections (from A). *=Necrotic area. Scale bar=200μm. (C-F) BPLER tumor serial sections from young and old mice stained for: Ki67 (C; brown), MECA32 (D; brown), αSMA (E; red) and Granulin (F; red). Cell nuclei counterstained with hematoxylin (blue). Scale bar=100μm. Graphs represent CellProfiler quantification of staining for indicated antigens. ~4–5 image fields per tumor with 4–5 tumors per group were analyzed (Ki67 and MECA32 n=28 per cohort; αSMA n=28 images for young, n=22 images for old; GRN n=15 images for young, n=16 images for old). (G) Human MCF7Ras breast tumor volume and growth rate in young (blue) and old (orange) nude mice (n=10 per cohort) (H) Representative H&E images of MCF7Ras tumor sections (from G). Scale bar=500μm. (I-L) MCF7Ras tumor serial sections from young and old mice stained for: Ki67 (I; brown), MECA32 (J; brown), αSMA (K; red) and Granulin (L; red). Cell nuclei counterstained with hematoxylin (blue). Scale bar=100μm. Graphs represent CellProfiler quantification of staining for indicated antigens. ~3–4 image fields per tumor with 3–4 tumors per group were analyzed. Values represent mean ± SEM and data were analyzed using Student’s t-test for significance.
Histopathologically, TNBC tumors from young mice appeared much like advanced adenocarcinoma, while tumors from old mice displayed areas of edema and necrosis; necrosis was visible in only 20% of the tumors from young mice but in 80% of those from old mice (Fig. 1B). Consistent with those observations, the tumors from young mice had a higher mitotic index (p=0.001) (Fig. 1C).
We next assessed the TME by immunohistochemistry and image analysis, excluding necrotic areas. Positive staining for mouse endothelial cell antigen (MECA32) was significantly lower (p=0.03) in the tumors from old mice (Fig. 1D). Tumor vascularization is facilitated by cancer-associated fibroblasts (myofibroblasts; CAFs) (22), which are alpha-smooth muscle actin (αSMA)-positive and associated with poor prognosis (27). Positive staining for αSMA was over 50% lower (p<0.001) in the tumors from old mice than those from young mice (Fig. 1E). Since blood vessel-associated pericytes also stain positively for αSMA, we compared the MECA32 and αSMA staining in serial sections and concluded that αSMA labeled distinct populations of myofibroblasts in addition to pericytes (Fig. 1D, E).
Various factors expressed by cells within the tumor microenvironment are known to activate tumor-supportive myofibroblasts (14,28). In particular, the hematopoietic cell derived secreted growth factor, granulin (GRN), induces formation of αSMA-rich stroma and supports outgrowth of indolent tumors in mouse models of TNBC (23). Therefore, we examined tumors for infiltration of GRN-positive cells and found that tumors from old mice had recruited significantly fewer (p=0.008) GRN+ cells than those from young mice (Fig. 1F).
The incidence of luminal breast cancer (LBC) is higher in older women, while the incidence of TNBC does not correlate with age; nevertheless, both subtypes are more aggressive in younger women (4,6). Therefore, we also tested a model of MCF7Ras LBC. Consistent with clinical observations, LBC tumors grew more robustly in young mice (Fig. 1G, H), were significantly more proliferative (p<0.001), and had higher microvessel density (p=0.016) than their old counterparts (Fig. 1I, J). Unlike TNBC, LBC tumors had increased SMA+ coverage with age (p<0.0001), and GRN expression was negligible in both young and old mice (Figs. 1K, L).
Collectively, these results indicated that age significantly influences tumor latency, growth kinetics, histopathology, and microenvironment in these models of TNBC and LBC. Importantly, these findings were consistent with clinical observations that GRN expression is associated with TNBC but not LBC (23), and suggested that GRN expression may be a function of age in the context of TNBC.
Triple-negative breast cancer progression in old mice
We next wanted to gain insights into recurrence rate differences that are commonly observed between young and older TNBC patients (2). We therefore used a model of TNBC progression that mimics the early phases of metastasis when patients harbor indolent disseminated tumor cells in the periphery at the time of their primary diagnosis. Using this model, we reported that HMLER-HR cells (weakly tumorigenic breast cancer cells oncotype-matched to BPLER cells) (29), which otherwise do not form lung or subcutaneous tumors in cancer-free mice, form adenocarcinomas when implanted into mice bearing distant BPLER tumors (25). Given the observed impact of host age on tumor growth and disease histopathology, we hypothesized that the ability of BPLER tumors to systemically instigate outgrowth of distant tumors might be impaired in old mice.
To test our hypothesis, we injected HMLER-HR cells (“distant tumors”) contralaterally to BPLER tumors (“primary tumors”) in either young or old mice (Fig. 2A). We reasoned that primary tumor burden should be comparable among cohorts in order to make conclusions about systemic stimulation of distant tumor formation. Hence, we harvested tissues at different time points (28–92 d in young; 45–98 d in old) in order to obtain primary tumors of equivalent average size in both young (96.5 ± 18.1 mm3) and old (114.9 ± 13.9 mm3) cohorts (Fig. 2B, S1A).
Figure 2. Systemic promotion of tumor progression is attenuated in old mice.
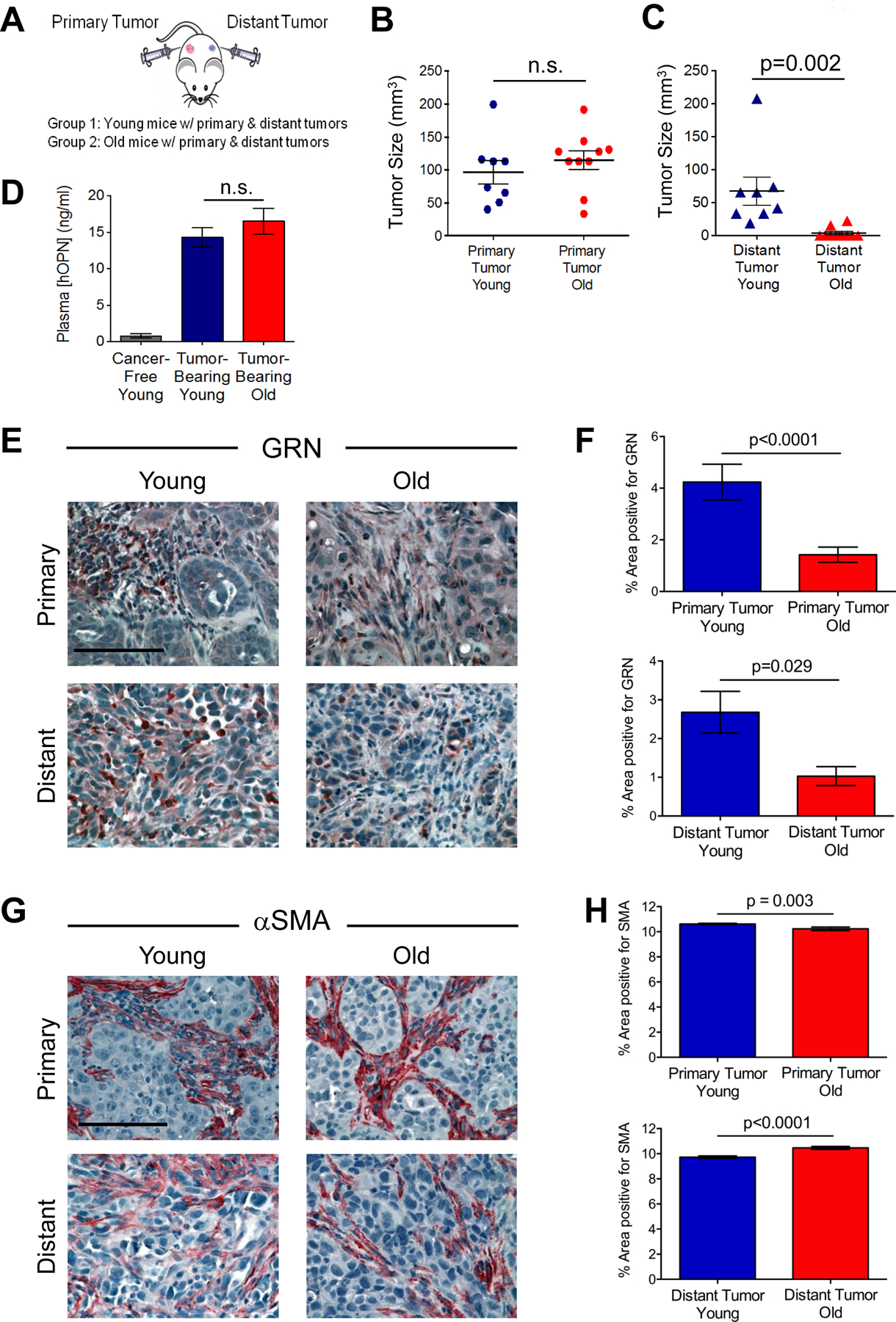
(A) Experimental design to test cancer progression in young and old mice. Distant HMLER-HR tumor cells were injected into young and old mice bearing primary BPLER tumors. Mice were sacrificed at various time points in order to obtain comparably sized BPLER primary tumors between young and old cohorts. (B) Size of primary BPLER tumors from young (n=8) and old (n=10) mice. (C) Size of distant HMLER-HR tumors from young and old mice bearing primary BPLER tumors. (D) Plasma levels of human tumor-derived OPN (hOPN) in young and old mice and cancer-free young mice as a control (n=6 per cohort). (E) Representative images of GRN (red) stained primary tumors (top) and distant tumors (bottom) from young and old mice. Cell nuclei counterstained with hematoxylin (blue); scale bar=100 μm. (F) Quantification of GRN-positive area as a percentage of total area (from E); n=22 images primary young; n=38 images primary old; n=28 images distant young; n=12 images distant old. (G) Representative images of αSMA (red) stained primary tumors (top) and distant tumors (bottom) from young and old mice. Cell nuclei counterstained with hematoxylin (blue); scale bar=100 μm. (H) Quantification of αSMA-positive area as a percentage of total area (from g); n=81 images primary young; n=57 images primary old; n=41 images distant young; n=21 images distant old. Data represent mean ± SEM and analyzed using Student’s t-test for significance. n.s. not significant. See also Figures S1–2.
The distant tumors in young mice formed with 100% incidence (8/8) when injected opposite primary TNBC tumors (Fig. 2C). However, despite similar primary tumor size, only 20% of the distant tumors (2/10) grew in the old cohort (Fig. 2C). The average size of the resulting distant tumors at the various experimental end points (Fig. S1A) was significantly smaller in the old mice (~3.8 mm3) than in the young mice (~68 mm3) (Fig. 2C). We made similar observations when we conducted an experiment over the course of 70 days (Fig. S2A–C).
TNBC systemic instigation of distant tumors is dependent on secretion of the cytokine osteopontin (OPN) into the circulation by TNBC primary tumors (25). Indeed, elevated OPN plasma levels are correlated with poor prognosis (30). In order to determine whether size-matched primary TNBC tumors had equal potential to systemically instigate growth of distant tumors in young and old mice, we measured circulating levels of human and mouse OPN (hOPN and mOPN, respectively). Plasma levels of host mOPN were not significantly different between cancer-free and tumor-bearing young or old mice (Fig. S1B). As expected, plasma levels of hOPN were negligible in young cancer-free mice (Fig. 2D). Importantly, there were no differences in the plasma levels of tumor-secreted hOPN between young (14.3 ± 1.3 ng/ml) and old (16.5 ± 1.8 ng/ml) tumor-bearing mice (Fig. 2D), leading us to conclude that the primary TNBC tumors should have had equal instigating potential in young and old mice.
We next analyzed primary and distant tumors for evidence of systemically acting factors that are known to impact the microenvironment during TNBC progression (15). Hence, we first stained tumors for GRN, which had appeared to be affected by age in our earlier experiments (Fig. 1F). While GRN-positive staining was abundant in both primary and distant tumors from young mice, these levels were ~75% lower in the primary tumors and ~60% lower in the distant tumors of the old mice (Fig. 2E, F).
Since GRN induces formation of reactive stroma (23), we stained tumor sections for αSMA. Unlike the primary BPLER tumors observed in our earlier experiments (Fig. 1), αSMA-positive staining in these size-matched primary tumors was only modestly (although significantly) reduced in the old mice (Fig. 2G, H). Hence, a desmoplastic stroma eventually developed in primary tumors from old mice. In the distant tumors, we observed a subtle yet significant increase in αSMA staining in the tumors from old mice relative to those from young mice (Fig. 2G, H).
Interestingly, the distant tumor indolence observed in old mice occurred specifically in the context of TNBC. HMLER-HR cells injected contralaterally to a skin wound formed tumors efficiently in both young (6/8 injections) and old (4/8 injections) mice, with no significant difference in average tumor volume observed after 28 days (Fig. S2D, E). Likewise, systemic instigation of distant tumors was more effective in old mice with LBC than in those with TNBC (Fig. S2F). As we had observed in our earlier experiments (Fig. 1), distant tumor progression in old mice with LBC appeared to occur in a GRN-independent manner (Fig. S2G). Thus, when provided with pro-tumorigenic systemic conditions such as those stimulated by LBC or wound healing, distant HMLER-HR tumors can grow more efficiently in old mice.
Collectively, these results indicated that the ability of TNBC primary tumors to drive outgrowth of distant tumors was severely impaired in old mice. Moreover, histopathological observations suggested phenotypic differences in stromal components not only between young and old mice with TNBC, but also between different breast cancer subtypes.
Microenvironment of distant tumors from young and old mice
The fact that the distant tumor microenvironment from old hosts with TNBC was abundant with myofibroblasts yet displayed significantly lower GRN positivity suggested that stromal desmoplasia had occurred in a GRN-independent fashion in this cohort. Indeed, CAFs are heterogeneous and arise via different mechanisms (27,31).
We previously reported a gene expression signature of GRN-treated normal human mammary fibroblasts that is similar to CAFs isolated from patient breast adenocarcinomas; these expression signatures were generated by comparing normal to GRN-treated fibroblasts (23). Here, we wished to know whether gene expression signatures differed between GRN-dependent and GRN-independent CAFs. Hence, we prepared GRN-dependent CAFs by treating normal human mammary fibroblasts with GRN, and generated GRN-independent CAFs by exposing the same fibroblasts to MCF7Ras LBC tumor cells (24) (Fig. 3A). Importantly, both types of CAFs express αSMA and support primary tumor growth (22–24).
Figure 3. Age affects tumor stroma.
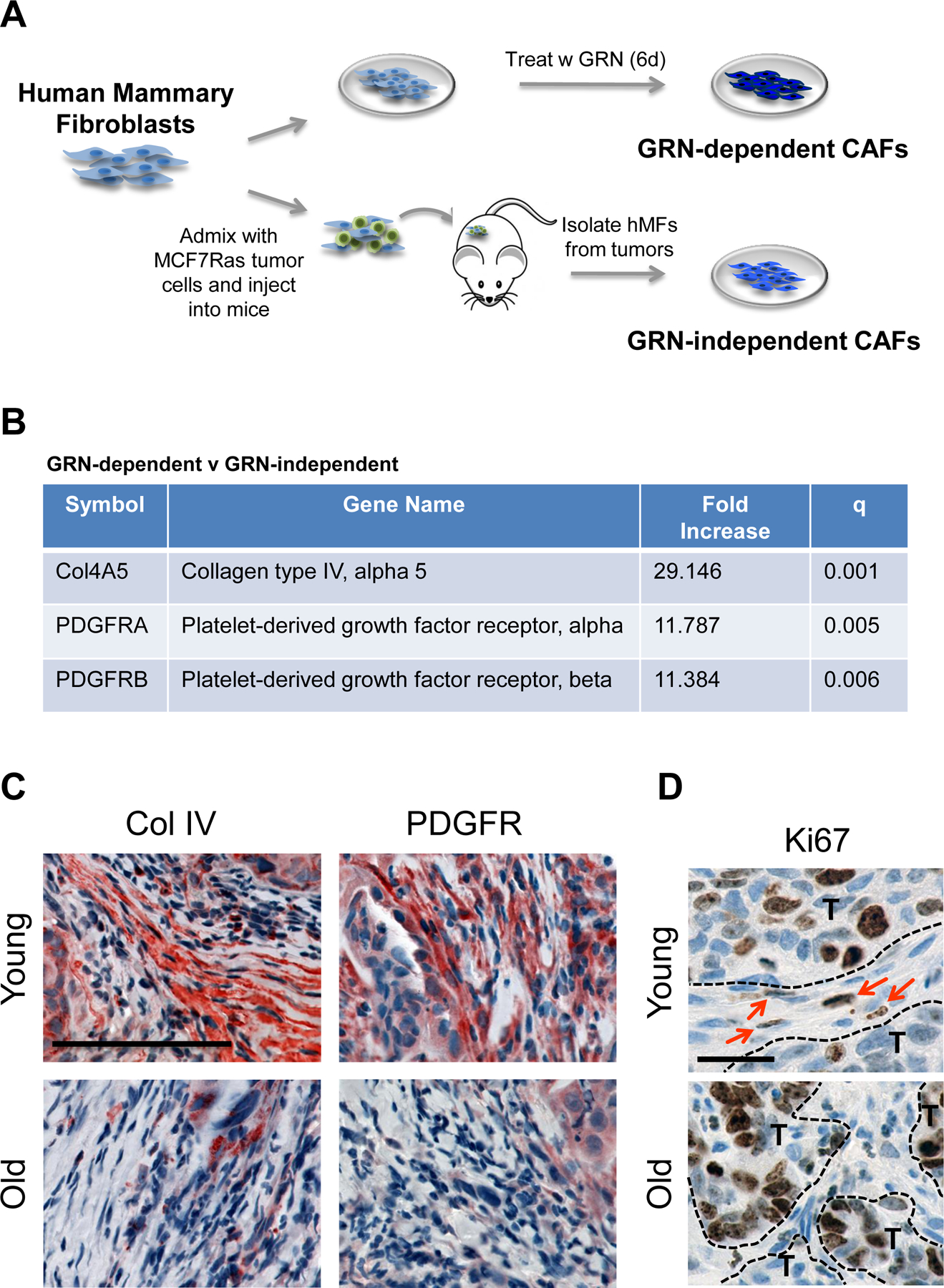
(A) Derivation of granulin (GRN)-dependent and GRN-independent cancer associated fibroblasts (CAFs) from normal human mammary fibroblasts (hMFs). (B) Table shows select genes upregulated in GRN-dependent v GRN-independent CAFs (GEO GSE75333) (C) Collagen IV (red, left) and PDGFR (red, right) staining in the distant tumors from young and old mice. Cell nuclei counterstained with hematoxylin. Scale bar=100 μm. (D) Ki67 (brown) staining in distant tumors from young and old mice; cell nuclei counterstained with hematoxylin. Dashed lines segregate tumor (T) from stroma; arrows indicate proliferating stromal cells. Scale bar=25 μm.
Gene expression profiling revealed significant differences between GRN-dependent and GRN-independent CAFs (GEO GSE75333). Most notably, the GRN-dependent CAFs expressed 29.1-fold higher levels of Collagen IV (Col IV alpha 5 isoform), 11.8-fold higher levels of platelet-derived growth factor receptor (PDGFR) A, and 11.4-fold higher levels of PDGFRB than their counterpart GRN-independent CAFs (Fig. 3B). Immunohistochemical analysis confirmed that both Col IV and PDGFR were more abundant in tumors from young mice than in those from old mice (Fig. 3C). Moreover, cells within the stromal compartment of tumors from young mice were proliferative, as measured by Ki67 staining, while those from old mice displayed no evidence of Ki67 positivity (Fig. 3D).
These findings were consistent with the notion that the CAFs present in TNBC tumors from young mice had formed in a GRN-dependent fashion, while those observed in tumors from old mice likely formed in a GRN-independent manner.
Age-dependent effects on pro-tumorigenic cells in the bone marrow
We further investigated the possibility that differences in reactive stroma between tumors from young and old mice with TNBC arose as a consequence of differential recruitment of GRN+ bone marrow derived cells (BMDCs). A subpopulation of Sca1+/cKit- hematopoietic progenitor cells expresses high levels of GRN and has been reported to infiltrate various tumors following mobilization from the bone marrow (32,33). In particular, these BDMCs promote formation of desmoplastic stroma and outgrowth of indolent tumors in mice with TNBC (23).
To determine if Sca1+/cKit- BMDCs were recruited into tumors in an age-dependent manner, we first stained distant tumors for Sca1 and GRN as hallmarks of these specialized BMDCs (Fig. S3A). While hematopoietic CD45+ cells were efficiently recruited to the distant tumors that formed in both young and old mice with TNBC (Fig. S3A), the numbers of Sca1+ cells were considerably higher in young mice than in old mice (Fig. 4A). Specifically, after normalizing for the number of Large-T antigen (LgT+) tumor cells, Sca1+ cells were ~9-fold more abundant in the tumors from young mice (Fig. 4A). Likewise, GRN+ cells were 3-fold more abundant in the tumors from young mice (Fig. 4A, S3A).
Figure 4. Age affects tumor-promoting hematopoietic bone marrow cells.
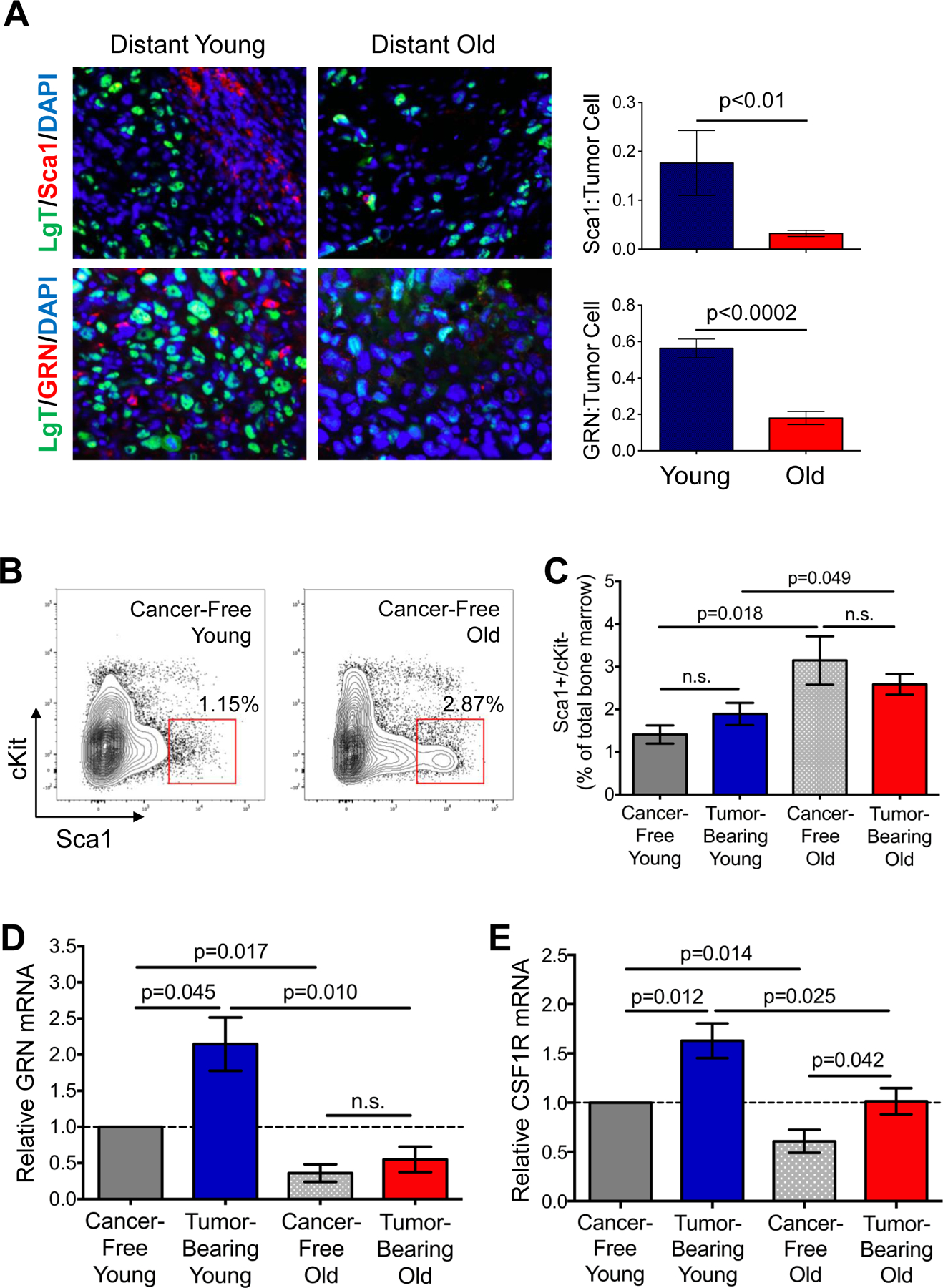
(A) Staining of distant tumors from young and old mice for LgT (green), Sca1 (red, top), or GRN (red, bottom). Cell nuclei counterstained with DAPI (blue). Graphs indicate number of Sca1+ cells or GRN+ cells recruited per LgT+ tumor cell. n=9 images for young; n=8 images for old. (B) Flow cytometry plots indicating gating strategy to isolate Sca1+/cKit- BMCs from cancer-free young (left) and old (right) mice. (C) Flow cytometric analysis of CD45+/Sca+/cKit- cells as a percent of total bone marrow in cancer-free and BPLER tumor-bearing young and old mice; n=4–6 per group. (D, E) Relative GRN (D) and CSF1R (E) mRNA in CD45+/Sca1+/cKit- BMCs isolated from indicated cohorts; values are relative to those for the identical cell population from cancer-free young mice (broken line); n=3 per group analyzed in triplicate. Data represented as mean ± SEM, analyzed using one-way ANOVA with Student’s t-test for significance; n.s.=not significant. See also Figure S3.
A number of reports have demonstrated that BMCs are rendered pro-tumorigenic in the marrow prior to their mobilization into the circulation and recruitment to tumors (19,25). Hence, we analyzed BMCs of cancer-free and TNBC tumor-bearing young and old mice. We found that the numbers of Sca1+/cKit- cells in the bone marrow were increased ~2-fold with age (p= 0.018; Fig. 4B, C). These findings were consistent with reports that Lin-/Sca1+/cKit- progenitor cells accumulate in the marrow of elderly humans and mice (34). Interestingly, TNBC did not significantly affect these numbers in either young or old mice (Fig. 4C).
Both age- and tumor-dependent associations became apparent when we assessed GRN expression in Sca1+/cKit- cells isolated from the marrow. First, GRN expression was increased ~2-fold in Sca1+/cKit- BMCs from young tumor-bearing mice compared to young cancer-free mice, consistent with our previous report (23) (p=0.045, Fig. 4D). Second, baseline levels of GRN in Sca1+/cKit-BMCs of cancer-free old mice were less than half that of their young counterparts (p=0.017; Fig. 4D). Interestingly, the levels of GRN did not increase upon tumor development in old mice as they had in the young cohort (Fig. 4D).
We observed similar age-dependent increases in numbers of Sca1+/cKit- BMCs, and decreases in GRN expression in these BMCs, in an immunocompetent Her2/Neu breast cancer mouse model (Fig. S3B,C). However, unlike these mice or the mice with TNBC, GRN expression in Sca1+/cKit- BMCs was not different between young and old mice with LBC tumors (Fig. S3D).
We also assessed whether the potential for Sca1+/cKit- BMC recruitment was impaired in old mice with TNBC, thus explaining why fewer of these cells were observed in tumors from old mice despite their enhanced numbers in the marrow. The CSF-1/CSF-1R axis is a predominant mechanism of hematopoietic cell mobilization and recruitment under normal and pathophysiological conditions (35–37). CSF-1R expression on the Sca1+/cKit- BMCs from old cancer-free mice was significantly reduced (p=0.014) relative to those from the young cancer-free mice (Fig. 4E). Significant increases in CSF-1R expression were observed in both young and old tumor-bearing mice when compared with their respective cancer-free counterparts (Fig. 4E). Nevertheless, these levels in tumor-bearing young mice significantly exceeded those of old tumor-bearing mice (p=0.025); in fact, CSF-1R expression levels in the BMCs of old mice with TNBC were comparable to those of the cancer-free young mice (Fig. 4E). No differences in Sca1+/cKit- BMC expression of CSF-1R were observed between young and old mice bearing LBC tumors (Fig. S3E).
Collectively, these results indicated that despite increased numbers of Sca1+/cKit- cells in the marrow of old cancer-free mice, these BMCs failed to respond to TNBC breast tumors that normally induce them to increase expression of GRN and CSF-1R in young mice.
Young BMCs rescue tumor growth and stromal desmoplasia in old mice
Our results suggested that the tumor-promoting function Sca1+/cKit- BMCs might be impaired in old mice. As noted earlier, Sca1+/cKit- BMCs are rendered pro-tumorigenic in the marrow prior to their mobilization; hence, BMCs harvested from the marrow can be tested for their tumor support function using in vivo assays (15). Therefore, we harvested BMCs from young and old mice bearing size-matched BPLER tumors (68.3 ± 18.3 mg for young and 75.0 ± 35.3 mg for old mice; p = n.s.), immediately admixed donor BMCs with indolent HMLER-HR cells, and injected the mixtures subcutaneously into either young or old recipient mice (Fig. 5A).
Figure 5. Bone marrow cells from young mice rescue tumor growth in old mice.
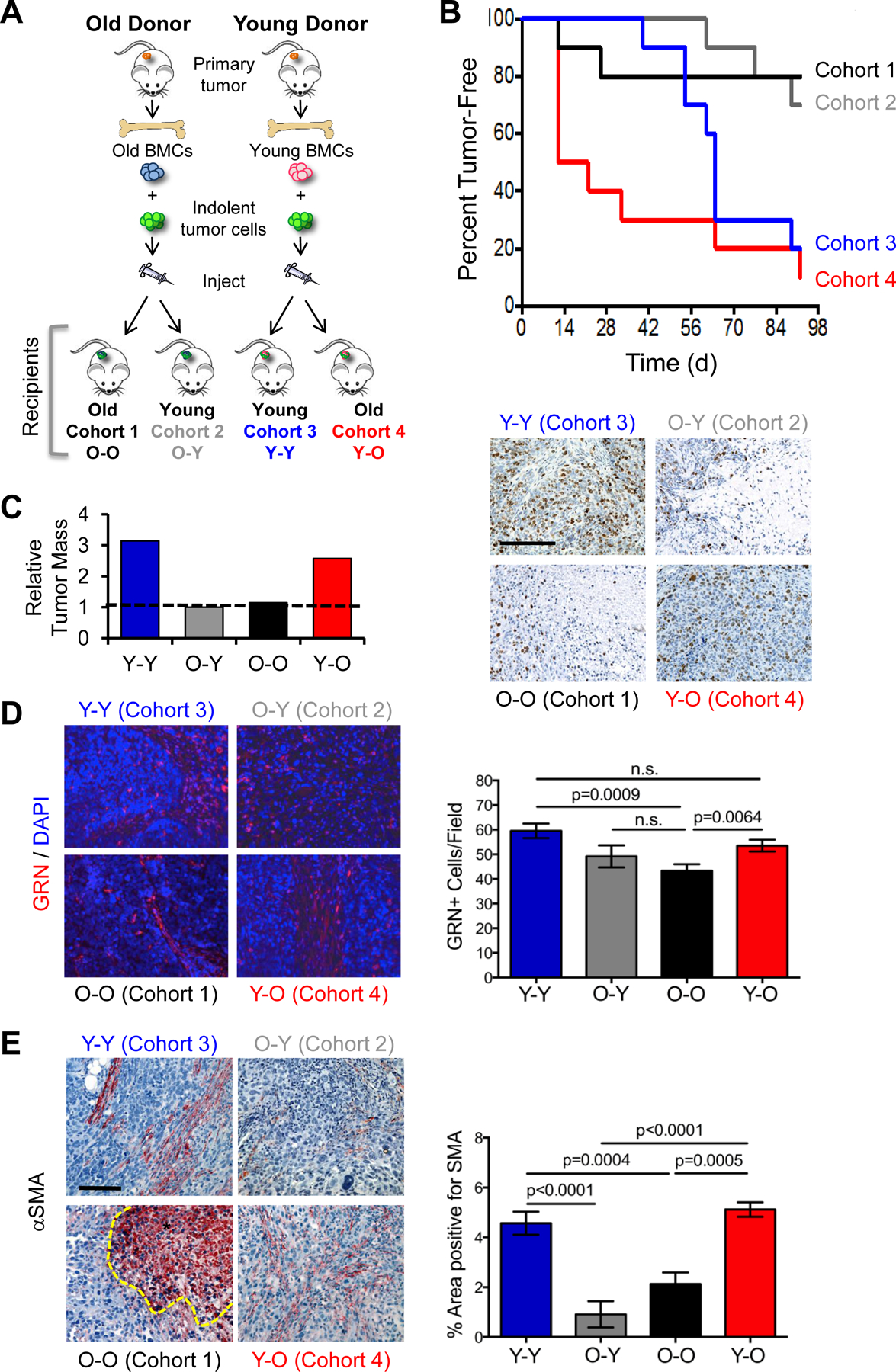
(A) Experimental protocol for BMC functional assay in young and old mice. BMCs were isolated from young or old mice bearing human breast tumors, mixed with HMLER-HR human tumor cells ex vivo, and subcutaneously injected into young or old secondary host mice. (B) Incidence of tumor formation in recipient cohorts during a 90-day time course (n=10/cohort). (C) Final tumor mass (graph) and images of tumors from (B) stained for Ki67 (brown); cell nuclei counterstained with DAPI (blue); scale bar=200μm (D) Tumors from (B) stained for GRN (red; 20x magnification). Cell nuclei counterstained with DAPI (blue). Graph represents average number of GRN-positive cells per field (n=4–5 fields per tumor, with 2–4 tumors per group). (E) Tumors from (B) stained for αSMA (red); cell nuclei were counterstained with hematoxylin (blue); scale bar=100 μm. *Denotes necrotic area with non-specific uptake of chromogen. Graph represents percent average area of SMA-positive staining (n=4 fields per tumor, with 2–4 tumors per group). Data represented as mean ± SEM; n.s.=not significant. Data analyzed using one-way ANOVA and Student’s t-test for significance. See also Figures S4, S5.
As expected, BMCs from young BPLER-bearing mice promoted HMLER-HR tumor growth in young recipient mice whereby 80% of the mice had formed tumors by the experimental end point (90d) (cohort 3, Fig. 5B). These tumors appeared invasive, proliferative, and displayed collagen-rich stroma (Fig. 5C, S4A). In contrast, BMCs from old mice with TNBC were significantly less efficient at promoting tumor growth, with only 20% of the old recipient mice (cohort 1) and 25% of the young recipient mice having formed tumors by the experimental end point (Fig. 5B). Tumors from both of these cohorts had only moderate mitotic index and little evidence of reactive stroma (Fig. 5C, S4A).
Strikingly, BMCs from young mice with TNBC rescued tumor growth in the old recipient mice (cohort 4, Fig. 5B). In old mice, 50% of the cohort developed tumors within the first 2 weeks of implantation and by the experimental end point, the incidence of tumor formation reached 90% (Fig. 5B). Final tumor mass was equivalent to that of the young mice (~3-fold greater than those mixed with old BMCs) and these tumors appeared much like those that had formed in young mice with respect to invasion, proliferation, and reactive stroma (Fig. 5C, S4A).
GRN+ cells were abundant in the tumors that formed in both young and old recipient mice (cohorts 3, 4; Fig. 5D). These numbers were significantly reduced in tumors that formed after admixture with BMCs from old donor hosts in both young and old recipients (cohorts 1, 2; Fig. 5D), consistent with our finding that GRN expression is not induced in the bone marrow of old mice with TNBC.
Most noticeably, αSMA-positive cells were abundant in the tumors that arose after admixture of young BMCs and implantation into young recipient mice (cohorts 3; Fig. 5E). When BMCs from old tumor-bearing mice were used for admixture, staining for αSMA was ~3–5-fold less prevalent in both young and old recipients (cohorts 1, 2) than it was when young BMCs were used (cohort 3) (Fig. 5E). Strikingly, BMCs from young mice with TNBC promoted formation of αSMA-enriched stroma in old recipient mice (cohort 4) to a level that was equivalent to that of the young mice (cohort 3) (Fig. 5E). We confirmed that tumor-promoting activity and the appearance of αSMA+ stromal cells depends on the presence of Sca1+ cells in the donor bone marrow when we tested the pro-tumorigenic function of other BMC subpopulations from young mice bearing BPLER tumors (Fig. S5).
Similar results were obtained using another human tumor xenograft, MDA-MB-435, whereby BMCs from young tumor-bearing donor mice rescued HMLER-HR tumor growth in old recipient mice (Fig. S4B). Hence, the fact that young BMCs promoted tumor growth, even in old hosts, suggested that BMCs are critical for TNBC progression and that hematopoietic age supersedes other aspects of physiological aging during TNBC progression from a state of indolence to one of aggressive growth.
Age- and tumor-dependent effects on gene expression of tumor-promoting BMCs
We next analyzed gene expression profiles corresponding to BMC tumorigenic potential by conducting gene expression and gene set enrichment analyses (GSEA) of the Sca1+/cKit- BMCs harvested from young and old mice that were either cancer-free or bearing size-matched TNBC tumors. Our analyses revealed biological processes and signaling pathways that were age-dependent, TNBC-dependent, or both (Fig. 6; Tables S1, S2; GEO GSE74120).
Figure 6. Gene set enrichment analysis of expression data from TP-BMCs of young and old cancer-free and TNBC tumor-bearing mice.
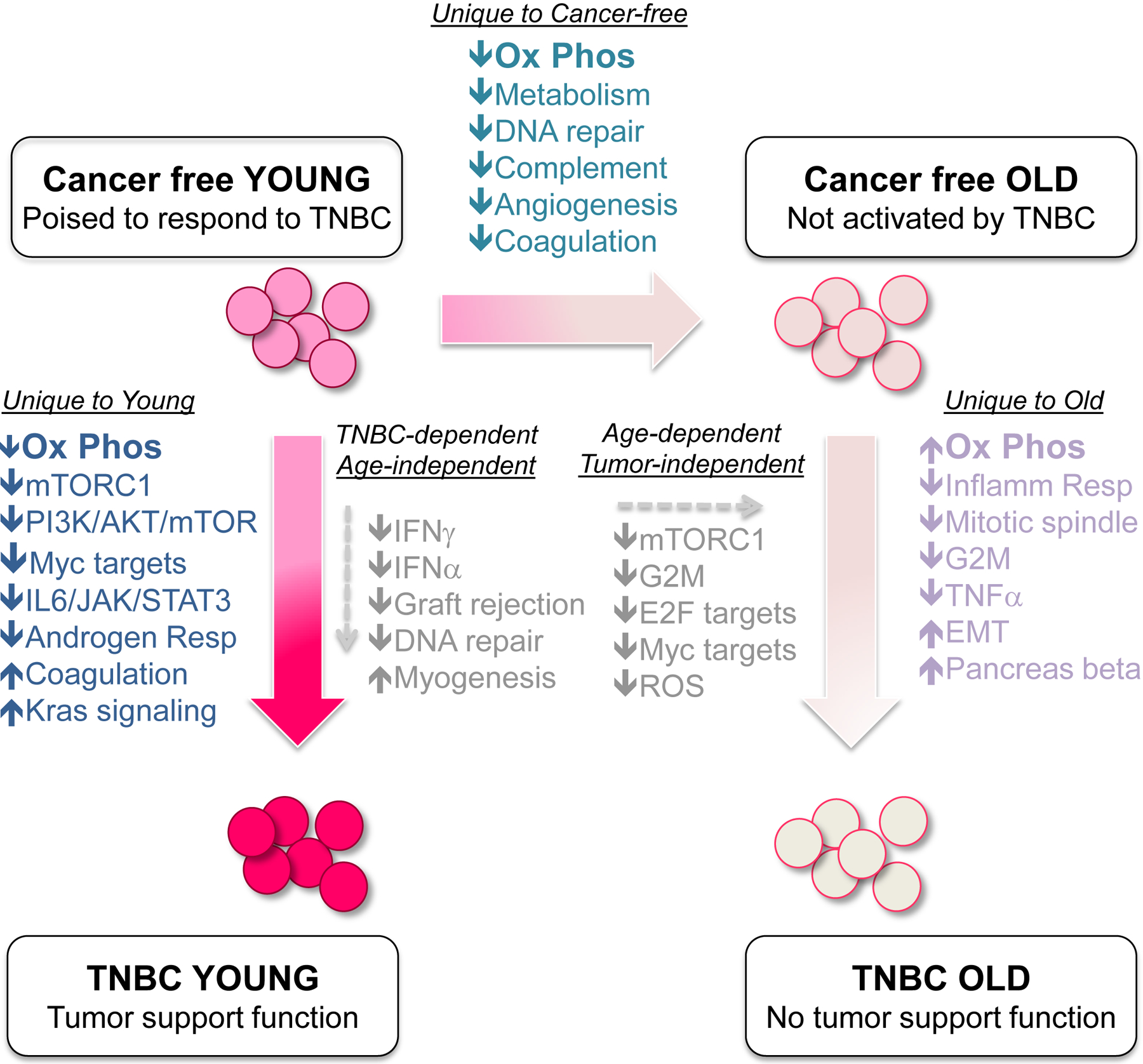
Gene expression profiling was performed on Sca1+/cKit- BMCs from indicated cohorts of mice and enrichment analyses conducted to identify groups of genes differentially represented (at statistical significance) when comparing various cohorts. Pathways unique to young mice when comparing cancer-free to TNBC are represented in dark blue text. Pathways uniquely altered with age in cancer-free animals are indicated in teal text. Pathways uniquely altered in the BMCs from old mice when comparing cancer-free to TNBC are listed in lavender text. Pathways shared in common when comparing either cancer-free to TNBC (regardless of age), or young to old (regardless of disease status) are represented in the center of diagram. See Figs. S6, S7, Tables S1 and S2; GEO GSE74120.
First, we compared the gene expression profiles of Sca1+/cKit- cells that are poised to respond to TNBC (i.e., from young cancer-free mice) to those that do not respond to TNBC (i.e., from old cancer-free mice). We observed overall decreases in gene expression with age (Fig. S6) and through GSEA, found that many of these genes regulate known age-related pathways such as oxidative phosphorylation, metabolism, and DNA repair (Fig. 6, Table S2). Expression of angiogenesis-related genes was also reduced with age, perhaps revealing a previously unknown function of these BMCs and explaining reduced vascularization observed in old mice (Fig. 1).
Second, tumor-dependent differences in gene expression were observed in Sca1+/cKit- BMCs from tumor-bearing mice compared to cancer-free controls. Common pathways that decreased in both young and old tumor-bearing cohorts were mostly related to immune surveillance and DNA repair (Fig. 6, Table S1). However, we also observed alterations to pathways unique to young or old mice (Fig. 6), suggesting that the age of the host governs the manner in which Sca1+/cKit- cells respond to systemic signals from the tumor - in young mice, such alterations rendered the BMCs tumor-supportive while in old mice, they did not. In support of this notion, while there were over 40 differentially expressed genes in Sca1+/cKit- cells comparing tumor-bearing young to tumor-bearing old mice (Fig. S7), GSEA did not reveal any uniquely tumor-dependent influences on the old BMC population (Fig. 6, Tables S1, S2).
These analyses demonstrated that age determines the ability of Sca1+/cKit-BMCs to become pro-tumorigenic in response to systemic signals imparted by TNBC. These findings also support the notion that expression profiling is useful for distinguishing BMCs that have tumor support function from those that do not.
Taken collectively, our results reveal the importance of age in governing the potential of bone marrow hematopoietic cells to promote TNBC progression and suggest that the microenvironment of TNBC is indeed different at the cellular and molecular level with age (Fig. 7).
Figure 7. Model of age-dependent triple-negative breast cancer progression.
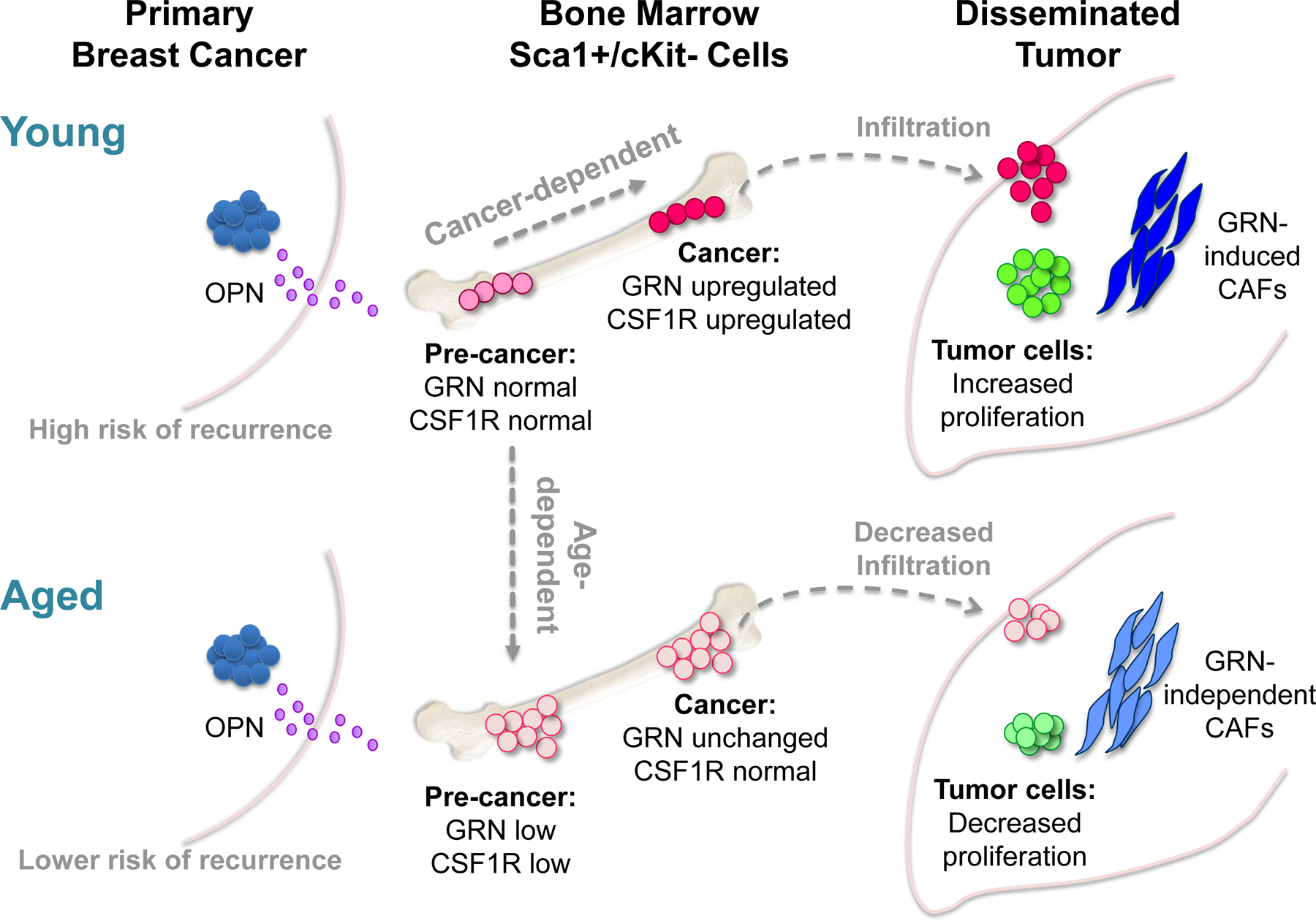
Our data indicate that age influences the tumorigenic capacity of Sca1+/cKit- BMCs in response to TNBC-associated signals, such as osteopontin (OPN). In young mice with TNBC, these BMCs undergo gene expression changes, including elevated levels of granulin (GRN) and colony stimulating factor 1 receptor (CSF1R), which render them pro-tumorigenic. When recruited to tumors, these BMCs establish a microenvironment that supports disease progression. With age, Sca1+/cKit- BMCs are increased in number but undergo distinct gene expression changes that render them unresponsive to TNBC. As a consequence, in old mice with TNBC, Sca1+/cKit- BMCs do not support tumor growth and show reduced infiltration into distant tumors; the resulting cancer-associated microenvironment is less efficient at supporting tumor progression.
DISCUSSION
The incidence of luminal breast cancers is higher in elderly patients than in young, while the incidence of TNBC does not significantly change with age (6,38). Importantly, though, recurrence free survival among all breast cancer subtypes, with the exception of Her2+ breast cancers, is lower in young patients than it is in older patients (4). In light of our findings, it is possible that TNBC is more reliant on age-dependent hematopoietic function than other breast cancer subtypes. In further support of this notion, we previously reported that BMCs from young mice bearing MCF7Ras LBC tumors (39) or PC3 prostate tumors (25) do not promote tumor growth.
Age-related gene expression profiles that correlate with pro-tumorigenic potential or function of BMCs, as we have defined here, may be useful to for screening bone marrow aspirates or blood samples. As such, it will be necessary to identify the homologous BMC population in human patients. In mice, the Sca1+/cKit-population is comprised of different functional cell types, which all have human counterparts, including T and B cell precursors, nuocytes (33), a subset of CD25-expressing cells that expand with age (34), and plasmacytoid dendritic cells (pDCs; (33)). While pDCs account for the vast majority of GRN expression in young cancer-free mice and humans (33), and pDCs have been implicated in tumor promotion (40), further examination is required to determine whether they play a role in TNBC progression with age. Moving forward, we will want to learn how tumor-promoting BMCs function in the context of the immune milieu. Indeed, age- and tumor-dependent changes to Sca1+/cKit- BMCs occurred in both nude and immunocompetent breast cancer models in our study.
Our own analysis of a published stromal gene expression profile obtained from breast cancer patient tumors (GSE9014; (41)) revealed a negative correlation between age and stromal GRN for patients with TNBC (Pearson correlation, r = −0.7; n=7), with a weaker negative correlation across all breast cancers (Pearson correlation r = −0.17; n=66). Moreover, when we analyzed GRN staining of tumor tissues from a cohort of breast cancer patients (23), we observed a trend toward increased GRN in the epithelial tumor cells with age in non-TNBC patients (Pearson correlation r = 0.361; p=0.064, n=27). While these trends are provocative, small numbers of TNBC patients in these cohorts prevent us from making solid conclusions about GRN+ infiltrating stromal cells in patient tumors. Information about stromal infiltrates with age and gene expression profiling of tumor-promoting hematopoietic BMCs with age are needed. The ability to detect tumor-supportive BMCs, using GRN expression as a biomarker for example, may thus help to identify both young and elderly patients most at risk for TNBC recurrence.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Mahnaz Paktinat of the Boston Children’s Hospital Heme/Onc-HSCI Flow Cytometry facility, Boston, MA; Per-Hernrik Edqvist and Fredrik Pontén of Uppsala University for analysis of patient tissue microarray; Shannan Hui of the HSPH Bioinformatics Core, Harvard School of Public Health, Boston, MA for assistance with computational analysis; and Kristin Tracy for technical assistance.
Financial support:
This work was supported by funds from the Knut and Alice Wallengerg’s Foundation grant 2012.0193 (BN); NIH NCI R01CA151518 and American Cancer Society Research Scholar Award (SAS); and NIH NCI R21CA182614 and the Department of Defense BCMRP Era of Hope Scholar Award W81XWH-14-1-0191 (SSM).
List of abbreviations:
- BMCs
bone marrow cells
- BMDCs
bone marrow derived cells
- LBC
luminal breast cancer
- αSMA
alpha-smooth muscle actin
- TME
tumor microenvironment
- TNBC
triple - negative breast cancer
Footnotes
Conflict of Interest: No potential conflicts of interest to disclose
REFERENCES
- 1.Edwards BK, Howe HL, Ries LA, Thun MJ, Rosenberg HM, Yancik R, et al. Annual report to the nation on the status of cancer, 1973–1999, featuring implications of age and aging on U.S. cancer burden. Cancer 2002;94(10):2766–92. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Q, Ma B, Kang M. A retrospective comparative study of clinicopathological features between young and elderly women with breast cancer. International journal of clinical and experimental medicine 2015;8(4):5869–75. [PMC free article] [PubMed] [Google Scholar]
- 3.Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, et al. Triple-negative breast cancer: risk factors to potential targets. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14(24):8010–8. [DOI] [PubMed] [Google Scholar]
- 4.Liedtke C, Rody A, Gluz O, Baumann K, Beyer D, Kohls EB, et al. The prognostic impact of age in different molecular subtypes of breast cancer. Breast cancer research and treatment 2015;152(3):667–73. [DOI] [PubMed] [Google Scholar]
- 5.Klepin HD, Pitcher BN, Ballman KV, Kornblith AB, Hurria A, Winer EP, et al. Comorbidity, chemotherapy toxicity, and outcomes among older women receiving adjuvant chemotherapy for breast cancer on a clinical trial: CALGB 49907 and CALGB 361004 (alliance). Journal of oncology practice / American Society of Clinical Oncology 2014;10(5):e285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA: a cancer journal for clinicians 2014;64(1):52–62. [DOI] [PubMed] [Google Scholar]
- 7.Hutchins LF, Unger JM, Crowley JJ, Coltman CA Jr., Albain KS. Underrepresentation of patients 65 years of age or older in cancer-treatment trials. The New England journal of medicine 1999;341(27):2061–7. [DOI] [PubMed] [Google Scholar]
- 8.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nature reviews Molecular cell biology 2007;8(9):715–22. [DOI] [PubMed] [Google Scholar]
- 9.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences 2000;908:244–54. [DOI] [PubMed] [Google Scholar]
- 10.Balliet RM, Capparelli C, Guido C, Pestell TG, Martinez-Outschoorn UE, Lin Z, et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: understanding the aging and cancer connection. Cell cycle 2011;10(23):4065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer research 2012;72(9):2251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grizzle WE, Xu X, Zhang S, Stockard CR, Liu C, Yu S, et al. Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mechanisms of ageing and development 2007;128(11–12):672–80. [DOI] [PubMed] [Google Scholar]
- 13.Chang EI, Loh SA, Ceradini DJ, Chang EI, Lin SE, Bastidas N, et al. Age decreases endothelial progenitor cell recruitment through decreases in hypoxia-inducible factor 1alpha stabilization during ischemia. Circulation 2007;116(24):2818–29. [DOI] [PubMed] [Google Scholar]
- 14.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature reviews Cancer 2009;9(4):239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nature cell biology 2014;16(8):717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. The Journal of experimental medicine 2000;192(9):1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America 2005;102(26):9194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beerman I, Maloney WJ, Weissmann IL, Rossi DJ. Stem cells and the aging hematopoietic system. Current opinion in immunology 2010;22(4):500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao D, Mittal V. The role of bone-marrow-derived cells in tumor growth, metastasis initiation and progression. Trends in molecular medicine 2009;15(8):333–43. [DOI] [PubMed] [Google Scholar]
- 20.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes & development 2001;15(1):50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer cell 2007;12(2):160–70. [DOI] [PubMed] [Google Scholar]
- 22.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121(3):335–48. [DOI] [PubMed] [Google Scholar]
- 23.Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. The Journal of clinical investigation 2011;121(2):784–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polanska UM, Acar A, Orimo A. Experimental generation of carcinoma-associated fibroblasts (CAFs) from human mammary fibroblasts. Journal of visualized experiments : JoVE 2011(56):e3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008;133(6):994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer cell 2013;24(2):182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehner C, Radisky DC. Triggering the landslide: The tumor-promotional effects of myofibroblasts. Experimental cell research 2013;319(11):1657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annual review of pathology 2006;1:119–50. [DOI] [PubMed] [Google Scholar]
- 29.Gupta PB, Proia D, Cingoz O, Weremowicz J, Naber SP, Weinberg RA, et al. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer research 2007;67(5):2062–71. [DOI] [PubMed] [Google Scholar]
- 30.Tuck AB, Chambers AF, Allan AL. Osteopontin overexpression in breast cancer: knowledge gained and possible implications for clinical management. J Cell Biochem 2007;102(4):859–68. [DOI] [PubMed] [Google Scholar]
- 31.Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochimica et biophysica acta 2013;1832(7):1070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palfree RG, Bennett HP, Bateman A. The Evolution of the Secreted Regulatory Protein Progranulin. PloS one 2015;10(8):e0133749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo G, Luc S, Marco E, Lin TW, Peng C, Kerenyi MA, et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell stem cell 2013;13(4):492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar R, Fossati V, Israel M, Snoeck HW. Lin-Sca1+kit- bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. Journal of immunology 2008;181(11):7507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Chen K, Zhu L, Pollard JW. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genesis 2006;44(7):328–35. [DOI] [PubMed] [Google Scholar]
- 36.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer research 2009;69(24):9498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery 2011;1(1):54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love RR, Duc NB, Dinh NV, Quy TT, Xin Y, Havighurst TC. Young age as an adverse prognostic factor in premenopausal women with operable breast cancer. Clinical breast cancer 2002;2(4):294–8. [DOI] [PubMed] [Google Scholar]
- 39.Kuznetsov HS, Marsh T, Markens BA, Castano Z, Greene-Colozzi A, Hay SA, et al. Identification of luminal breast cancers that establish a tumor-supportive macroenvironment defined by proangiogenic platelets and bone marrow-derived cells. Cancer discovery 2012;2(12):1150–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nature reviews Immunology 2015;15(8):471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nature medicine 2008;14(5):518–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.