
Fig. 4 |. Bioinformatics for enhanced calling.
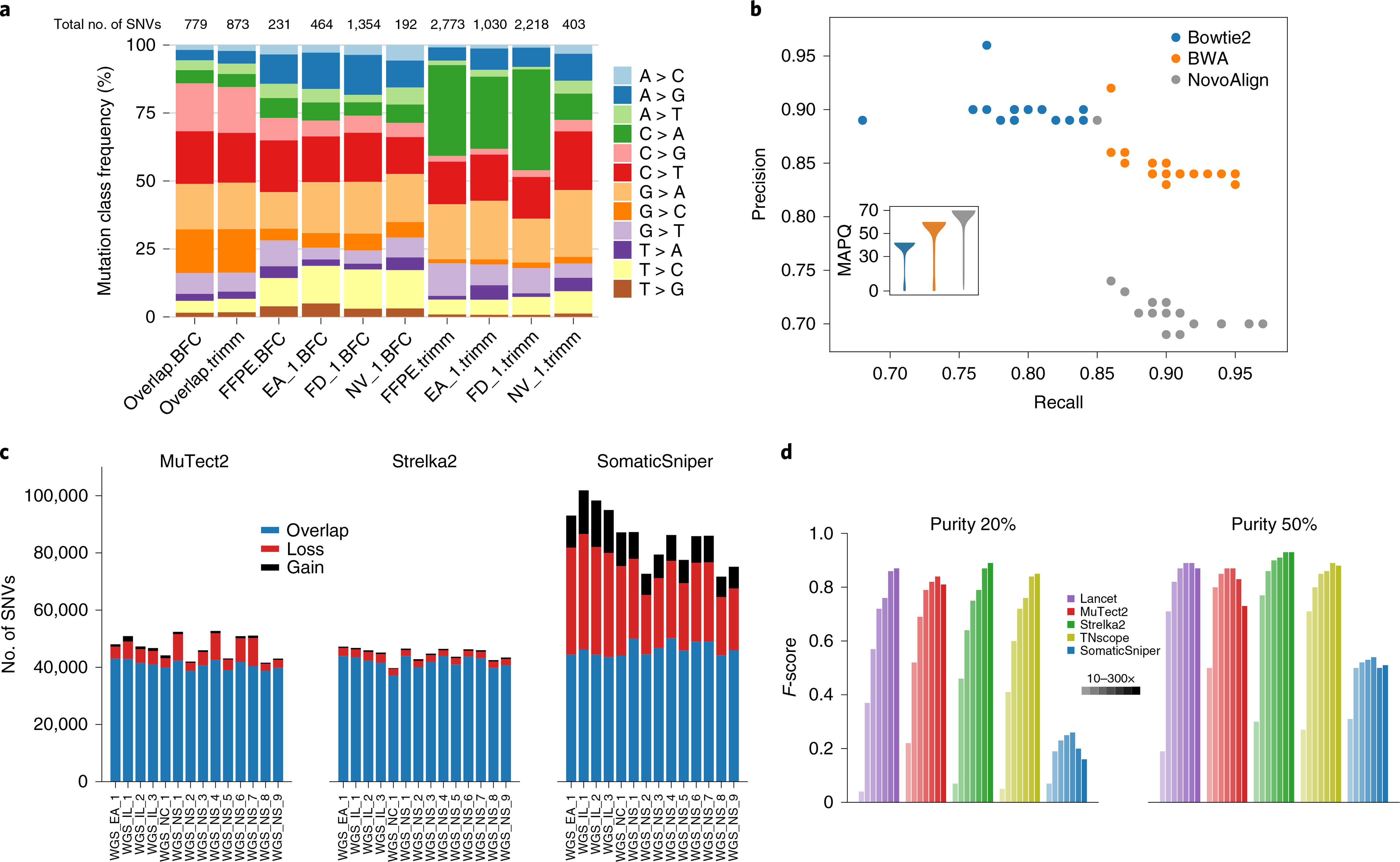
a, Distribution of mutation types called with Strelka2 on BWA alignments of four WES runs preprocessed by Trimmomatic or BFC. WES run on FFPE DNA (FFPE) or fresh DNA (EA_1, FD_1 and NV_1). Numbers of SNVs called from each process are shown at the top. Mutations shared across BFC datasets (overlap.BFC) and Trimmomatic datasets (overlap.trimm) are shown on the left. C > A and T > C artifacts were observed in the Trimmomatic and BFC datasets, respectively; both artifacts were minimized with repeats. b, Performance of mutation calling by Strelka2 on three alignments (Bowtie2, BWA and NovoAlign). Insert is a violin plot of mapping quality (MAPQ) scores from three alignments for an example WGS run. In total, 81 billion, 118 billion and 140 billion data points were used in violin plots for Bowtie2, BWA and NovoAlign, respectively. c, Effect of postalignment processing (indel realignment + BQSR) on mutation calling by MuTect2, Strelka2 and SomaticSniper). d, Effect of tumor purity (20 versus 50%) on five callers (Lancet, MuTect2, Strelka2, TNscope and SomaticSniper) with read coverage of 10×, 30×, 50×, 80×, 100×, 200× and 300×.