

Abstract
Melatonin is a hormone mainly produced by the pineal gland and MT1 is one of the two G protein-coupled receptors (GPCRs) mediating its action. Despite an increasing number of available GPCR crystal structures, the molecular mechanism of activation of a large number of receptors, including MT1, remains poorly understood. The purpose of this study is to elucidate the structural elements involved in the process of MT1’s activation using naturally occurring variants affecting its function. Thirty-six nonsynonymous variants, including 34 rare ones, were identified in MTNR1A (encoding MT1) from a cohort of 8687 individuals and their signaling profiles were characterized using Bioluminescence Resonance Energy Transfer-based sensors probing 11 different signaling pathways. Computational analysis of the experimental data allowed us to group the variants in clusters according to their signaling profiles and to analyze the position of each variant in the context of the three-dimensional structure of MT1 to link functional selectivity to structure. MT1 variant signaling profiles revealed three clusters characterized by (1) wild-type-like variants, (2) variants with selective defect of βarrestin-2 recruitment, and (3) severely defective variants on all pathways. Our structural analysis allows us to identify important regions for βarrestin-2 recruitment as well as for Gα12 and Gα15 activation. In addition to identifying MT1 domains differentially controlling the activation of the various signaling effectors, this study illustrates how natural variants can be used as tools to study the molecular mechanisms of receptor activation.
Keywords: Melatonin, Receptor, GPCR variants, Functional selectivity, Mechanisms of activation
Next-generation sequencing (NGS) is rapidly increasing our understanding of disease causes and susceptibilities in humans as well as of the adaptation and evolution of organisms. Human DNA contains 10,000 to 11,000 nonsynonymous genetic variants on average,1 although only few of them are expected to lead to disease, while the rest are considered neutral. Information on such variants in databases has been often limited to basic annotations like “neutral” or “deleterious”. Deleteriousness can be approximated with available information on sequence conservation and molecular functionality, but such data are rarely combined. Furthermore, even a strong association between a mutation and a clinical trait does not provide specific information on the corresponding changes in protein function and/or structure.2,3 Since just a single genetic variant can change the local structure of a protein or its interactions with other proteins, it can actually have various functional effects.4 Thus, genetic variants could be instrumental in deciphering molecular determinants and pathways.
Many proteins display several functions by interacting with different protein partners; however, most protein-interaction maps ignore structural details and the mechanisms of protein–protein interaction. This is the case for many G protein-coupled receptors (GPCRs), which can interact with different G proteins, GPCR-regulated kinases (GRKs), and βarrestins, triggering different signaling pathways. The molecular determinants controlling these different functions are still generally not well understood. It is now known that different ligands can preferentially activate specific GPCR signaling pathways leading to distinct physiological effects. Variants at specific positions in the receptor structure can also modify the activation mechanism and thus the signaling profile of a receptor5 by inhibiting one or several pathways or, conversely, by promoting others. This concept can be referred to as genetically defined functional selectivity or biased signaling.6
We took advantage of the abundance of genetic variants in MTNR1A (encoding the melatonin receptor 1 [MT1]) to explore the link between structure and functional selectivity. Melatonin (MLT) is mainly released by the pineal gland in circadian and seasonal manners.7 The MT1 is one out of two melatonin receptors involved in the physiological action of this neuro-hormone.8 MT1 expression has been reported in several tissues and organs9 including the retina,10,11 brain,12 pancreas, and immune system.13,14 MT1 is involved in various physiological functions including circadian entrainment,15 sleep,16 dopamine level regulation,17 and glycemic homeostasis.18
In the present study, we identified 36 MTNR1A variants and investigated their capacity to activate seven G protein subtypes and to recruit βarrestin-2. We found that these variants can be clustered in three distinct functional groups providing insights into specific structural features of the MT1 underlying functional selectivity.
Results
Signaling Profile of Wild-Type MT1
To obtain a comprehensive profiling of the signaling repertoire of MT1, we used bioluminescence resonance energy transfer (BRET)-based biosensors to assess G protein activation measuring Gα protein dissociation from the Gβγ complex19 (Figure S1A). We measured βarrestin-2 recruitment to the activated receptor measuring its proximity with the plasma membrane using enhanced bystander BRET (ebBRET)20 (Figure S1B). Melatonin stimulation in cells expressing the wild-type (WT) human MT1 leads to the activation of Gαi1, Gαi2, Gαi3, GαoA, GαoB, Gα12, and Gα15 (Figure 1). This is consistent with previous findings suggesting a predominant coupling to Gαi for this receptor. Gα15, an atypical member of the Gαq/11 family, has been shown to couple in a promiscuous manner to many distinct GPCRs.21 Because the human embryonic kidney 293 (HEK293) cells used do not express Gα15, its potential activation by MT1 was assessed by probing the downstream activation of protein kinase C (PKC) using a BRET-based PKC activity sensor22 (Figure S1C) upon Gα15 expression. The data show that no MLT-promoted PKC activation could be detected in the absence of Gα15, but the addition of Gα15 resulted in a robust PKC activation illustrating the ability of MT1 to functionally engage this protein (Figure 1).
Figure 1.
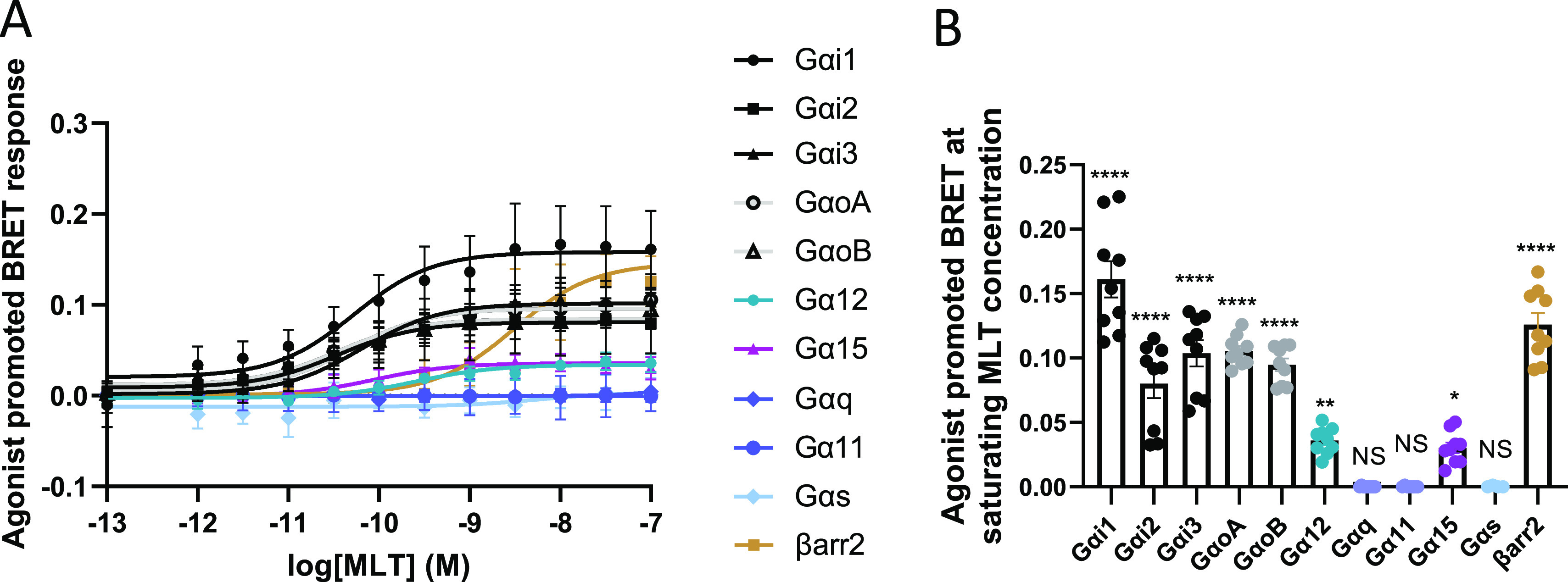
MT1-WT activates Gαi/o, Gα12, Gα15, and recruits βarrestin-2. (A) MLT concentration curves for MT1-WT. Data were plotted using nonlinear regression curves with a fixed Hill slope equal to 1. (B) MT1 response for Gαi1, Gαi2, Gαi3, GαoA, GαoB, Gα12, Gαq, Gα11, Gα15, and Gαs activation and βarrestin-2 recruitment at saturated MLT concentration (100 nM) of the corresponding curves in A. Statistical analysis was performed using one-way ANOVA followed by a Dunnett’s posthoc test. Data represents the mean ± SEM of 5 to 9 experiments. Data were fitted in GraphPad Prism 9. * p ≤ 0.05, ** p ≤ 0.01, **** p ≤ 0.0001. βarr2: βarrestin-2.
Identification of MT1 Variants and Cell Surface Expression
To identify natural variants, we sequenced the two exons of the MTNR1A gene in 8687 unrelated European individuals. We accurately identified 36 genetic variants, including 34 rare variants with a minor allele frequency (MAF) of <1% (Table S1). The two frequent variants are nonsynonymous variants. Among the 34 rare variants, we found 2 nonsense variants introducing STOP codons (p.W93*, p.Y170*), one variant on a donor splice site (c.184 + 1G > T), one frameshift variant (c.999_1014del/p.W333*), one loss of STOP codon variant (c.1053A > T/p.*351Yext* 18 (YKSTTFRVRWTRCARPRS)) and 29 nonsynonymous variants (Table S1). As we previously found that rare deleterious variants in MTNR1B (encoding the MT2 receptor) were significantly associated with increased risk of type 2 diabetes,23 we assessed the association between the coding variants of MTNR1A and the risk of type 2 diabetes or obesity. Neither the frequent MTNR1A variants (i.e., p.G166E-rs28383653 and p.A266 V-rs28383652) nor the 34 rare MTNR1A variants were associated with the risk of type 2 diabetes or obesity in both children and adults (Table S2).
Then, we determined the functional properties of all 32 nonsynonymous MT1 variants in comparison to the MT1-WT in terms of cell surface expression, G protein activation, βarrestin-2 recruitment, and binding of the radioligand 2(125I)-iodomelatonin (125I-MLT).
Cell surface expression of MT1 variants was assessed by enzyme-linked immunosorbent assay (ELISA), detecting the Flag tag located at the extracellular N-terminus of the receptor. Figure S2 shows that similar expression levels were observed for all variants compared to MT1-WT, indicating that the results of the functional assays for the different MT1 variants would not be severely affected by differences in the number of receptors at the cell surface.
Signaling Profiles of MT1 Variants
Melatonin concentration–response curves were generated for each MT1 variant and MT1-WT, and both the EC50 (potency) and the maximal agonist-mediated response (maximal efficacy; Emax) were determined for βarrestin-2 recruitment and for those G proteins for which activation by MT1-WT was observed: Gαi1, Gαi2, Gαi3, GαoA, GαoB, Gα12, and Gα15 (Table 1, Figure S3, and Table S3). Each signaling pathway was studied using a different biosensor, and consequently, comparison of BRET intensities can only be done between receptors for the same biosensor but not between different pathways for a same variant. Representative examples of the signaling profiles for the MT1-WT, and three melatonin-responsive variants are shown in Figure 2 and clearly indicate the differential impact of variants including selective effects on the potency and/or efficacy toward the different pathways.
Table 1. Comparison of Efficacies and Potencies of the Different MT1 Receptors to Activate Gα Proteins and Recruit βarrestin-2a.
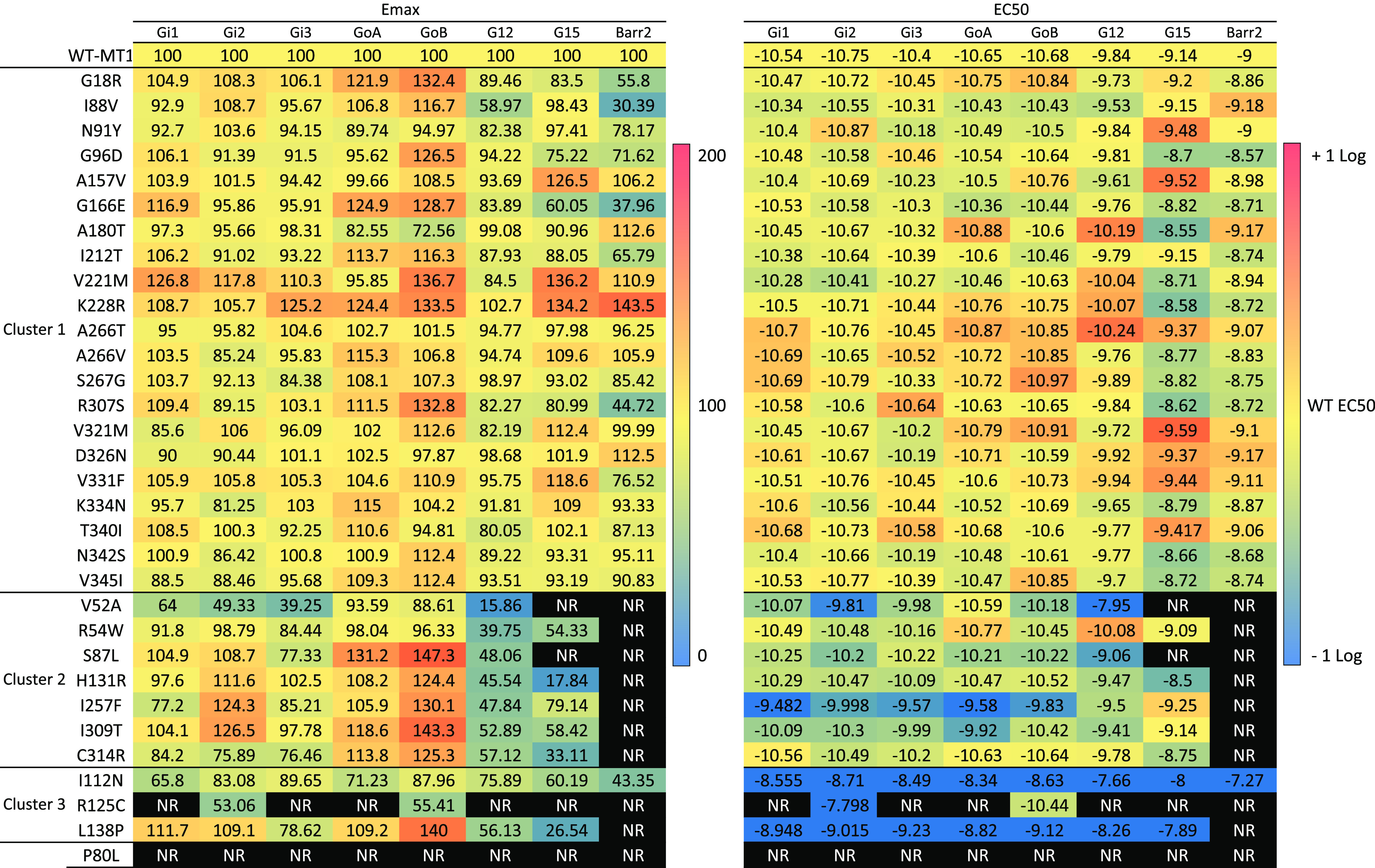
Functional profiling of Gα protein activations and βarrestin-2 recruitment by MT1-WT and MT1 variants. Data represents the mean ± SEM of 3-16 independent experiments with repeats in quadruplicate. NR (No response) denotes that the experimental parameter could not be determined due to lack of a concentration-response curve. The data were analyzed by comparing independent curves.
Figure 2.
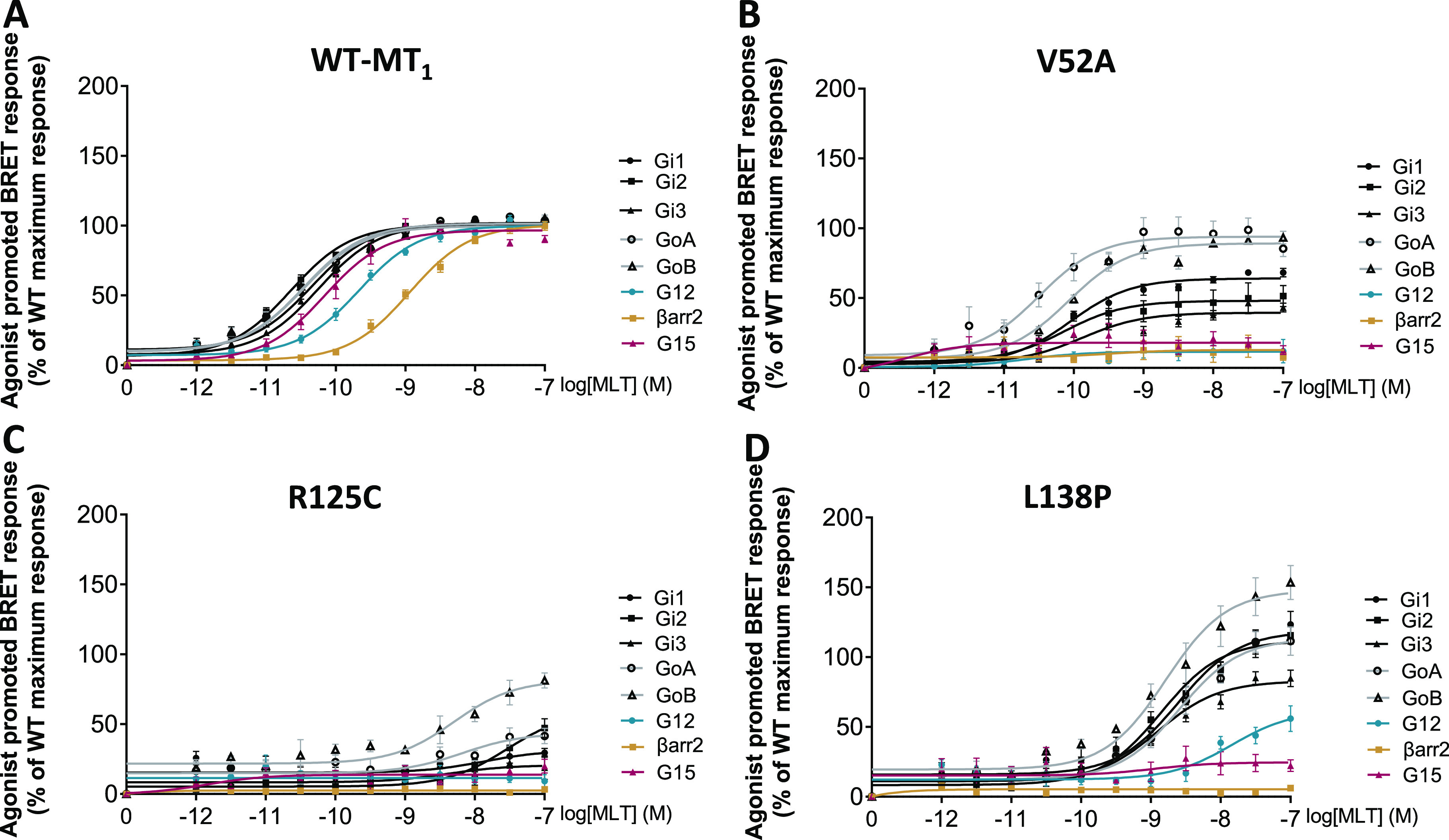
Melatonin concentration–response curves for G protein activations and βarrestin-2 recruitment of the MT1-WT and representative receptor variants with distinct signaling profiles. MLT concentration curves for (A) MT1-WT, (B) V52A, (C) R125C, and (D) L138P receptors. Data were plotted using nonlinear regression curves with a fixed Hill slope equal to 1. Data points represent means ± SEM of 14 to 16 experiments (A) or 3 to 5 experiments (B, C, D). βarr2: βarrestin-2, WT: wild-type. See also Figure S1. Data were fitted in GraphPad Prism 9.
To provide a visual representation of the relative signaling profiles of each variant for the different signaling pathways, we used a radial graph representation including the Emax and the EC50 for each pathway (Figure S4). We used a non-negative matrix factorization (NMF) and K-means clustering method to divide the variants into groups according to their functional impact similarities and differences. The different signaling profiles could be grouped in three distinct clusters. Figure 3 presents the superposition of the individual signaling profiles of each member of a given cluster providing a global view of the cluster signaling characteristics.
Figure 3.

Radial graph representing the different variant profiles compiled in 3 clusters. Compilation of the different MT1 variant radial graph profiles in three clusters. On each radius, maximal agonist-induced efficacy (Max) and potency (EC50) are indicated. MT1-WT profile is represented in red and mutant profiles are in blue. The values were normalized to fit a −1 to +1 scale using the following formula: (variant – WT)/(variant + WT) as indicated in the methods. A loss of potency or efficacy for a specific protein results in the decrease of the blue area. Data were fitted in Microsoft Excel 2016.
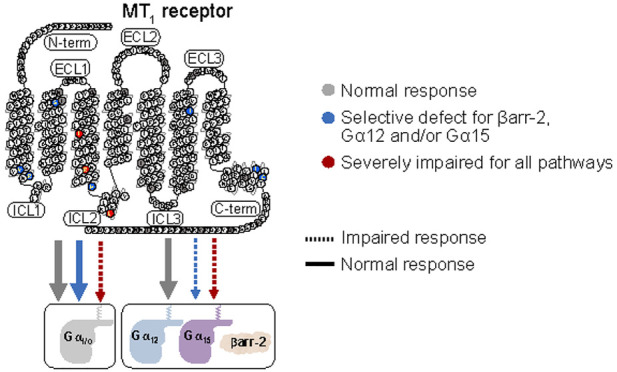
Cluster 1 comprises 21 variants distributed over transmembrane (TM) domains 2, 3, 4, 5, and 6, helix 8, the N-terminus (Nterm), extracellular loops (ECL) 2 and 3, and the C-terminus (Cterm) (p.G18RNterm, p.I88V2.65, p.N91Y2.68, p.G96D3.21, p.A157V4.55, p.G166E4.64, p.A180TECL2, p.I212T5.63, p.V221M5.72, p.K228R6.25, p.A266TECL3, p.A266VECL3, p.S267GECL3, p.R307S8.55, p.V321MCterm, p.D326NCterm, p.V331FCterm, p.K334NCterm, p.T340ICterm, p.N342SCterm, and p.V345ICterm) (superscripts represent Ballesteros–Weinstein nomenclature24) that did not significantly impact any of the signaling pathways considered in response to MLT. Cluster 2 comprises seven variants (p.V52A1.57, p.R54W1.59, p.S87L2.64, p.H131R3.56, p.I257F6.54, p.I309T8.57, and p.C314RCterm) that cause a total loss of βarrestin-2 recruitment and a decrease of Emax and/or EC50 for some Gα proteins, the most impacted being Gα12; however, for each of these variants, the activation of at least one G protein is not negatively impacted by the mutation. Cluster 3 comprises three variants (p.I112N3.37, p.R125C3.50, p.L138PICL2) (intracellular loop (ICL)) with a total loss of βarrestin-2 recruitment and a global reduction in the activation of all G protein pathways.
To exclude the possibility that differences in the signaling properties, in particular EC50 values, could result from reduced binding affinity to MLT, we determined the dissociation constant (Kd) of the melatonin receptor-specific radioligand 125I-MLT for the 20 variants with decreased EC50 values and for the melatonin-unresponsive p.P80L2.57. Kd values were not statistically different between the variants and the MT1-WT, except for p.P80L2.57, which showed a complete loss of binding capacity (Table 2). Our cell surface ELISA data show that p.P80L2.57 expresses properly at the cell surface excluding a defect in trafficking (Figure S2). The p.P802.57 amino acid is part of a YPYP motif conserved in melatonin receptors. Our observation is consistent with previous studies suggesting that this motif located in transmembrane helix 2 (TM2) is crucial for both stability and activation of MT1.25
Table 2. MT1 Variant Receptors Bind to Melatonin as well as the MT1-WT Except P80La.
| receptors | Kd (pM) ± SEM |
|---|---|
| Wild Type | 101 ± 9.3 |
| V52A | 99 ± 6.9 |
| R54W | 178 ± 25 |
| P80L | No binding |
| S87L | 136 ± 19 |
| I88V | 79 ± 4.5 |
| G96D | 81 ± 8.7 |
| I112N | 244 ± 75 |
| R125C | 227 ± 61 |
| H131R | 62 ± 11 |
| L138P | 171 ± 61 |
| G166E | 63 ± 1.2 |
| I212T | 101 ± 5.0 |
| I257F | 168 ± 7.2 |
| A266V | 224 ± 43 |
| S267G | 186 ± 54 |
| R307S | 40 ± 2.5 |
| I309T | 75 ± 5.6 |
| C314R | 48 ± 7.8 |
| K334N | 395 ± 68 |
| N342S | 140 ± 43 |
| V345I | 69 ± 1.8 |
Melatonin dissociation constant measured by 2-(125I)Iodomelatonin binding experiment on MT1 variants with at least one signaling defect. Data represents means ± SEM of at least three experiments.
Evolutionary Action Analysis of MT1 Variants
To assess whether the extent of the functional impact of the naturally occurring variants could be estimated by the evolutionary impact of the individual substitution, we scored the Evolutionary Action (EA) of the nonsynonymous variants. EA is a bioinformatics algorithm designed to predict the impact of mutational variants26 in which the EA score varies from 0, for variations with the most benign impact, to 100, for those with the most adverse effects. As expected, cluster 1 is comprised of variants of low EA scores, cluster 2 of medium scores, and cluster 3 of high scores. Figure 4 shows the statistically significant correlation (R2 = 0.580, P < 0.0001) between EA scores and the experimentally determined signaling phenotypic impact for each of the variants, indicating that the severity of the functional outcome of SNPs can be reasonably well predicted using trace evolution-based methods such as EA.
Figure 4.

Evolutionary action of MT1 variants and correlation with signaling impairment. (A) An evolutionary action (EA) score was calculated for each variant. EA ranges from 0 to 100 with a score of 0 predicted to be benign and a score of 100 predicted to be highly impactful or detrimental to protein function. Scores are colored based on EA (EA = 0 (blue) to EA = 100 (red)). (B) Functional defects of every MT1 variant were correlated with the EA score. The 3 clusters were represented with the cluster 1 in blue, the cluster 2 in yellow, and the cluster 3 in red. Data were fitted in GraphPad Prism 9 by linear regression analysis, P and R2 values obtained for the overall correlation.
Structural Determinants of the Functional Impact of MT1 Variations
To investigate the structural basis of the functional impact of the MT1 variants, we analyzed the position and topology of the individual mutated residues of Clusters 1–3 on the three-dimensional models of the active form of MT1 coupled to Gαi or βarrestin-2 (see Methods).
Variants in Cluster 1 (with no impact on the receptor signaling profile) are spread throughout the structure (Figure 5A,B), appearing mostly at the surface of the extra- and intracellular domains and generally far from the ligand-binding pocket25,27 or from any of the known GPCR activation microswitches.
Figure 5.

Location of the mutated residues and topology of Clusters 1–3. (A) Distribution of the mutated residues depicted in a 2D snake plot of the MT1 receptor. Residues in Cluster 1 are colored gray, in Cluster 2 blue, and in Cluster 3 red; these colors are preserved in all panels. (B) Location of Cluster 1 mutations in the three-dimensional structure of inactive MT1. The receptor is colored in a rainbow spectrum (TM1 blue - TM7/helix8 red). (C) Location of Cluster 2 mutations in active MT1 bound to βarrestin-2. (D) Location of Cluster 3 mutations in active MT1 bound to βarrestin-2 and Gαi. (E) Mutation I112N3.37 is located near the key functional PIF (or triad) motif. (F) Mutation P80L2.57 is located near the binding site (shown as gray spheres) of melatonin (shown as sticks).
The seven variants in Cluster 2 (p.V52A1.57, p.R54W1.59, p.S87L2.64, p.H131R3.56, p.I257F6.54, p.I309T8.57, and p.C314RCterm) are associated with a total loss of βarrestin-2 recruitment and a decrease of different Gα activations, especially for Gα12 and/or Gα15 Emax (Table 1 and Figure S3), but at least one Gα protein is not impacted. Interestingly, four of these variants are located at the interface between the cytoplasmic side of TM1 (p.V52A1.57 and p.R54W1.59) and helix 8 (H8) (p.I309T8.57 and p.C314RCterm) (Figure 5C). The short amphipathic H8 lies adjacent to the inner leaflet of the plasma membrane, anchored through a palmitoylated cysteine (p.C314 in MT1). In other GPCRs, H8 has been shown to play a crucial role in the interaction of the receptor with βarrestin-2—leading to receptor internalization28 and its palmitoylation has been shown to enhance the recruitment of βarrestin-2.29 The structural models show that both H8 and ICL1 of MT1 can interact with the finger loop of βarrestin-2 (Figure 5C). Thus, we hypothesize that p.V52A1.57, p.R54W1.59, p.I309T8.57, and p.C314RCterm variants affect βarrestin-2 recruitment by altering the local environment of H8 in MT1. Both p.I309T8.57 and p.C314RCterm affect directly H8 by weakening hydrophobic interactions with nearby phospholipids or by removing the palmitoylation site, respectively. On the other hand, p.V52A1.57 and p.R54W1.59—located at the interface between TM1 and ICL1—would affect H8 indirectly. These results are consistent with findings on other GPCRs; for instance, hydrophobic amino acids in H8 of the mouse odorant receptor mOR-S6 have been suggested to form a hydrophobic core with TM1 responsible for the proper positioning of H8, and mutations of these residues increase the flexibility of H8 and destabilize its structure.30 Not surprisingly, the loss of βarrestin-2 recruitment by these variants leads to a loss of internalization of the receptor (Figure S5) as measured by ebBRET between the RlucII-fused receptors and rGFP located in early endosomes20 In addition, in Cluster 2, the p.H131R3.56 variant is located at the interface between the cytoplasmic end of TM3 and ICL2, at a location where it could interact directly with βarrestin-2 (Figure 5C). This variation also places a positively charged residue next to the highly conserved p.Y1263.51, which might alter its function by establishing a spurious cation−π interaction. Interestingly, this region has been proposed to be a secondary site for βarrestin interaction in other GPCRs.31,32 The p.I257F6.54 variant is located at the extracellular side of TM6 (Figure 5C), a site already shown to be important for the active melatonin-bound form of MT1, favoring the conformational changes necessary for signal transmission.33 Clement et al. proposed that, similarly to rhodopsin,34 ECL2 of MT1 stabilizes TM5, TM6, and TM7 upon activation, particularly through the interaction between ECL2 and p.I2576.54. Consequently, we suggest that the variation p.I257F6.54 alters the interactions with ECL2 decreasing the stability of the MT1 active form. Furthermore, p.I2576.54 is located one turn above the highly conserved proline-kink at p.P2536.50, a key residue in shaping TM6. The extracellular sides of TM6 and TM7—near position p.I2576.54—form important interactions to maintain the fold of the receptor,35 and TM7 is known to be important for the interaction between GPCRs and βarrestin.36 Thus, the p.I257F6.54 variant might also destabilize the TM6/7 interface and prevent the appropriate location of TM7 impairing interactions between MT1 and βarrestin-2. The last variant (p.S87L2.64) of Cluster 2 is located away from known potential functional sites (Figure 5C), which makes it difficult to rationalize its effect from a structural point of view.
We propose that residues from Cluster 2 contribute to an allosteric network between the binding pocket and the domains involved in the engagement of βarrestin-2. The fact that these variations do not affect the efficacy or potency of MT1 to activate Gαi/o family members rules out the possibility that the functional defects result from a more general misfolding of the receptor. Regarding this specific loss of βarrestin-2 recruitment and Gα12 and/or Gα15 activation, we cannot exclude the possibility that a modest nonspecific structural impairment has a more important impact on the pathways that are less efficiently coupled to the receptor. Indeed, the lower potency of MT1 to activate Gα12 and Gα15 vs the Gαi/o family members suggests that these pathways are indeed less efficiently coupled to the receptor.
The burden of the seven variants of Cluster 2 were not found to be associated with an increased risk of type 2 diabetes or obesity (Table S1 and Table S2).
The three variants of Cluster 3 (p.I112N3.37, p.R125C3.50, and p.L138PICL2) (Figure 5D) have a more severe effect on the signaling profile of MT1. On one hand, as in Cluster 2, they completely abolish βarrestin-2 recruitment (Figure S1), but moreover, they result in loss of potency (EC50)—but not in efficacy (Emax) for p.I112N3.37 and p.L138PICL2—in all signaling pathways compared to MT1-WT. The possibility that this loss of potency could be due to a loss of binding affinity for MLT could be ruled out, since we observe no difference in binding affinity for these three variants compared to MT1-WT (Table 2). We suggest instead, that these variants lead to a destabilized active state and, thus, to a less efficient activation.
The variant p.L138PICL2 of cluster 3 is located in ICL2, which forms a short α-helix in several GPCRs when activated that can then interact with G proteins37 and βarrestins (Figure 5D). The exchange of a leucine for a proline is expected to either disrupt the α-helix or at least prevent an optimal position, which would lead to a less efficient interaction with G proteins and βarrestin-2 and the observed decrease in potency.
The variant p.I112N3.37 is located in the middle of TM3 (Figure 5E) near residues p.I1153.40 and p.P1995.50, which are part of a key switch (the “triad” or PIF motif) for GPCR activation.38 The p.I112N3.37 variant would alter the packing and physicochemical properties of this region, leading to a less efficient activation of the PIF motif and an impaired transduction of the conformational changes through TM6 and TM5 that lead to the formation of the active state. As in p.L138PICL2, the p.I112N3.37 variant can still activate G proteins (its Emax is unaffected), but these events are less efficient most likely due to an impaired conformational change in TM5 and TM6 leading to the observed decrease in potency.
The p.R125C3.50 variant (Figure 5D) changes a key residue of the D/NRY motif, known to be essential for GPCR function and which is the only natural variants directly involved in the binding of βarrestins and G proteins. The p.R125C3.50 variant shows a total loss of signaling capacity, except for GαoB which remains well activated by this variant, an observation that remains without explanation at the current state.
The burden of the three variants of Cluster 3 were not associated with an increased risk of type 2 diabetes or obesity (Table S2). The p.P80L2.57 variant, with a completely impaired MLT binding (Figure 5F), is located in the YPYP motif (residues p.Y792.56 to p.P822.59) that shapes the ligand binding pocket by modulating a kink in TM2.25,39 Thus, the proline to leucine substitution most likely modifies the local structure of TM2 and disrupts the binding pocket, leading to its inability to bind MLT. This variant was very rare in the Genome Aggregation Database (gnomAD; v2.1.1) with a MAF below 0.00005 and was found in a normal-glucose, obese individual from the present study.
Discussion
Here, we took advantage of the large diversity of naturally selected variants in the coding region of the human MTNR1A gene encoding the MT1 receptor to obtain mechanistic insights into the function of this receptor. Notably, all impactful mutants show a loss-of-function phenotype, and no gain of function for the pathways engaged by the WT receptor was observed. The possible gain of function for the pathways not engaged by the WT receptor was not assessed. One mutant, p.P80L2.57, was a complete loss-of-function mutant as it could not bind MLT. Based on the functional profiles of the MLT-responsive mutants, their location in the MT1 structure, and the general knowledge about the role of specific regions in receptor function, we were able to classify these impactful natural variants into two categories: those with severely impaired overall signaling locating near regions identified as key regions of general GPCR activation and those with major defects on more specific signaling functions, i.e., βarrestin-2 recruitment, that are located in distinct regions of ICLs and H8.
Our study reports the first exhaustive characterization of the G protein activation profile of the MT1 receptor. Upon MLT stimulation, this receptor can activate a broad set of G proteins (Gαi1, Gαi2, Gαi3, GαoA, GαoB, Gα12, and Gα15) in addition to the recruitment of βarrestin-2 in transfected HEK293 cells. These results are consistent with the notion that MT1 inhibits cyclic adenosine monophosphate (cAMP) production through pertussis-toxin-sensitive G proteins.40 Here, we provide direct evidence for the ability of MT1 to engage the entire family of Gαi/o proteins. We also show that in the Gαq/11 family only Gα15 (but not Gαq or Gα11) can be directly activated. Notably, Gα15 is an atypical member of the Gαq/11 family that has been found to be activated by a large number of GPCRs.41 The lack of observed coupling to Gαq and Gα11 is not due to a lack of sensitivity of the biosensors or the HEK293 cells used, since coupling to more conventional Gαq/11 family members (Gαq and Gα11) can be measured in HEK293 cells for other GPCRs.42,43 Coupling of MT1 to Gα15 is likely to be of physiological relevance as MT1 expression has been reported in Gα15-expressing lymphocytes in which MLT promotes diacylglycerol production.44 MLT-promoted activation of Gα15 appears to be specific to MT1, as it was not observed for MT2 in our previous study using the same biosensors.5 However, given the high sequence similarity between MT1 and MT2 for cluster 2 residues, no conclusion on the determinants of Gα12 and Gα15 coupling could be drawn. Our study also reveals for the first time the coupling of MT1 to Gα12, a G protein linked to the activation of small G proteins of the Rho family and cytoskeleton modulation. Coupling to Gα12 appears to be specific to MT1, as it was also not observed for MT2.5 Coupling of MT1 to Gα12 is consistent with previous studies reporting cytoskeleton changes upon stimulation of MT1.45 In conclusion, our study shows that MT1 couples not only to Gαi/o proteins, like MT2, but shows a more expended profile by coupling in addition to Gα12 and Gα15 proteins.
Previous reports showed that frequent and rare variants in the MTNR1B gene encoding the MT2 melatonin receptor are associated with type 2 diabetes (T2D) risk.23 The most extensively studied frequent rs10830963 variant, located in the unique intron, affects insulin secretion, most likely during the development of prediabetic fasting hyperglycemia.46 Carriers of a rare variant, located in the coding region, display a number of adverse sleep, circadian, and caloric intake phenotypes that might participate in their T2D risk.47 We therefore evaluated whether the genetic MT1 variants identified in our cohort of 8687 Europeans could be associated with increased risks of T2D or obesity. We were unable to detect any association either in children or in adults. The absence of association with obesity is consistent with previous observations that did not find strong support for a major role of MLT in body weight regulation and lipid metabolism in adult humans.46 They are contrasting with rodent data showing that MLT has a beneficial effect on body weight regulation by limiting body weight gain and adipose tissue mass48 as well as the phenotype of MT1 knockout mice that develop selective leptin resistance in the arcuate nucleus, an important hypothalamic center for body weight control.49 Studies in rodents indicate also a general beneficial role of melatonin on glucose homeostasis49 and MT1 knockout mice show systemic insulin resistance, most likely because of decreased insulin sensitivity and impaired hepatic glucose production.50,51 There exists some information in humans supporting the beneficial role of melatonin on glucose regulation, but the role of MT1 remains unknown (see ref (46) for review). Our data on nonsynonymous MTNR1A gene variants or on genetically loss-of-function variants (i.e., p.W93*, p.Y170*, c.184 + 1G > T, c.999_1014del/p.W333*; Table S2) do not support a major role of MT1 in T2D development. Further studies, including copy number variations, will be necessary to fully address this point, as the MTNR1A gene shows an exceptionally high number of gene duplications compared to other GPCR genes.2 In conclusion, these observations indicate important species-specific differences concerning the role of MT1 in T2D and obesity development between humans and rodents. Existing data are compatible with nonredundant functions of both MLT receptors in humans with a predominant role of MT2 in the regulation in glucose homeostasis. Whether the variants significantly impacting G protein signaling and/or βarrestin recruitment could be associated with specific phenotypes in humans remains to be determined.
The signaling profiles obtained for 31 naturally occurring MT1 variants reveals different signaling signatures that allowed us to classify them in three different clusters: Cluster 1 with no effect in the signaling profiles; Cluster 2 showing a major reduction in βarrestin-2 recruitment and different Gα protein activations, in particular Gα12 and/or Gα15; and Cluster 3 with severe impairing of overall signaling. Interestingly, EA scores matched the experimental results, which cross-validates the measured signaling profiles and the predictive power of evolutionary-based approaches.
The analysis of the location of these different clusters in three-dimensional structural models of active MT1 allowed us to propose specific mechanisms for the effect of the variations on receptor activation and signaling. The effects in Cluster 2 can be explained in most of the cases by alterations in the structure/dynamics of H8 that impair receptor-βarrestin-2 interactions. On the other hand, each variant in Cluster 3 is located near key regions for GPCR activation (the helical ICL2, the D/NRY motif, and the PIF motif); these variations may thus alter the signaling profile by altering the environment of these activation switches and impairing their function.
Interestingly, we found specific mutations that have a greater impact on βarrestin-2 recruitment, exemplifying how distinct receptor domains and allosteric interaction networks are involved in functional selectivity, i.e., the selective engagement of distinct signaling pathways. Remarkably, we found natural MT1 variants with signaling defects in apparently healthy people. However, the lack of detectable disease association may be due to the rare frequency of the variants that prevent robust statistical analysis. Alternatively, as MLT can act through two receptors—MT1 and MT2—one could think that MT2 could compensate for any loss of MT1 activity. However, it has been recently shown that MT2 can only activate Gαi/o family members and to recruit βarrestin-2.5 Thus, such compensation from MT2 would only lead to partial signaling in response to MLT production. Most striking defects would be expected in individuals carrying MT1 defective variants in those cells/tissues in which MT2 is unable to compensate the MT1 defect, either because of the absence of MT2 expression or because of a MT1-specific function. Prominent examples are lymphocytes and MCF-7 human breast cancer cells, both expressing exclusively MT1 receptors. In addition, differential roles of MT1 and MT2 in sleep and circadian rhythm regulation have been suggested.16 Based on our observation that several variant carriers show a complete loss of βarrestin-2 recruitment to MT1, the study of such variants in these biological system should be informative on the role of MT1-mediated βarrestin-2 signaling.
In conclusion, we have shown that sequencing and the functional characterization of rare variants combined to their computational analysis in relation with structural modeling represent valuable tools to identify key functional regions or residues in a receptor and allow prediction and rationalization of the effect of mutations on functional selectivity.
Methods
Study Participants
We investigated 8687 blood DNA samples from several population studies: (1) 3443 participants from the D.E.S.I.R. 9-year prospective study including middle-aged men and women from western France;52 (2) 2642 participants who were recruited and followed up either by the CNRS UMR8199 (Lille, France), by the Department of Nutrition of Hotel-Dieu Hospital (Paris, France), or by the Centre d’Etude du Polymorphisme Humain (CEPH, Saint-Louis Hospital, Paris, France);53,54 (3) 1567 participants who were recruited from the Department of Endocrinology of the Corbeil-Essonnes Hospital (Corbeil-Essonnes, France);53 (4) 631 participants from the French Haguenau regional cohort study;55 (5) 404 participants from the French Fleurbaix-Laventie Ville Santé study.56 The study protocols were approved by local ethics committees. All participants older than 18 years signed an informed consent form. Oral assent from children or adolescents was obtained and parents (or legal guardian) signed an informed consent form.
In participants older than 18 years, obesity was defined as body mass index (BMI) ≥ 30 kg.m–2, while normal weight was defined as BMI < 25 kg.m–2. In children and adolescents younger than 18 years old, obesity was defined as BMI-for-age ≥95th percentile, while normal weight was defined as BMI-for-age <85th percentile according to the Centers for Disease Control and Prevention (CDC) growth charts. Type 2 diabetes (T2D) was defined as fasting plasma glucose ≥7.0 mmol L–1, and/or use of hyperglycemia treatment; while normal glucose was defined as fasting plasma glucose <6.1 mmol L–1 without hyperglycemia treatment.
Next-Generation Sequencing, Detection of MTNR1A Coding Variants, and Their Association Analyses
NGS targeting the coding exons of MTNR1A (NM_005958.4) was done as previously described.57 In brief, target enrichment was performed according to the manufacturer’s protocol (NimbleGen SeqCap EZ) for Illumina sequencing. Sequence reads were mapped to the human genome (hg19/GRCh37) using Burrows-Wheeler Aligner (version 0.7.13). The variant calling was performed using Genome Analysis ToolKit (GATK; version 3.3). The annotation of variants was performed using the Ensembl Perl Application Program Interfaces (version 75). In the participants, all MTNR1A coding variants had a coverage higher than 8 reads and a QUAL score higher than 50. No variant had more than 5% of missing genotype (i.e., with a depth of coverage below 8 reads or a QUAL score below 50) across the participants, and no participant had more than 5% of missing genotypes (i.e., with a depth of coverage below 8 reads or a QUAL score below 50) across MTNR1A. The location of each variant was determined relative to the translation initiation codon using Human Genome Variation Society (HGVS) nomenclature for the description of variants. Among adults, we performed a case-control study for obesity adjusted for age, sex, the presence of T2D and genetic ancestry (controls were normal-weight adults), and a case-control study for T2D adjusted for age, sex, BMI, and genetic ancestry (controls were normal glucose adults with age above 40). Among children and adolescents, we performed a case-control study for obesity adjusted for age and sex (controls were normal-weight children/adolescents). Ancestry assessment was done as previously described.57 The rare variants (with a MAF below 1%) were analyzed as a single cluster using MiST. MiST provides a score statistic S(π) for the mean effect (π) of the cluster, and a score statistic S(τ) for the heterogeneous effect (τ) of the cluster. The overall p-value is the combined p-values Pπ and Pτ, from the Fisher’s procedure. The frequent variants (with a MAF above 1%) were separately analyzed using a standard logistic regression adjusted for covariates defined above.
Cell Culture and Transfection
HEK293T cells were grown in complete medium (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 4.5 g·L–1 glucose, 100 U mL–1 penicillin, 0.1 mg mL–1 streptomycin, and 1 mM glutamine) (Invitrogen, CA). Transient transfections were performed using PEI (Sigma-Aldrich), according to manufacturer’s instructions.
Analysis of Cell Surface Receptor Expression
ELISAs were performed using the mouse antiflag antibody (Sigma-Aldrich, cat# F3165, RRID: AB_259529) and a horseradish peroxidase-conjugated mouse immunoglobulin G (IgG) whole-sheep secondary antibody (Sigma-Aldrich, cat# A6782, RRID: AB_258315).
Bioluminescence Resonance Energy Transfer (BRET) Measurement
Gαi1/i2/i3/oA/oB activations was assessed by measuring change of BRET between RlucII-Gαi1/i2/i3/oA/oB and GFP10-Gγ2 upon treatment of HEK293 cells cotransfected to express receptor and BRET sensors with MLT (Sigma-Aldrich).5,19 For RlucII-Gαs, RlucII-Gαz, RlucII-Gαq, and RlucII-Gα12, we used the appropriate RlucII-tagged Gα subunits,5 GFP10-Gγ1 was used given that optimal responsive window is generally obtained using these BRET sensor pairs for these Gα proteins. In all cases, the BRET pairs were used in the presence of untagged Gβ1 protein. Gα15 activation by receptors was assessed using a unimolecular biosensor. Since MT1 stimulation by melatonin does not activate PKC in our HEK293T cells in the absence of Gα15 but does so in its presence, we used the PKC activation as a surrogate assay to measure Gα15 activation by MT1. PKC activation was assessed by measuring BRET with this unimolecular biosensor. The BRET2-based PKC sensor consists of GFP10, 2 phospho-sensing domains (FHA1 and FHA2), a long and flexible linker, 2 specific phospho-substrate sequences, a RlucII, and the diacyl glycerol (DAG)-binding “C1b” domain of PKCδ added to increase signal specificity. Once phosphorylated, the phospho-substrates interact with their respective phospho-sensing domain, which stabilizes a conformation characterized by a higher BRET signal. βarrestin-2 recruitment was assessed by measuring BRET between RlucII-βarrestin-2 and rGFP-CAAX (prenylation CAAX box of KRas) demonstrating the recruitment of the βarrestin-2 to the plasma membrane.20 Transfected cells were plated in 96-well white microplates (100 μL per well; Greiner). Forty-eight hours after transfection, cells were washed with Dulbecco’s phosphate-buffered saline, and then Tyrode’s buffer was added. After the addition of MLT, the cells were incubated for 10 min at 37 °C, and coelenterazine 400a (2.5 μM; NanoLight Technology) was added 5 min before BRET reading in a Synergy Neo Microplate reader (BioTek) (acceptor filter: 515 ± 15 nm; donor filter: 410 ± 40 nm).
To monitor receptor internalization, ebBRET was measured between MT1-WT or variants (R54W, I309T, or C314R) fused to RlucII and the endosomal compartment marker rGFP-FYVE.20 Prolume purple (2.5 μM; NanoLight Technology) is added for 6 min followed by MLT stimulation and readings are recorded on a Mithras (Berthold scientific) microplate reader (acceptor filter: 515 ± 10 nm; donor filter: 400 ± 35 nm).
Net BRET represents the BRET signal minus the signal in the absence of a GFP10-tagged sensor. ΔBRET refers to the difference in net BRET recorded from cells treated with agonist and cells treated with vehicle. For the agonist concentration–response curves, the responses for the variants were expressed as a % of the response observed for the WT receptor.
Evolutionary Action
The evolutionary action (EA) of a mutation on fitness is given by26
| 1 |
where ϕ is the fitness phenotype, γ is the genotype sequence, and f is the evolutionary function between them: f(γ) = ϕ. Although the function f describes the fitness landscape and is unknown, its derivative is formally given by eq 1, the terms of which can be approximated from evolutionary information. Specifically, dy is the magnitude of mutation, which can be calculated in terms of log odds of amino acid substitutions in the case of a coding mutation, and ∇f is the gradient of the fitness landscape, which can be calculated from the functional sensitivity to mutations at each sequence positions, which can be calculated by the coupling between sequence variations and phylogenetic divergences.58 In practice, the EA score is normalized by percentile rank from 0 to 100, where a variant is relatively more deleterious as the score close to 100. This approach tends to perform well against other predictors of variant effect in blind challenges.59 To predict the effect of each variant within the MT1 receptor, EA score is calculated using MTNR1A multiple sequence alignment retrieved from the nonredundant protein sequence (nr) database (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Statistical Analysis of Functional Analyses
Data are presented as means ± SEM of indicated “n” independent experiments, performed at least in triplicate to ensure the reliability of single values. The level of probability used in this article is defined as follows: NS: nonsignificant, *p < 0.05, **p < 0.01, ***p < 0.001 relative to control.
LogEC50 and agonist-induced Emax values were obtained following nonlinear regression (curve fit) with four parameters of data from a minimum of eight different concentrations per experiment, repeated at least three times independently. All assays performed in this study were previously validated and demonstrated the robustness and variability of the procedure using this number of independent experiments for concentration–response curves. Data and statistical analysis were performed using GraphPad Prism software version 6.0.
For each assay, the agonist-induced Emax value for every MT1 variant was normalized as a percentage of the maximal MLT-stimulated response of the WT receptor (set at 100) monitored in parallel with the receptor variant. The dose–response curves were fitted to an operational model of agonism designed by Kenakin and Christopoulos60,61 to obtain log(τ/KA) values for the WT receptor and its variants. Normally, an agonist is set as a reference agonist, against which within-pathway comparisons for the same receptor to other agonists can be made and expressed as Δlog(τ/KA). Here, within-pathway comparisons were made between MT1 variants and the WT receptor. Normalized difference was calculated on values corresponding to agonist-induced Emax, and logEC50 to fit a–1 to +1 scale using the following formula: (variant – WT)/(variant + WT). In the case of logEC50, before normalization, the antilogs were first calculated and then were fitted to the aforementioned formula. Positive and negative values represent mutations with better or worse responses, respectively, than those of the WT receptor. Subsequently, logEC50, agonist-induced Emax, and Δlog(τ/KA) values were expressed as means ± SEM of the indicated number of experiments (n). Statistical analysis for logEC50 and agonist-induced Emax was performed by comparing independent fits with a global fit that shared the selected parameter.
Computational Analysis
To quantify similarities between signaling perturbations resulting from each mutation, we adapted the NMF and K-means clustering process originally described in ref (6). We performed this analysis on an input matrix of 31 mutations and 24 signaling parameters spanning 8 signaling pathways (Gαi1/Gαi2/Gαi3/GαoA/GαoB/Gα12/Gα15/βarrestin-2) in response to melatonin. The mean and STD for each phenotypic measure were used to propagate experimental error through the clustering process. Explicitly, 1000 input matrices were generated by independently sampling from the experimental error (Mean + STD) for each phenotypic data point. Each of these sampled input matrices were then processed separately using the NMF/K-means method. The values for each phenotype were scaled to avoid bias introduced by differences in phenotype scale using
for every mutation i and every parameter j, where maximumj and minimumj values were specific to each parameter column (e.g., EC50, Emax, τ/Ka). In this way, every parameter was normalized between 0 and 1. The dimensionality of these 1000 input matrices were then independently reduced using NMF to identify the key signaling patterns. K-means clustering was used, as described, to cluster the [mutation × feature] (31 × K) basis vector. As previously described, clustering results for each sampled matrix were averaged across K = 2 to K = 6, where K is the number of features for NMF and the number of clusters for K-means (KNMF = KK-means). Final cluster assignment, quantifying mutation similarity, was the average over all 1000 sampled matrices. Clustering frequency was converted into distance matrix using Pearson’s correlation and visualized as a heatmap and dendrogram.
Radioligand Binding Experiments
Radioligand saturation binding experiments were performed in transient transfected HEK293T cell lines. Saturation binding assays were carried out on crude membrane preparations with increasing concentrations of 2-[125I]MLT (PerkinElmer Life Sciences) to determine dissociation constants (Kd) and total melatonin receptor expression (Bmax) of wild-type and mutant MT1 receptors using PRISM software (GraphPad). Nonspecific binding was determined using 10 μM melatonin.
Three-Dimensional Modeling
The location of the mutant variants in the inactive state of MT1 was mapped onto the crystal structure of MT1 bound to ramelteon (PDBid: 6ME2).25 The location of the mutant variants in the active state of MT1 was mapped onto a three-dimensional model of the receptor built by homology modeling using as templates (i) the cryo-EM structure of the human mu-opioid receptor (33% sequence similarity) in complex with Gαi (PDBid: 6DDE)62 for the transmembrane bundle (TM1–TM7; residues 21–290), and (ii) the crystal structure of the human adenosine A2A receptor (26% sequence similarity) (PDBid: 2YDO)63 for the last 18 residues of the model comprising ICL4 and helix 8 (residues 291–308). We chose A2A receptor as a template because, although it couples to Gαs, it has a high sequence similarity with MT1 in the transmembrane domain (see above) and, importantly, is phylogenetically close to it (the melatonin receptor cluster and the MECA receptor cluster are neighbors in the α group of class A GPCRs64). A structure-based sequence alignment between the templates and MT1 was obtained from the GPCRdb.65 This initial alignment was manually refined using Chimera66 to adjust the gaps in the loop regions. Using this alignment and the structural templates, 3D models of MT1 in an active conformations were built using Modeler ver. 9.16.67 The cysteine bridge between p.C1003.25 in TM3 and p.C177ecl2.50 in the second ECL was explicitly defined during model building. All models were subjected to 300 iterations of variable target function method (VTFM) optimization and thorough (md_level = refine.slow) molecular dynamics and simulated annealing optimization within Modeler, and scored using the discrete optimized protein energy potential.67 The 20 best-scoring models were analyzed visually, and a suitable model (in term of low score and structure of the loops) was selected.
To visualize the arrangement of the MT1/Gαi and MT1/arrestin complexes, the structures of Gαi (PDBid: 6DDE)62 and visual βarrestin-1 (PDBid: 5W0P)68 were structurally aligned onto the model of active MT1 (using the receptor transmembrane domains for the fit) with molecular graphics software (The PyMOL Molecular Graphics System, Version 2.2.3, Schrödinger, LLC.).
Acknowledgments
The authors are grateful to Dr. Monique Lagacé for her critical reading of the manuscript.
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- NGS
next-generation sequencing
- GRKs
GPCR-regulated kinases
- MT1
melatonin receptor 1
- MT2
melatonin receptor type 2
- MLT
melatonin
- BRET
bioluminescence resonance energy transfer
- ebBRET
enhanced bystander BRET
- WT
wild-type
- MAF
minor allele frequency
- ELISA
enzyme linked immunosorbent assay
- Emax
maximal efficacy
- NMF
non-negative matrix factorization
- TM
transmembrane
- Nterm
N-terminus
- ECL
extracellular loops
- Cterm
C-terminus
- ICL
intracellular loop
- Kd
dissociation constant
- EA
evolutionary action
- H8
helix 8
- EC50
potency
- BMI
body mass index
- CDC
Centers for Disease Control and Prevention
- T2D
Type 2 diabetes
- GATK
Genome Analysis ToolKit
- HGVS
Human Genome Variation Society
- DAG
diacyl glycerol
- CAAX
prenylation CAAX box of KRas
- GFP
green fluorescent protein
- rGFP
Renilla GFP
- Rluc
Renilla luciferase
- nr
nonredundant
- VTFM
variable target function method
- βarr-2
βarrestin-2
- HEK293 cells
human embryonic kidney 293 cells
- PKC
protein kinase C
- cAMP
cyclic adenosine monophosphate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00157.
BRET-based biosensors used (Figures S1), cell surface expression of MT1 variants (Figure S2), concentration–response curve and radial graph of MT1 variants grouped in 3 clusters (Figure S3–S4), kinetics of MT1 internalization (Figure S5), functional characterization and association of MTNR1A coding variants to diabetes or obesity (Tables S1–S3) PDF)
Author Present Address
⬡ Dept. of Molecular Pathobiology, New York University College of Dentistry, New York, New York 10010, United States of America
Author Present Address
⬢ The Wellcome-Wolfson Institute for Experimental Medicine, Queen’s University Belfast, Belfast, BT9 7BL, United Kingdom
Author Contributions
AH, CLG, BP, RJ, and MBouvier conceived and designed experiments. AH and WG made constructions of variant plasmids. AH performed most of the experiments to characterize the receptors’ functionality with the exception of binding done by WG. BS contributed to the BRET experiments. EH and OL did the clustering analysis. XD built and analyzed the 3D models. AB and PF conceived and designed the next-generation sequencing study and analyzed the genetic variants. MC and MBoissel performed statistical analyses related to genetic data. GC, RR, and BB contributed DNA samples from patients. AH, CLG, BP, and MBouvier analyzed data. PF, MBouvier, and RJ obtained the funding. AH, RJ, and MBouvier wrote the manuscript that was edited and/or approved by all authors.
This work was supported by the Fondation de la Recherche Médicale (Equipe FRM EQU201903008055) (RJ), Agence Nationale de la Recherche (ANR-2011-BSV1-012-01 “MLT2D” and ANR-2011-META “MELABETES (PF, RJ); ANR-19-CE16-0025 “mitoGPCR” (RJ); ANR-10-LABX-46 “European Genomics Institute for Diabetes“ (PF) and ANR-10-EQPX-07-01 “LIGAN-PM“ (PF)), Fonds de Recherche du Québec – Santé (26657 “MELA-BETES”) (MBouvier), Institut National de la Santé et de la Recherche Médicale (INSERM) (PF, RJ), Centre National de la Recherche Scientifique (CNRS) (PF, RJ); Canadian Institute of Health Research Foundation grant (# FDN-148431) (MBouvier), National Institutes of Health (NIH) (# R01-GM066099) (OL). AH was supported by a doctoral fellowship from University of Montreal. WG was supported by a doctoral fellowship from the Chinese Scholarship Council (China). BP was supported by a Postdoctoral Fellowship from Diabetes Canada, an Early-Career Small Grant for Basic Scientists from Diabetes UK, and a Welcome Trust Seed Award (215229/Z/19/Z). MBouvier holds the Canada Research Chair in Signal Transduction and Molecular Pharmacology.
The authors declare the following competing financial interest(s): M.B. is the president of the scientific advisory board of Domain Therapeutics which licenced-in some of the BRET-based biosensors used in the present study for their commercial use. All other authors declare no competing interests.
Notes
Data availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information. All biosensors are available for academic noncommercial studies through regular Material Transfer Agreements and can be requested by email: michel.bouvier@umontreal.ca.
Notes
Ethics approval: The study protocols were approved by local ethics committees. All participants older than 18 years signed an informed consent form. Oral assent from children or adolescents was obtained and parents (or legal guardian) signed an informed consent form.
Supplementary Material
References
- Abecasis G. R.; Altshuler D.; Auton A.; Brooks L. D.; Durbin R. M.; Gibbs R. A.; Hurles M. E.; McVean G. A. A map of human genome variation from population-scale sequencing. Nature 2010, 467 (7319), 1061–1073. 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Chavali S.; Masuho I.; Jahn L. J.; Martemyanov K. A.; Gloriam D. E.; Babu M. M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172 (1–2), 41–54. 10.1016/j.cell.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoever C.; Venkatakrishnan A.; Paggi J. M.; Heydenreich F. M.; Laurin S.-A.; Masureel M.; Tanigawa Y.; Venkataraman G.; Bouvier M.; Dror R.; Rivas M. A. Medical relevance of common protein-altering variants in GPCR genes across 337,205 individuals in the UK Biobank study. bioRxiv 2019, 876250. 10.1101/2019.12.13.876250. [DOI] [Google Scholar]
- DePristo M. A.; Weinreich D. M.; Hartl D. L. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 2005, 6 (9), 678–687. 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- Karamitri A.; Plouffe B.; Bonnefond A.; Chen M.; Gallion J.; Guillaume J. L.; Hegron A.; Boissel M.; Canouil M.; Langenberg C.; Wareham N. J.; Le Gouill C.; Lukasheva V.; Lichtarge O.; Froguel P.; Bouvier M.; Jockers R. Type 2 diabetes-associated variants of the MT2 melatonin receptor affect distinct modes of signaling. Sci. Signaling 2018, 11 (545), eaan6622 10.1126/scisignal.aan6622. [DOI] [PubMed] [Google Scholar]
- Schonegge A. M.; Gallion J.; Picard L. P.; Wilkins A. D.; Le Gouill C.; Audet M.; Stallaert W.; Lohse M. J.; Kimmel M.; Lichtarge O.; Bouvier M. Evolutionary action and structural basis of the allosteric switch controlling beta2AR functional selectivity. Nat. Commun. 2017, 8 (1), 2169. 10.1038/s41467-017-02257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Yu Y.; Shen Y.; Liu Q.; Zhao Z.; Sharma R.; Reiter R. J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. (Lausanne, Switz.) 2019, 10, 249. 10.3389/fendo.2019.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockers R.; Delagrange P.; Dubocovich M. L.; Markus R. P.; Renault N.; Tosini G.; Cecon E.; Zlotos D. P. Update on melatonin receptors: IUPHAR Review 20. Br. J. Pharmacol. 2016, 173 (18), 2702–2725. 10.1111/bph.13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecon E.; Oishi A.; Jockers R. Melatonin receptors: molecular pharmacology and signalling in the context of system bias. Br. J. Pharmacol. 2018, 175 (16), 3263–3280. 10.1111/bph.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujieda H.; Scher J.; Hamadanizadeh S. A.; Wankiewicz E.; Pang S. F.; Brown G. M. Dopaminergic and GABAergic amacrine cells are direct targets of melatonin: immunocytochemical study of mt1 melatonin receptor in guinea pig retina. Vis Neurosci 2000, 17 (1), 63–70. 10.1017/S0952523800171068. [DOI] [PubMed] [Google Scholar]
- Scher J.; Wankiewicz E.; Brown G. M.; Fujieda H. MT(1) melatonin receptor in the human retina: expression and localization. Invest Ophthalmol Vis Sci. 2002, 43 (3), 889–897. [PubMed] [Google Scholar]
- Ng K. Y.; Leong M. K.; Liang H.; Paxinos G. Melatonin receptors: distribution in mammalian brain and their respective putative functions. Brain Struct. Funct. 2017, 222 (7), 2921–2939. 10.1007/s00429-017-1439-6. [DOI] [PubMed] [Google Scholar]
- Guerrero J. M.; Reiter R. J. Melatonin-immune system relationships. Curr. Top. Med. Chem. 2002, 2 (2), 167–179. 10.2174/1568026023394335. [DOI] [PubMed] [Google Scholar]
- Skwarlo-Sonta K.; Majewski P.; Markowska M.; Oblap R.; Olszanska B. Bidirectional communication between the pineal gland and the immune system. Can. J. Physiol. Pharmacol. 2003, 81 (4), 342–349. 10.1139/y03-026. [DOI] [PubMed] [Google Scholar]
- Dubocovich M. L.; Hudson R. L.; Sumaya I. C.; Masana M. I.; Manna E. Effect of MT1 melatonin receptor deletion on melatonin-mediated phase shift of circadian rhythms in the C57BL/6 mouse. J. Pineal Res. 2005, 39 (2), 113–120. 10.1111/j.1600-079X.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- Gobbi G.; Comai S. Differential Function of Melatonin MT1 and MT2 Receptors in REM and NREM Sleep. Front. Endocrinol. (Lausanne, Switz.) 2019, 10, 87. 10.3389/fendo.2019.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benleulmi-Chaachoua A.; Hegron A.; Le Boulch M.; Karamitri A.; Wierzbicka M.; Wong V.; Stagljar I.; Delagrange P.; Ahmad R.; Jockers R. Melatonin receptors limit dopamine reuptake by regulating dopamine transporter cell-surface exposure. Cell. Mol. Life Sci. 2018, 75 (23), 4357–4370. 10.1007/s00018-018-2876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Qiao B.; Zhan Y.; Peng W.; Chen Z. J.; Sun L.; Zhang J.; Zhao L.; Gao Q. Association between genetic variations in MTNR1A and MTNR1B genes and gestational diabetes mellitus in Han Chinese women. Gynecol. Obstet. Invest. 2013, 76 (4), 221–227. 10.1159/000355521. [DOI] [PubMed] [Google Scholar]
- Gales C.; Van Durm J. J.; Schaak S.; Pontier S.; Percherancier Y.; Audet M.; Paris H.; Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 2006, 13 (9), 778–786. 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Namkung Y.; Le Gouill C.; Lukashova V.; Kobayashi H.; Hogue M.; Khoury E.; Song M.; Bouvier M.; Laporte S. A. Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 2016, 7, 12178. 10.1038/ncomms12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G.; Marshall F.; Rees S. G16 as a universal G protein adapter: implications for agonist screening strategies. Trends Pharmacol. Sci. 1996, 17 (7), 235–237. 10.1016/0165-6147(96)10026-2. [DOI] [PubMed] [Google Scholar]
- Namkung Y.; LeGouill C.; Kumar S.; Cao Y.; Teixeira L. B.; Lukasheva V.; Giubilaro J.; Simões S. C.; Longpré J.-M.; Devost D.; Hébert T. E.; Piñeyro G.; Leduc R.; Costa-Neto C. M.; Bouvier M.; Laporte S. A. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signaling 2018, 11 (559), eaat1631 10.1126/scisignal.aat1631. [DOI] [PubMed] [Google Scholar]
- Bonnefond A.; Clement N.; Fawcett K.; Yengo L.; Vaillant E.; Guillaume J. L.; Dechaume A.; Payne F.; Roussel R.; Czernichow S.; Hercberg S.; Hadjadj S.; Balkau B.; Marre M.; Lantieri O.; Langenberg C.; Bouatia-Naji N.; Charpentier G.; Vaxillaire M.; Rocheleau G.; Wareham N. J.; Sladek R.; McCarthy M. I.; Dina C.; Barroso I.; Jockers R.; Froguel P. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012, 44 (3), 297–301. Meta-Analysis of Glucose and Insulin-Related Traits Consortium (MAGIC); 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H., [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in neurosciences; Elsevier: 1995; Vol. 25; pp 366–428. [Google Scholar]
- Stauch B.; Johansson L. C.; McCorvy J. D.; Patel N.; Han G. W.; Huang X. P.; Gati C.; Batyuk A.; Slocum S. T.; Ishchenko A.; Brehm W.; White T. A.; Michaelian N.; Madsen C.; Zhu L.; Grant T. D.; Grandner J. M.; Shiriaeva A.; Olsen R. H. J.; Tribo A. R.; Yous S.; Stevens R. C.; Weierstall U.; Katritch V.; Roth B. L.; Liu W.; Cherezov V. Publisher Correction: Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 2019, 569 (7756), E6. 10.1038/s41586-019-1209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P.; Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014, 24 (12), 2050–2058. 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala D.; Lodola A.; Bedini A.; Spadoni G.; Rivara S. Homology models of melatonin receptors: challenges and recent advances. Int. J. Mol. Sci. 2013, 14 (4), 8093–8121. 10.3390/ijms14048093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feierler J.; Wirth M.; Welte B.; Schussler S.; Jochum M.; Faussner A. Helix 8 plays a crucial role in bradykinin B(2) receptor trafficking and signaling. J. Biol. Chem. 2011, 286 (50), 43282–43293. 10.1074/jbc.M111.256909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest P. G.; Bouvier M. Palmitoylation of the V2 vasopressin receptor carboxyl tail enhances beta-arrestin recruitment leading to efficient receptor endocytosis and ERK1/2 activation. J. Biol. Chem. 2003, 278 (42), 41541–41551. 10.1074/jbc.M306589200. [DOI] [PubMed] [Google Scholar]
- Sato T.; Kawasaki T.; Mine S.; Matsumura H. Functional Role of the C-Terminal Amphipathic Helix 8 of Olfactory Receptors and Other G Protein-Coupled Receptors. Int. J. Mol. Sci. 2016, 17 (11), 1930. 10.3390/ijms17111930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S.; Oakley R. H.; Kim K. M.; Caron M. G.; Barak L. S. A beta-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem. 2006, 281 (5), 2932–2938. 10.1074/jbc.M508074200. [DOI] [PubMed] [Google Scholar]
- Raman D.; Osawa S.; Gurevich V. V.; Weiss E. R. The interaction with the cytoplasmic loops of rhodopsin plays a crucial role in arrestin activation and binding. J. Neurochem. 2003, 84 (5), 1040–1050. 10.1046/j.1471-4159.2003.01598.x. [DOI] [PubMed] [Google Scholar]
- Clement N.; Renault N.; Guillaume J. L.; Cecon E.; Journe A. S.; Laurent X.; Tadagaki K.; Coge F.; Gohier A.; Delagrange P.; Chavatte P.; Jockers R. Importance of the second extracellular loop for melatonin MT1 receptor function and absence of melatonin binding in GPR50. Br. J. Pharmacol. 2018, 175 (16), 3281–3297. 10.1111/bph.14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S.; Hornak V.; Yan E. C.; Syrett N.; Goncalves J. A.; Hirshfeld A.; Ziliox M.; Sakmar T. P.; Sheves M.; Reeves P. J.; Smith S. O.; Eilers M. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat. Struct. Mol. Biol. 2009, 16 (2), 168–175. 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Deupi X.; Lebon G.; Tate C. G.; Schertler G. F.; Babu M. M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494 (7436), 185–194. 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Kang Y.; Zhou X. E.; Gao X.; He Y.; Liu W.; Ishchenko A.; Barty A.; White T. A.; Yefanov O.; Han G. W.; Xu Q.; de Waal P. W.; Ke J.; Tan M. H.; Zhang C.; Moeller A.; West G. M.; Pascal B. D.; Van Eps N.; Caro L. N.; Vishnivetskiy S. A.; Lee R. J.; Suino-Powell K. M.; Gu X.; Pal K.; Ma J.; Zhi X.; Boutet S.; Williams G. J.; Messerschmidt M.; Gati C.; Zatsepin N. A.; Wang D.; James D.; Basu S.; Roy-Chowdhury S.; Conrad C. E.; Coe J.; Liu H.; Lisova S.; Kupitz C.; Grotjohann I.; Fromme R.; Jiang Y.; Tan M.; Yang H.; Li J.; Wang M.; Zheng Z.; Li D.; Howe N.; Zhao Y.; Standfuss J.; Diederichs K.; Dong Y.; Potter C. S.; Carragher B.; Caffrey M.; Jiang H.; Chapman H. N.; Spence J. C.; Fromme P.; Weierstall U.; Ernst O. P.; Katritch V.; Gurevich V. V.; Griffin P. R.; Hubbell W. L.; Stevens R. C.; Cherezov V.; Melcher K.; Xu H. E. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523 (7562), 561–567. 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Arlow D. H.; Borhani D. W.; Jensen M. O.; Piana S.; Shaw D. E. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (12), 4689–4694. 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Fung J. J.; Pardon E.; Casarosa P.; Chae P. S.; Devree B. T.; Rosenbaum D. M.; Thian F. S.; Kobilka T. S.; Schnapp A.; Konetzki I.; Sunahara R. K.; Gellman S. H.; Pautsch A.; Steyaert J.; Weis W. I.; Kobilka B. K. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469 (7329), 175–180. 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X.; Dolker N.; Lopez-Rodriguez M. L.; Campillo M.; Ballesteros J. A.; Pardo L. Structural models of class a G protein-coupled receptors as a tool for drug design: insights on transmembrane bundle plasticity. Curr. Top. Med. Chem. 2007, 7 (10), 991–998. 10.2174/156802607780906799. [DOI] [PubMed] [Google Scholar]
- Morgan P. J.; Davidson G.; Lawson W.; Barrett P. Both pertussis toxin-sensitive and insensitive g-proteins link melatonin receptor to inhibition of adenylate cyclase in the ovine pars tuberalis. J. Neuroendocrinol. 1990, 2 (6), 773–776. 10.1111/j.1365-2826.1990.tb00639.x. [DOI] [PubMed] [Google Scholar]
- Giannone F.; Malpeli G.; Lisi V.; Grasso S.; Shukla P.; Ramarli D.; Sartoris S.; Monsurro V.; Krampera M.; Amato E.; Tridente G.; Colombatti M.; Parenti M.; Innamorati G. The puzzling uniqueness of the heterotrimeric G15 protein and its potential beyond hematopoiesis. J. Mol. Endocrinol. 2010, 44 (5), 259–269. 10.1677/JME-09-0134. [DOI] [PubMed] [Google Scholar]
- Mende F.; Hundahl C.; Plouffe B.; Skov L. J.; Sivertsen B.; Madsen A. N.; Luckmann M.; Diep T. A.; Offermanns S.; Frimurer T. M.; Bouvier M.; Holst B. Translating biased signaling in the ghrelin receptor system into differential in vivo functions. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (43), E10255–E10264. 10.1073/pnas.1804003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B.; Beautrait A.; Aguila B.; Charles R.; Escher E.; Claing A.; Bouvier M.; Laporte S. A. Differential beta-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signaling 2012, 5 (221), ra33 10.1126/scisignal.2002522. [DOI] [PubMed] [Google Scholar]
- Garcia-Perganeda A.; Pozo D.; Guerrero J. M.; Calvo J. R. Signal transduction for melatonin in human lymphocytes: involvement of a pertussis toxin-sensitive G protein. J. Immunol 1997, 159 (8), 3774–3781. [PubMed] [Google Scholar]
- Jarzynka M. J.; Passey D. K.; Ignatius P. F.; Melan M. A.; Radio N. M.; Jockers R.; Rasenick M. M.; Brydon L.; Witt-Enderby P. A. Modulation of melatonin receptors and G-protein function by microtubules. J. Pineal Res. 2006, 41 (4), 324–336. 10.1111/j.1600-079X.2006.00371.x. [DOI] [PubMed] [Google Scholar]
- Karamitri A.; Jockers R. Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol. 2019, 15 (2), 105–125. 10.1038/s41574-018-0130-1. [DOI] [PubMed] [Google Scholar]
- Imam A.; Winnebeck E. C.; Buchholz N.; Froguel P.; Bonnefond A.; Solimena M.; Ivanova A.; Bouvier M.; Plouffe B.; Charpentier G.; Karamitri A.; Jockers R.; Roenneberg T.; Vetter C. Circadian, Sleep and Caloric Intake Phenotyping in Type 2 Diabetes Patients With Rare Melatonin Receptor 2 Mutations and Controls: A Pilot Study. Front. Physiol. 2020, 11, 564140. 10.3389/fphys.2020.564140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owino S.; Buonfiglio D. D. C.; Tchio C.; Tosini G. Melatonin Signaling a Key Regulator of Glucose Homeostasis and Energy Metabolism. Front. Endocrinol. (Lausanne, Switz.) 2019, 10, 488. 10.3389/fendo.2019.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonfiglio D.; Tchio C.; Furigo I.; Donato J. Jr.; Baba K.; Cipolla-Neto J.; Tosini G. Removing melatonin receptor type 1 signaling leads to selective leptin resistance in the arcuate nucleus. J. Pineal Res. 2019, 67 (2), e12580 10.1111/jpi.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owino S.; Sanchez-Bretano A.; Tchio C.; Cecon E.; Karamitri A.; Dam J.; Jockers R.; Piccione G.; Noh H. L.; Kim T.; Kim J. K.; Baba K.; Tosini G. Nocturnal activation of melatonin receptor type 1 signaling modulates diurnal insulin sensitivity via regulation of PI3K activity. J. Pineal Res. 2018, 64 (3), e12462. 10.1111/jpi.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Alcantara S.; Baba K.; Tosini G. Removal of melatonin receptor type 1 induces insulin resistance in the mouse. Obesity 2010, 18 (9), 1861–1863. 10.1038/oby.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkau B. [An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): epidemiologic data on the insulin resistance syndrome]. Rev. Epidemiol Sante Publique 1996, 44 (4), 373–375. [PubMed] [Google Scholar]
- Sladek R.; Rocheleau G.; Rung J.; Dina C.; Shen L.; Serre D.; Boutin P.; Vincent D.; Belisle A.; Hadjadj S.; Balkau B.; Heude B.; Charpentier G.; Hudson T. J.; Montpetit A.; Pshezhetsky A. V.; Prentki M.; Posner B. I.; Balding D. J.; Meyre D.; Polychronakos C.; Froguel P. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445 (7130), 881–885. 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- Meyre D.; Delplanque J.; Chevre J. C.; Lecoeur C.; Lobbens S.; Gallina S.; Durand E.; Vatin V.; Degraeve F.; Proenca C.; Gaget S.; Korner A.; Kovacs P.; Kiess W.; Tichet J.; Marre M.; Hartikainen A. L.; Horber F.; Potoczna N.; Hercberg S.; Levy-Marchal C.; Pattou F.; Heude B.; Tauber M.; McCarthy M. I.; Blakemore A. I.; Montpetit A.; Polychronakos C.; Weill J.; Coin L. J.; Asher J.; Elliott P.; Jarvelin M. R.; Visvikis-Siest S.; Balkau B.; Sladek R.; Balding D.; Walley A.; Dina C.; Froguel P. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 2009, 41 (2), 157–159. 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- Leger J.; Levy-Marchal C.; Bloch J.; Pinet A.; Chevenne D.; Porquet D.; Collin D.; Czernichow P. Reduced final height and indications for insulin resistance in 20 year olds born small for gestational age: regional cohort study. BMJ. 1997, 315 (7104), 341–347. 10.1136/bmj.315.7104.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romon M.; Lafay L.; Bresson J. L.; Oppert J. M.; Borys J. M.; Kettaneh A.; Charles M. A. Relationships between physical activity and plasma leptin levels in healthy children: the Fleurbaix-Laventie Ville Sante II Study. Int. J. Obes. 2004, 28 (10), 1227–1232. 10.1038/sj.ijo.0802725. [DOI] [PubMed] [Google Scholar]
- Baron M.; Maillet J.; Huyvaert M.; Dechaume A.; Boutry R.; Loiselle H.; Durand E.; Toussaint B.; Vaillant E.; Philippe J.; Thomas J.; Ghulam A.; Franc S.; Charpentier G.; Borys J. M.; Levy-Marchal C.; Tauber M.; Scharfmann R.; Weill J.; Aubert C.; Kerr-Conte J.; Pattou F.; Roussel R.; Balkau B.; Marre M.; Boissel M.; Derhourhi M.; Gaget S.; Canouil M.; Froguel P.; Bonnefond A. Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat. Med. 2019, 25 (11), 1733–1738. 10.1038/s41591-019-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O.; Bourne H. R.; Cohen F. E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257 (2), 342–358. 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Katsonis P.; Lichtarge O. CAGI5: Objective performance assessments of predictions based on the Evolutionary Action equation. Hum. Mutat. 2019, 40 (9), 1436–1454. 10.1002/humu.23873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T.; Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery 2013, 12 (3), 205–216. 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Watson C.; Muniz-Medina V.; Christopoulos A.; Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3 (3), 193–203. 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; Schertler G. F. X.; Granier S.; Weis W. I.; Dror R. O.; Manglik A.; Skiniotis G.; Kobilka B. K. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558 (7711), 547–552. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G.; Tate C. G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474 (7352), 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R.; Lagerstrom M. C.; Lundin L. G.; Schioth H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63 (6), 1256–72. 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Isberg V.; de Graaf C.; Bortolato A.; Cherezov V.; Katritch V.; Marshall F. H.; Mordalski S.; Pin J. P.; Stevens R. C.; Vriend G.; Gloriam D. E. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36 (1), 22–31. 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Webb B.; Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc Protein Sci. 2016, 86, 1–37. 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- Zhou X. E.; He Y.; de Waal P. W.; Gao X.; Kang Y.; Van Eps N.; Yin Y.; Pal K.; Goswami D.; White T. A.; Barty A.; Latorraca N. R.; Chapman H. N.; Hubbell W. L.; Dror R. O.; Stevens R. C.; Cherezov V.; Gurevich V. V.; Griffin P. R.; Ernst O. P.; Melcher K.; Xu H. E. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170 (3), 457–469. 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.