

Abstract
Doxorubicin is a chemotherapeutic drug widely utilized in cancer treatment. An enzyme critical to doxorubicin metabolism is the cytosolic sulfotransferase (SULT) SULT1C4. This study investigated the functional impact of SULT1C4 single nucleotide polymorphisms (SNPs) on the sulfation of doxorubicin by SULT1C4 allozymes. A comprehensive database search was performed to identify various SULT1C4 SNPs. Ten nonsynonymous SULT1C4 SNPs were selected, and the corresponding cDNAs, packaged in pGEX-2TK expression vector, were generated via site-directed mutagenesis. Respective SULT1C4 allozymes were bacterially expressed and purified by affinity chromatography. Purified SULT1C4 allozymes, in comparison with the wild-type enzyme, were analysed for sulphating activities towards doxorubicin and 4-nitrophenol, a prototype substrate. Results obtained showed clearly differential doxorubicin-sulphating activity of SULT1C4 allozymes, implying differential metabolism of doxorubicin through sulfation in individuals with distinct SULT1C4 genotypes.
Keywords: doxorubicin, cytosolic sulfotransferase, single nucleotide polymorphisms, SNPs, sulfation, SULT, SULT1C4
Graphical Abstract

Doxorubicin is an anthracycline anticancer agent commonly used to treat a wide variety of cancers including Hodgkin's and non-Hodgkin's lymphomas, sarcoma, ovarian, breast, gastric, lung and paediatric cancers (1, 2). Despite being recognized as one of the most effective chemotherapeutic agents, the use of doxorubicin is complicated by cardiomyopathy and heart failure, which develop in a dose-dependent manner for reasons not fully understood (3). The majority of patients treated with doxorubicin therefore live on with cardiac damage (4, 5). The dosage of doxorubicin that results in toxic responses has been shown to be highly variable between patients. For instance, a dosage of 1,000 mg/m2 may be tolerated by some, while a dosage of merely 200 mg/m2 may cause acute cardiotoxicity in others (6). These findings indicate that no ‘safe’ doxorubicin dose where cardiac damage will not occur can be defined, and interindividual variations should be accounted for in the treatment using doxorubicin. To better understand both the therapeutic and adverse effects of doxorubicin in different individuals, it is essential to better understand the metabolism and deactivation mechanisms to which the drug is subject. Previous studies have shown the involvement of glucuronidation, sulfation and demethylation in the metabolism of doxorubicin (7, 8).
Sulfation is a key process for the biotransformation and excretion of a variety of xenobiotics, including drugs (9). Sulfation is known to be catalyzed by the cytosolic sulfotransferases (SULTs) (10). In humans, there are 13 distinct SULTs that are classified into four families: SULT1, SULT2, SULT4 and SULT6 (11). It has been reported that of the 13 human SULTs, SULT1C4 is the only one capable of sulphating doxorubicin (12). In view of the involvement of sulfation in the metabolism of doxorubicin (7), it is conceivable that polymorphisms of the gene encoding SULT1C4 may lead to SULT1C4 allozymes with differential sulphating activity towards doxorubicin, thereby impacting the pharmacokinetics and, consequently, efficacy of the drug, as well as its adverse effects, in patients with distinct SULT1C4 genotypes.
In this study, a comprehensive database search for nonsynonymous single nucleotide polymorphisms (SNPs) of human SULT1C4 gene was carried out. Ten nonsynonymous SULT1C4 SNPs were selected and the corresponding cDNAs were generated. Respective SULT1C4 allozymes were bacterially expressed, affinity-purified and characterized with respect to their differential sulphating activity towards doxorubicin and 4-nitrophenol (4-NP), a prototype substrate.
Materials and Methods
Materials
Doxorubicin was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). 4-NP, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), adenosine-5ʹ-triphosphate, dimethyl sulphoxide (DMSO), dithiothreitol (DTT), isopropyl-1-thio-β-d-galactopyranoside (IPTG) and silica gel thin-layer chromatography (TLC) plates were obtained from Sigma Chemical Company (St. Louis, MO, USA). QIAprep Spin Miniprep Kits were products of QIAGEN (Germantown, MD, USA). PrimeSTAR® Max DNA polymerase was a product of Takara Bio (Mountain View, CA, USA). X-ray films were sourced from Products International Corporation (Mt Prospect, IL, USA). Carrier-free sodium [35S]sulphate was obtained from American Radiolabeled Chemicals (St. Louis, MO, USA). 3ʹ-Phosphoadenosine-5ʹ-phospho[35S]sulphate (PAP[35S]) was synthesized as previously described using recombinant human bifunctional PAPS synthase (13). EcoLume scintillation cocktail was obtained from MP Biomedicals (Solon, OH). Oligonucleotide primers were synthesized by Eurofins Genomics (Louisville, KY, USA). Protein molecular weight markers were from Bioland Scientific LLC (Paramount, CA, USA). Glutathione–Sepharose was a product of GE Healthcare Life Sciences (Pittsburgh, PA, USA). All other chemicals used were of the highest grade commercially available.
Identification and analysis of human SULT1C4 SNPs
SNP databases located at the National Center for Biotechnology Information (NCBI) and the UniProt Knowledgebase (UniProtKB) websites were comprehensively searched for SULT1C4 genotypes. SULT1C4 SNPs identified were analysed and categorized based on the locations of the nucleotide variations.
Preparation of SULT1C4 cDNAs encoding selected SULT1C4 allozymes
Polymerase chain reactions (PCRs) were performed to generate cDNAs encoding SULT1C4 allozymes using PrimeSTAR® Max DNA polymerase in conjunction with specific mutagenic primers. Wild-type SULT1C4 cDNA packaged in pGEX-2TK prokaryotic expression vector was used as a template. Specific sense and antisense mutagenic primers for selected SULT1C4 allozymes were designed, synthesized and used in individual PCRs. PCR-amplification conditions adopted were an initial 30 s for denaturation, followed by 12 cycles of 30 s at 94°C, 1 min at 55°C and 15 min at 72°C. Afterwards, the wild-type template was digested by Dpn I endonuclease and ‘mutated’ SULT1C4 cDNA packaged in pGEX-2TK was transformed into NEB 5-alpha Escherichia coli competent cells. Colony PCR followed by agarose gel electrophoresis was performed to verify the presence of (mutated) SULT1C4-pGEX-2TK plasmid. The positive colonies were subjected to plasmid extraction and purification using a Miniprep Kit. Individual ‘mutated’ SULT1C4 cDNA/pGEX-2TK plasmids prepared were subjected to nucleotide sequencing to verify the ‘mutations’.
pGEX-2TK harbouring mutated SULT1C4 cDNAs were individually transformed into BL21 (DE3) competent cells for the expression of recombinant SULT1C4 allozymes. Colony PCR followed by agarose gel electrophoresis were performed to identify the positive colonies. One litre of LB medium containing 100 µg/ml of ampicillin was used for bacterial growth. Transformed BL21 cells were grown at 37°C until they reached A600 nm = 0.3 followed by an overnight induction with 0.1 mM IPTG. Afterwards, the cells were collected by centrifugation and resuspended in 25 ml of ice-cold lysis buffer (containing 150 mm NaCl, 10 mM Tris–HCl, pH 8. 0 and 1 mM ethylenediamine tetraacetic acid). An Aminco French press was used to homogenize the cell suspension, followed by centrifugation for 20 min at 10,000 × g. The supernatant collected was fractionated with 1 ml of Glutathione–Sepharose resin. Following a 20-min fractionation, ice-cold lysis buffer was used to wash away unbound proteins. The resin bound with glutathione S-transferases fusion protein was treated with 10 unit/ml of bovine thrombin in a thrombin digestion buffer (containing 1 M Tris–HCl, pH 8.0, 4 M NaCl and 0.25 M CaCl2) and to cleave off the recombinant SULT1C4 allozyme. Following a 15-min digestion with constant agitation, the suspension was subjected to centrifugation. Supernatant containing purified SULT1C4 allozyme collected was analysed by sodium dodecyl sulphate (SDS)–polyacrylamide gel electrophoresis (PAGE) in order to assess the purity of the recombinant SULLT1C4 allozyme. Thereafter, Bradford protein assay was performed to measure the protein concentration.
Enzymatic assay
The sulphating activity of SULT1C4 allozymes towards 4-NP or doxorubicin was determined using an established enzymatic essay. The reaction mixture consisted of 50 mM HEPES at pH 7.4, 1 mM DTT, 14 µM sulphate donor (PAP[35S]) and the substrate (4-NP or doxorubicin) dissolved in DMSO. Reactions were carried out for 10 mins at 37°C, then terminated by heating at 100°C for 3 min. TLC analysis was performed on the reaction mixture (1 µl) using a TLC plate and a solvent solution of n-butanol/isopropanol/formic acid/water at a ratio of 8:2:1:1 (by volume). The [35S]sulphated product was visualized on the plate by means of autoradiography using an X-ray film. Afterwards, the identified product was cut out, the product eluted in 0.5 ml water and liquid scintillation counting performed.
Data analysis
GraphPad prism software (GraphPad Software, San Diego, CA) was used to analyse the data and to determine kinetic parameters using nonlinear regression. One-way analysis of variance was used to compare the specific activities of wild-type SULT1C4 versus SULT1C4 allozymes, followed by Dunnett’s post hoc analysis. P-values <0.05 indicated statistical significance.
Results
Identification and analysis of human SULT1C4 SNPs
The National Center for Biotechnology Information (NCBI) and the UniProt Knowledgebase (UniProtKB) databases were searched for SULT1C4 SNPs. In total, 2,849 SULT1C4 SNPs were identified and categorized as the following: 4 in the 3ʹ-spliced region, 3 in the 5ʹ-spliced region, 345 in the 3ʹ-untranslated region, 179 in the 5ʹ-untranslated region, 2,004 in introns, 74 synonymous coding SNPs (cSNPs), 203 nonsynonymous cSNPs, 17 frame shift SNPs, 10 nonsense SNPs and 10 stop-gained SNPs. Supplementary Table S1 shows the 203 nonsynonymous SULT1C4 cSNPs and their respective code numbers, positions of nucleotide change, as well as corresponding amino acid changes. Ten nonsynonymous SULT1C4 cSNPs were selected for further investigation on the basis of the location of the SNP (within or in proximity to the dimerization motif, PAPS-binding site, active site and/or substrate-binding site) and the chemical nature of the altered amino acid residues [polar to (or from) nonpolar, acidic to (or from) basic and turn-inducing to (or from) nonturn-inducing residues], which may potentially affect protein conformation and function. The designated names and their SNP ID number for these 10 SNPs are: SULT1C4-T59R [Reference SNP (rs) rs368540945], SULT1C4-H115R (rs752596847), SULT1C4-A136P (rs748668211), SULT1C4-L156P (rs769869249), SULT1C4-S235L (rs750911106), SULT1C4-F236L (rs749518195), SULT1C4-D237N (rs372071845), SULT1C4-M263L (rs774975544), SULT1C4-R264k (rs762573156) and SULT1C4-V277M (rs141659142). The location of the selected SNPs and the altered amino acid residues in the structure of the SULT1C4 enzyme are depicted in Fig. 1. Table I shows the selected SULT1C4 cSNPs, their allele frequencies and the mutagenic primers designed for the PCR-amplification of the cDNAs encoding the corresponding SULT1C4 allozymes.
Fig. 1.

Ribbon diagram of the structure of human SULT1C4-4-nitrophenol-PAP complex showing the locations of amino acid residues involved in the SULT1C4 cSNPs. PAP and 4-nitrophenol molecules in the structure are shown by bond structures. The amino acid sequence of the human SULT1C4 demonstrating residues reported to be involved in substrate-binding, PAPS-binding, C-terminal dimerization motif region and/or catalysis. The figure was generated using Maestro 12.6 (Schrödinger, LLC) and the reported crystal structure of SULT1C4 (Protein Data Bank code: 2GWH) (30).
Table I.
List of human SULT1C4 cSNPs, their minor allele frequencies and mutagenic primer sets designed for the PCR-amplification of the corresponding cDNAs
| hSULT1C4 Allozyme | Mutagenic primer | Minor allele frequency |
|---|---|---|
| hSULT1C4-Thr59Arg | 5ʹ-ATCCTAAAGCAGGAACAACATGGACTCAGGAGATAT3ʹ | (0.00002–0.0001) |
| 5ʹ-ACTATCTCCTGAGTCCATGTTGTTCCTGCTTTAGGAT-3ʹ | ||
| hSULT1C4-His115Arg | 5ʹ-CACGGATCCTGAAAACACATCTTCCCTTTCACTTGCT-3ʹ | 0.00001 |
| 5ʹ-AGCAAGTGAAAGGGAAGATGTGTTTTCAGGATCCGTG-3ʹ | ||
| hSULT1C4-Ala136Pro | 5ʹ-TGTAAGATAATCTATGTAGCAAGAAATCCCAAGGACA-3ʹ | 0.00002 |
| 5ʹ-TGTCCTTGGGATTTCTTGCTACATAGATTATCTTACA-3ʹ | ||
| hSULT1C4-Leu156Pro | 5ʹ-AAAGAATGAATAAAGCTCTTCCTGCTCCAGGAACATG-3ʹ | 0.00002 |
| 5ʹ-CATGTTCCTGGAGCAGGAAGAGCTTTATTCATTCTTT-3ʹ | ||
| hSULT1C4-Ser235Leu | 5ʹ-AAATTGTCCATTACACTTCGTTTGATGTCATGAAACA-3ʹ | 0.00002 |
| 5ʹ-TGTTTCATGACATCAAACGAAGTGTAATGGACAATTT-3ʹ | ||
| hSULT1C4-Phe236Leu | 5ʹ-ATTGTCCATTACACTTCGTTTGATGTCATGAAACAGA-3ʹ | 0.00001 |
| 5ʹ-TCTGTTTCATGACATCAAACGAAGTGTAATGGACAAT-3ʹ | ||
| hSULT1C4-Asp237Asn | 5ʹ-GTCCATTACACTTCGTTTGATGTCATGAAACAGAATC-3ʹ | 0.0001 |
| 5ʹ-GATTCTGTTTCATGACATCAAACGAAGTGTAATGGAC-3ʹ | ||
| hSULT1C4-Met263Leu | 5ʹ-CACTCCATTTCTCCATTCATGAGAAAAGGGGCAGTGG-3ʹ | 0.00003 |
| 5ʹ-CCACTGCCCCTTTTCTCATGAATGGAGAAATGGAGTG-3ʹ | ||
| hSULT1C4-Arg264Lys | 5ʹ-CCATTTCTCCATTCATGAGAAAAGGGGCAGTGGGAGA-3ʹ | 0.00002 |
| 5ʹ-TCTCCCACTGCCCCTTTTCTCATGAATGGAGAAATGG-3ʹ | ||
| hSULT1C4-Val277Met | 5ʹ-TGGAAGAAACACTTCACCGTGGCTCAGAATGAGAGAT-3ʹ | (0.0001–0.0022) |
| 5ʹ-ATCTCTCATTCTGAGCCACGGTGAAGTGTTTCTTCCA-3ʹ |
Note: The bold underlined letters indicate altered/mutated nucleotides (SNPs).
Preparation of recombinant human SULT1C4 allozymes
Recombinant SULT1C4 allozymes were bacterially expressed and purified using Glutathione–Sepharose affinity chromatography as described in Materials and Methods section. Figure 2 shows the SDS-gel electrophoretic pattern of the 10 purified SULT1C4 allozymes together with the wild-type enzyme. All 10 SULT1C4 allozymes, as well as the wild-type enzyme, appeared highly homogeneous and all migrated at ∼35.5 kDa position, which is in accordance with their predicted molecular weights.
Fig. 2.

SDS-gel electrophoretic pattern of the purified human SULT1C4 allozymes. SDS–PAGE was performed on a 12% gel, followed by Coomassie Blue staining. Samples analysed in lanes 1 through 11 correspond to SULT1C4-WT (wild-type), SULT1C4-T59R, SULT1C4-H115R, SULT1C4-A136P, SULT1C4-L156P, SULT1C4-S235L, SULT1C4-F236L, SULT1C4-D237N, SULT1C4-M263L, SULT1C4-R264K and SULT1C4-V277M. Positions of protein molecular weight markers are indicated on the left.
Characterization of the sulphating activity of human SULT1C4 allozymes towards 4-NP and doxorubicin
Concentration dependence of the sulfation of 4-NP (a prototypic substrate) and doxorubicin by wild-type SULT1C4 was first evaluated (Fig. 3). The sulfation of 4-NP and doxorubicin by SULT1C4 appeared to follow Michaelis–Menten kinetics until reaching respective substrate concentrations of 10 and 2,000 µM. At higher concentrations, significant substrate inhibition was observed. Kinetic constants (Km, Vmax and Vmax/Km) were determined using GraphPad Prism 7 and the Michaelis–Menten equation with nonlinear regression (Table II). Based on the Km value determined for wild-type SULT1C4, three substrate concentrations, one well below Km, one near Km and one well above Km, were selected to examine the sulphating activity of SULT1C4 allozymes.
Fig. 3.

Kinetic analysis for the sulfation of the tested substrates by wild-type human SULT1C4. (A) and (B) plots represent the nonlinear Michaelis–Menten enzyme kinetics for 4-NP and doxorubicin, respectively. Data shown represent calculated mean ± standard deviation derived from three experiments.
Table II.
Kinetic parameters of wild-type SULT1C4 with 4-NP and doxorubicin as substratesa
| Substrate | K m (µM) | V max (nmol/min/mg) | V max/Km (ml/min/mg) |
|---|---|---|---|
| 4-NP | 0.57 ± 0.06 | 0.75 ± 0.03 | 1.320 |
| Doxorubicin | 249.93 ± 32.28 | 1.07 ± 0.05 | 0.004 |
Data shown represent calculated mean ± standard deviation derived from three experiments.
Doxorubicin as the substrate
At the tested concentrations of 50, 250 and 1,000 µM doxorubicin, all 10 SULT1C4 allozymes showed differential sulphating activities compared with the wild-type enzyme (Fig. 4). Five of the 10 allozymes (SULT1C4-T59R, SULT1C4-H115R, SULT1C4-L156P, SULT1C4-F236L and SULT1C4-R264k) showed no detectable sulphating activity towards doxorubicin, with the exception of a drastically reduced activity detected for SULT1C4-F236L at the mid (250 µM) substrate concentration. Meanwhile, two allozymes (SULT1C4-M263L and SULT1C4-S235L) showed 72% and 21% lower sulphating activities at low substrate concentration (50 µM), 38% lower activities (for both allozymes) at mid concentration (250 µM) and 70% and 62% lower activities at high substrate concentration (1,000 µM). The remaining three allozymes (SULT1C4-A136P, SULT1C4-D237N and SULT1C4-V277M) consistently showed higher sulphating activities than the wild-type enzyme, with SULT1C4-D237N exhibiting the highest activities, by 66%, 52% and 26%, respectively, at 50, 250 and 1,000 µM substrate concentrations.
Fig. 4.
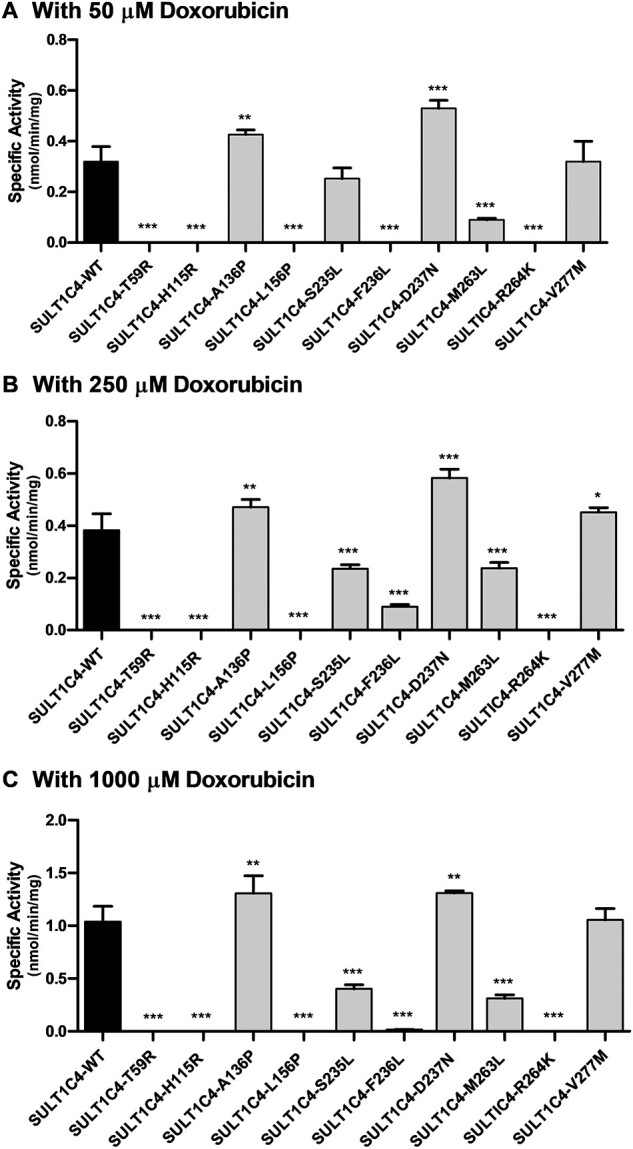
Specific activities of the sulfation of doxorubicin human SULT1C4 allozymes. Concentrations of doxorubicin used in the enzymatic assays were 50 µM (A), 250 µM (B) and 1,000 µM (C). Specific activity refers to nmol doxorubicin sulphated/min/mg of purified allozyme. Data shown represent mean ± standard deviation derived from three determinations. WT refers to wild-type SULT1C4. Data that were statistically significant with regards to the SULT1C4-wild-type is: *P < 0.05.
4-NP as the substrate
At the tested concentrations of 0.1, 0.5 and 2 µM 4-NP, all 10 SULT1C4 allozymes showed similar patterns of sulphating activity (Fig. 5). Three of the 10 allozymes (SULT1C4-T59R, SULT1C4-H115R and SULT1C4-L156P) displayed no detectable activity, while two (SULT1C4-F236L and SULT1C4-R264k) showed barely detectable activity. Meanwhile, the sulphating activities of SULT1C4-D237N and SULT1C4-V277M were consistently lower than that of the wild-type enzyme, being lower by 41% and 19%, respectively. Of the remaining three allozymes, SULT1C4-M263L and SULT1C4-S235L displayed sulphating activities comparable to those of the wild-type at low (0.1 µM) and high (2 µM) substrate concentrations, with notably lower activity (25% and 27%, respectively) at the mid (0.5 µM) substrate concentration. Only one allozyme (SULT1C4-A136P) displayed sulphating activities higher than those of the wild-type enzyme at all three substrate concentrations, with a 30% increase at low (0.1 µM) substrate concentration.
Fig. 5.
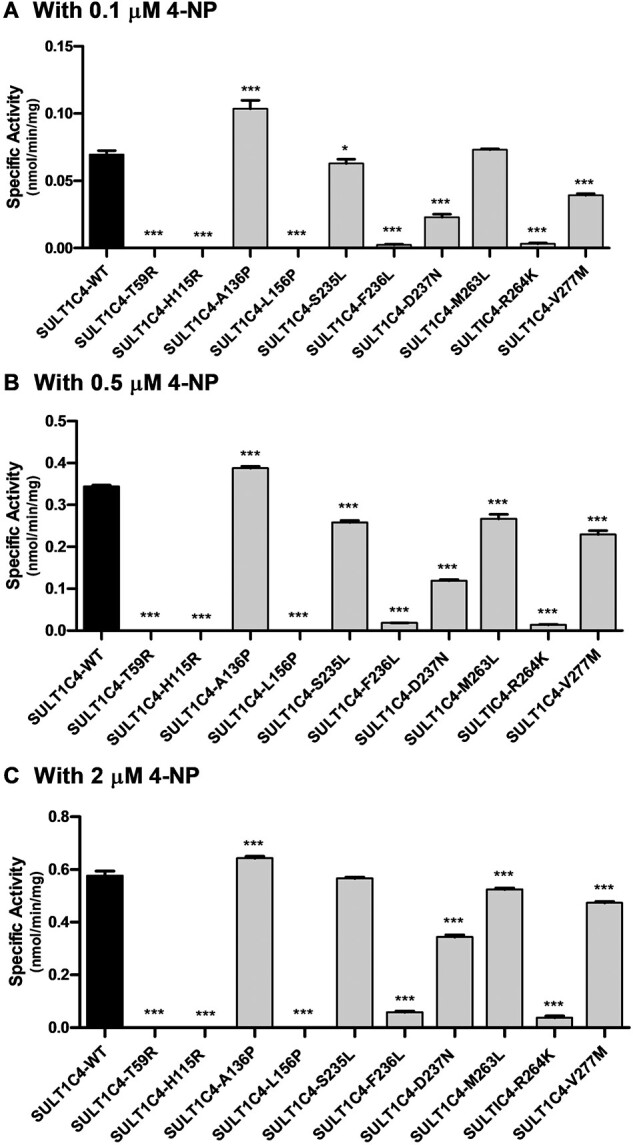
Specific activities of the sulfation of 4-NP human SULT1C4 allozymes. Concentrations of 4-NP used in the enzymatic assays were 0.1 µM (A), 0.5 µM (B) and 2 µM (C). Specific activity refers to nmol 4-NP sulphated/min/mg of purified allozyme. Data shown represent mean ± standard deviation derived from three determinations. WT refers to wild-type SULT1C4. Data that were statistically significant with regards to the SULT1C4-wild-type is: *P < 0.05.
Discussion
The chemotherapeutic drug doxorubicin is widely used in the treatment of numerous cancers, including prostate cancer, breast cancer and multiple myeloma (1, 2). However, the pharmacokinetic parameters of doxorubicin showed considerable variation from patient to patient (14). Whether and how genetic variation may affect doxorubicin response has received attention only recently (2). An important aspect is the possible impact of genetic polymorphisms of the genes coding for enzymes involved in the metabolism of doxorubicin, which may affect the efficacy and risk of toxicity of doxorubicin for individual patients.
Sulphate conjugation has been reported to be a major pathway for the metabolism of doxorubicin (7). A recent study has revealed that human SULT1C4 is the only enzyme that is responsible for the sulfation of doxorubicin (12). It is an interesting question whether the genetic polymorphisms of the coding gene may result in SULT1C4 allozymes with differential doxorubicin-sulphating activity. A systematic database search performed in the current study revealed 203 nonsynonymous cSNPs of the SULT1C4 gene. Ten of these SNPs were selected for experimental analysis of the encoded proteins. The corresponding cDNAs were produced through site-directed mutagenesis. The resultant recombinant SULT1C4 allozymes were bacterially expressed and affinity-purified. In characterizing the purified SULT1C4 allozymes, kinetic studies on the sulfation of doxorubicin and 4-NP (a prototypic substrate) were first performed using wild-type SULT1C4. Three substrate concentrations (one well below the Km, one close to the Km and one well above the Km; cf. Table II) were then used to analyse the differential sulphating activities of SULT1C4 allozymes towards doxorubicin and 4-NP (cf. Figs 4 and 5).
The crystal structure of human SULT1C4 has been solved (15). A number of amino acid residues integral to the enzyme function have been identified, including: the 5ʹ-phosphosulfate-binding loop (52TYPKAGT58), the PAPS/PAP-binding regions (55KAGTTW60, 234TSFDVM239, 262FMRKG266, Arg137, Ser145 and Tyr200), the catalytic histidine His115, the substrate-binding residues (Lys113 and Thr114) (15), the C-terminal dimerization motif region (272KKHFTVAQNE281) (16) and the β-sheet in the N-terminal region, which is important for proper folding of the protein (15). In the current study, nonsynonymous cSNPs that affect these residues or those in the critical regions mentioned above were selected.
Five (SULT1C4-T59R, SULT1C4-H115R, SULTL156P, SULT1C4-F236L and SULT1C4-R264k) of the 10 allozymes tested showed a drastic decrease, if not a total loss, of their catalytic activities towards doxorubicin and 4-NP. The loss of sulphating activity for SULT1C4-T59R was possibly due to the substitution of a polar uncharged threonine with a positively charged arginine. This change likely disrupted the hydrogen bonding of the enzyme to PAPS, the cosubstrate of the enzyme. SULT1C4-H115R also displayed a complete loss of sulphating activity. In this allozyme, replacement of the catalytic histidine with arginine likely affected deprotonation of the substrate, thereby suppressing the dissociation of the sulphuryl group from PAPS. It has been proposed that in human SULT1E1, the catalytic histidine accepts a proton from the phenyl group of 17β-estradiol and thereby facilitates nucleophilic attack on the sulphur atom of PAPS (17, 18). The loss of sulphating activity observed for SULT1C4-H115R is consistent with this proposition and similar findings on other SULTs in which the catalytic histidine is replaced (18–21). The amino acid substitution of SULT1C4-L156P, another allozyme shown to lose the sulphating activity, does not fall in regions known to be key for enzyme functions. However, the introduction of a turn-inducing proline residue may affect the overall conformation of this allozyme and result in the loss of its activity (22).
Two other SULT1C4 allozymes (SULT1C4-F236L and SULT1C4-R264K) displayed barely detectable activities towards 4-NP at all tested concentrations and with virtually no activity towards doxorubicin. In SULT1C4-F236L, a residue with an aromatic side-chain (phenylalanine) is replaced by one with an aliphatic side-chain (leucine). This side-chain difference may explain the dramatic decrease in enzyme activity. In another allozyme, SULT1C4-R264K, Arg264, has been identified as a conserved residue located within the PAPS-binding region (23). In SULT2B1b, this residue has been shown to form a hydrogen bond with the negatively charged O3P phosphate oxygen of PAPS (24). Therefore, the substitution of this arginine with lysine in SULT1C4-R264K can be expected to impair PAPS binding.
Two other allozymes (SULT1C4-S235L and SULT1C4-M263L) exhibited higher yet still markedly reduced sulphating activity. In SULT1C4-S235L, the affected serine is conserved in all SULT family members. Studies on SULT2B1b have suggested that the oxygen of the carbonyl group of this serine residue forms a hydrogen bond with the adenine group of PAPS (24). In SULT1C4-S235L, the replacement of this serine with leucine therefore may prevent this hydrogen bonding, which unsurprisingly exerted a negative effect on the sulphating activity of this allozymes. On the other hand, the amino acid substitution in SULT1C4-M263L is located in the PAPS/PAP-binding region. The substitution of methionine with leucine may similarly disrupt the hydrogen bonding of the enzyme with the cosubstrate PAPS. The small variations in the sulphating activity of SULT1C4-M263L towards doxorubicin and 4-NP could be related to the differences in the chemical structure of the two substrates.
In contrast to the seven SULT1C4 allozymes discussed above, three others (SULT1C4-A136P, SULT1C4-D237N and SULT1C4-V277M) displayed increased sulphating activity towards doxorubicin. In SULT1C4-A1365P, the replacement of alanine (a nonturn-inducing residue) with proline (a turn-inducing residue) may result in a kink in the peptide chain. Such a conformational change may reinforce the interaction between the adjacent residue R138 and PAPS, which may be the reason for the significant increase in the sulphating activity towards both substrates, as compared to the wild-type enzyme. For SULT1C4-D237N and SULT1C4-V277M, the amino acid substitutions share some key characteristics. In SULT1C4-D237N, both the substituted aspartate and the substituting asparagine are polar amino acid residues. Aspartate-237 has been reported to be involved in PAPS binding (15). The replacement with asparagine residue that carries similarly a polar side-chain may retain and even strengthen the interaction with PAPS, thereby stabilizing the PAPS binding and leading to an increase in sulfation activity towards doxorubicin. On the other hand, the same substitution caused a reduction in sulphating activity with 4-NP. Although D237 in SULT1C4 is not known to be involved in the substrate-binding pocket, it may interact with other residues, causing conformational changes in SULT1C4 protein, which may disrupt the binding of 4-NP. On the other hand, the differential sulphating activities of SULT1C4-D237N towards doxorubicin and 4-NP could also be attributed to differences in the chemical structures of these two substrates. In the case of SULT1C4-V277M, both the substituted valine and substituting methionine are nonpolar residues. The substituted valine-277 has been reported to be involved in the dimerization of SULT enzyme (16). Whether its replacement with methionine affect the dimerization of SULT1C4 subunits and led to the increased sulphating activity towards doxorubicin remains to be clarified. The similarities may underscore the intact and even slightly enhanced sulphating activities observed for these allozymes.
As noted above, previous studies have demonstrated that SULT enzymes, including SULT1C4, are present in dimeric form (16, 25, 26). In view of the heterozygosity of the SULT1C4 gene, it is an interesting question whether the formation of heterodimeric SULT1C4 proteins, consisting of a wild-type monomer and a ‘mutated’ monomer, may occur in cells (27). Interestingly, heterodimeric SULT protein products have been reported for three SULTs isolated from rat liver (28). Further studies are warranted in order to clarify whether clarify heterodimeric SULT1C4 may indeed occur and how such heterodimeric structure may affect its sulphating activity. Another issue is in regard to the level of expression and the stability of the SULT1C4 allozymes. As noted in Fig. 2, the band intensity of several purified SULT1C4 allozymes appeared weaker in comparison with that of the wild-type enzyme. Future studies are warranted in order to clarify whether nonsynonymous SULT1C4 cSNPs may also affect the expression level and stability of SULT1C4 protein products. Finally, it is important to point out that, of the 10 SULT1C4 allozymes, six (SULT1C4-T59R, SULT1C4-S235L, SULT1C4-F236L, SULT1C4-M236L, SULT1C4-D237N and SULT1C4-R264K) contain amino acid variations within the PAPS/PAP-binding regions. It is possible that these amino acid changes may affect the affinity of these SULT1C4 allozymes for PAPS, a cosubstrate for the SULT-mediated sulfation reaction (29). Future studies are warranted in order to clarify whether the Km for PAPS varies among these SULT1C4 allozymes, in comparison with the wild-type enzyme.
In summary, the current study represented a first attempt to systematically investigate how genetic polymorphisms of human SULT1C4 gene may affect the sulphating activity of SULT1C4 allozymes towards doxorubicin. Activity data obtained revealed significant differences in the sulphating activities of these allozymes studied. These results provide crucial insights into the functional relevance of SULT1C4 polymorphisms. Pending further studies, such information may eventually aid in the prediction of pharmacokinetic profiles and toxicity risk for doxorubicin and possibly other drugs as well.
Supplementary Data
Supplementary Data are available at JB Online.
Funding
This work was supported in part by a grant from National Institutes of Health [Grant # R03HD071146].
Conflict of Interest
None declared.
Supplementary Material
Abbreviations
- DMSO
dimethyl sulphoxide
- DTT
dithiothreitol
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl-1-thio-β-d-galactopyranoside
- 4-NP
4-nitrophenol
- PAPS
3ʹ-phosphoadenosine 5ʹ-phosphosulfate
- PCRs
polymerase chain reactions;
- SDS–PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- SNPs
single nucleotide polymorphisms
- SULT
cytosolic sulfotransferase
- TLC
thin-layer chromatography.
References
- 1. Gewirtz D.A. (1999) A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 57, 727–741 [DOI] [PubMed] [Google Scholar]
- 2. Thorn C.F., Oshiro C., Marsh S., Hernandez-Boussard T., McLeod H., Klein T.E., Altman R.B. (2011) Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet. Genomics 21, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefrak E.A., Piťha J., Rosenheim S., Gottlieb J.A. (1973) A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 32, 302–314 [DOI] [PubMed] [Google Scholar]
- 4. van den Anker J.N. (2015) How to improve the safe and effective use of doxorubicin in children with cancer. Clin. Pharmacokinet. 54, 1091–1093 [DOI] [PubMed] [Google Scholar]
- 5. Von Hoff D.D., Layard M.W., Basa P., Davis H.L. Jr., Von Hoff A.L., Rozencweig M., Muggia F.M. (1979) Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 91, 710–717 [DOI] [PubMed] [Google Scholar]
- 6. Chang V.Y., Wang J.J. (2018) Pharmacogenetics of chemotherapy-induced cardiotoxicity. Curr. Oncol. Rep. 20, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takanashi S., Bachur N. (1976) Adriamycin metabolism in man: evidence from urinary metabolites. Drug Metab. Dispos. 4, 79–87 [PubMed] [Google Scholar]
- 8. Lal S., Mahajan A., Chen W.N., Chowbay B. (2010) Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr. Drug Metab. 11, 115–128 [DOI] [PubMed] [Google Scholar]
- 9. Weinshilboum R., Otterness D. (1994) Sulfotransferase enzymes in Handbook of Experimental Pharmacology (Kaufman F.C., ed.) pp. 4578, Springer-Verlag, Berlin, Heidelberg. [Google Scholar]
- 10. Coughtrie M.W. (2002) Sulfation through the looking glass–recent advances in sulfotransferase research for the curious. Pharmacogenomics J. 2, 297–308 [DOI] [PubMed] [Google Scholar]
- 11. Freimuth R.R., Wiepert M., Chute C.G., Wieben E.D., Weinshilboum R.M. (2004) Human cytosolic sulfotransferase database mining: identification of seven novel genes and pseudogenes. Pharmacogenomics J. 4, 54–65 [DOI] [PubMed] [Google Scholar]
- 12. Luo L., Zhou C., Hui Y., Kurogi K., Sakakibara Y., Suiko M., Liu M.C. (2016) Human cytosolic sulfotransferase SULT1C4 mediates the sulfation of doxorubicin and epirubicin. Drug Metab. Pharmacokinet. 31, 163–166 [DOI] [PubMed] [Google Scholar]
- 13. Yanagisawa K., Sakakibara Y., Suiko M., Takami Y., Nakayama T., Nakajima H., Takayanagi K., Natori Y., Liu M.C. (1998) cDNA cloning, expression, and characterization of the human bifunctional ATP sulfurylase/adenosine 5ʹ-phosphosulfate kinase enzyme. Biosci. Biotechnol. Biochem. 62, 1037–1040 [DOI] [PubMed] [Google Scholar]
- 14. Jacquet J.M., Bressolle F., Galtier M., Bourrier M., Donadio D., Jourdan J., Rossi J.F. (1990) Doxorubicin and doxorubicinol: intra- and inter-individual variations of pharmacokinetic parameters. Cancer Chemother. Pharmacol. 27, 219–225 [DOI] [PubMed] [Google Scholar]
- 15. Allali-Hassani A., Pan P.W., Dombrovski L., Najmanovich R., Tempel W., Dong A., Loppnau P., Martin F., Thornton J., Thonton J., Edwards A.M., Bochkarev A., Plotnikov A.N., Vedadi M., Arrowsmith C.H. (2007) Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol. 5, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petrotchenko E.V., Pedersen L.C., Borchers C.H., Tomer K.B., Negishi M. (2001) The dimerization motif of cytosolic sulfotransferases. FEBS Lett. 490, 39–43 [DOI] [PubMed] [Google Scholar]
- 17. Negishi M., Pedersen L.G., Petrotchenko E., Shevtsov S., Gorokhov A., Kakuta Y., Pedersen L.C. (2001) Structure and function of sulfotransferases. Arch. Biochem. Biophys. 390, 149–157 [DOI] [PubMed] [Google Scholar]
- 18. Pedersen L.C., Petrotchenko E., Shevtsov S., Negishi M. (2002) Crystal structure of the human estrogen sulfotransferase-PAPS complex: evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J. Biol. Chem. 277, 17928–17932 [DOI] [PubMed] [Google Scholar]
- 19. Chen G. (2004) Histidine residues in human phenol sulfotransferases. Biochem. Pharmacol. 67, 1355–1361 [DOI] [PubMed] [Google Scholar]
- 20. Kakuta Y., Petrotchenko E.V., Pedersen L.C., Negishi M. (1998) The sulfuryl transfer mechanism. Crystal structure of a vanadate complex of estrogen sulfotransferase and mutational analysis. J. Biol. Chem. 273, 27325–27330 [DOI] [PubMed] [Google Scholar]
- 21. Liu M.C., Suiko M., Sakakibara Y. (2000) Mutational analysis of the substrate binding/catalytic domains of human M form and P form phenol sulfotransferases. J. Biol. Chem. 275, 13460–13464 [DOI] [PubMed] [Google Scholar]
- 22. Betts M.J., Russell R.B. (2003) Amino acid properties and consequences of substitutions in Bioinformatics for Geneticists (Barnes R.M., Gray C.I., eds.) pp. 289–316, John Wiley & Sons Ltd., Chichester, England. [Google Scholar]
- 23. Dong D., Ako R., Wu B. (2012) Crystal structures of human sulfotransferases: insights into the mechanisms of action and substrate selectivity. Expert Opin. Drug Metab. Toxicol. 8, 635–646 [DOI] [PubMed] [Google Scholar]
- 24. Lee K.A., Fuda H., Lee Y.C., Negishi M., Strott C.A., Pedersen L.C. (2003) Crystal structure of human cholesterol sulfotransferase (SULT2B1b) in the presence of pregnenolone and 3ʹ-phosphoadenosine 5ʹ-phosphate. Rationale for specificity differences between prototypical SULT2A1 and the SULT2BG1 isoforms. J. Biol. Chem. 278, 44593–44599 [DOI] [PubMed] [Google Scholar]
- 25. Guidry A.L., Tibbs Z.E., Runge-Morris M., Falany C.N. (2017) Expression, purification and characterization of human cytosolic sulfotransferase (SULT) 1C4. Horm. Mol. Biol. Clin. Investig. 29, 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tibbs Z.E., Rohn-Glowacki K.J., Crittenden F., Guidry A.L., Falany C.N. (2015) Structural plasticity in the human cytosolic sulfotransferase dimer and its role in substrate selectivity and catalysis. Drug Metab. Pharmacokinet. 30, 3–20 [DOI] [PubMed] [Google Scholar]
- 27. Monzo M., Brunet S., Urbano-Ispizua A., Navarro A., Perea G., Esteve J., Artells R., Granell M., Berlanga J., Ribera J.M., Bueno J., Llorente A., Guardia R., Tormo M., Torres P., Nomdedeu J.F., Montserrat E., Sierra J, CETLAM (2006) Genomic polymorphisms provide prognostic information in intermediate-risk acute myeloblastic leukemia. Blood 107, 4871–4879 [DOI] [PubMed] [Google Scholar]
- 28. Kiehlbauch C.C., Lam Y.F., Ringer D.P. (1995) Homodimeric and heterodimeric aryl sulfotransferases catalyze the sulfuric acid esterification of N-hydroxy-2-acetylaminofluorene. J. Biol. Chem. 270, 18941–18947 [DOI] [PubMed] [Google Scholar]
- 29. Alherz F.A., El Daibani A.A., Abunnaja M.S., Bairam A.F., Rasool M.I., Sakakibara Y., Suiko M., Kurogi K., Liu M.C. (2019) Effect of SULT2B1 genetic polymorphisms on the sulfation of dehydroepiandrosterone and pregnenolone by SULT2B1b allozymes. Mol. Cell. Endocrinol. 496, 110535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrödinger. (2021). Schrödinger Release 2021-1: Maestro, Schrödinger. LLC, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.