

Abstract
The proinflammatory cytokine IL‐1β mediates high levels of immune activation observed during acute and chronic human immunodeficiency virus 1 (HIV‐1) infection. Little is known about the mechanisms that drive IL‐1β activation during HIV‐1 infection. Here, we have identified a crucial role for abortive HIV‐1 RNAs in inducing IL‐1β in humans. Abortive HIV‐1 RNAs were sensed by protein kinase RNA‐activated (PKR), which triggered activation of the canonical NLRP3 inflammasome and caspase‐1, leading to pro‐IL‐1β processing and secretion. PKR activated the inflammasome via ROS generation and MAP kinases ERK1/2, JNK, and p38. Inhibition of PKR during HIV‐1 infection blocked IL‐1β production. As abortive HIV‐1 RNAs are produced during productive infection and latency, our data strongly suggest that targeting PKR signaling might attenuate immune activation during acute and chronic HIV‐1 infection.
Keywords: Abortive HIV‐1 RNA, Human immunodeficiency virus 1, Inflammasome activation, Pattern recognition receptor, Protein kinase RNA‐activated
The processing of pro‐IL1β into bioactive IL‐1β is regulated by PKR‐dependent recognition of abortive HIV‐1 RNA, an HIV‐1‐derived RNA transcript produced during chronic and latent HIV‐1 infection. These results suggest that abortive HIV‐1 RNA plays an important role in the induction of inflammation observed in HIV‐1‐infected individuals.
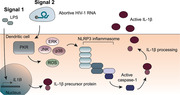
Introduction
Untreated human immunodeficiency virus 1 (HIV‐1) infection is characterized by a sustained increase in immune activation [1]. High levels of cytokines and chemokines that contribute to high levels of immune activation are a strong predictor of disease progression [2]. Although combination antiretroviral therapy successfully reduces viral replication levels, high immune inflammation levels persist [3]. It is unclear how immune inflammation in combination antiretroviral therapy‐treated people living with HIV‐1 is triggered.
The proinflammatory cytokine interleukin‐1 beta (IL‐1β) plays a central role in virtually all inflammatory conditions and as such also in HIV‐1 infection, via the induction of TNF and IL‐6 expression and attraction of various innate immune cells, leading to sustained proinflammatory responses [4]. It has been reported that IL1B and CASP1 gene expression in gut‐associated lymphoid tissue from HIV‐1 progressors was elevated compared to HIV‐1 controllers [5]. Although IL‐1β is produced as a result of HIV‐1 infection [6, 7], much remains undetermined about how IL‐1β production is regulated during different phases of HIV‐1 infection. Generation of bioactive IL‐1β is a multistep process that is tightly regulated at the level of transcription, post‐translational processing as well as extracellular release [4]. PRR‐induced signaling in response to a plethora of (viral) PAMPs triggers IL1B transcription. Following translation, processing of the pro‐IL‐1β protein is required for generation of mature, bioactive IL‐1β [4, 8], which is directed by inflammasome complexes that are comprised of NOD‐like receptor (NLR) members, such as NLRP1, NOD‐like receptor family pyrin domain containing 3 (NLRP3), or NLRC4, together with adaptor protein apoptosis‐associated speck‐like adaptor protein (ASC) and pro‐caspase‐1. Once procaspase‐1 is cleaved into active caspase‐1 upon inflammasome assembly and activation, pro‐IL‐1β cleavage follows and mature IL‐1β is secreted from the cell [8].
Different HIV‐1‐specific PAMPs have been identified during different stages of HIV‐1 infection, for example, the envelope protein gp120 binds to DC‐specific intercellular adhesion molecule‐3‐grabbing nonintegrin (DC‐SIGN) on the DC surface, while genomic HIV‐1 single‐stranded RNA (ssRNA) becomes available upon entry, cDNA after reverse transcription and HIV‐1‐related transcripts after integration within the host genome [9, 10, 11, 12, 13]. These HIV‐1‐specific PAMPs are recognized by different PRRs and could potentially trigger IL‐1β responses. For example, recognition of synthesized HIV‐1 genomic ssRNA by cytosolic RIG‐I, a PRR from the RIG‐I‐like receptor family, has been shown to result in IL‐1β responses via induction of IL1B transcription in response to different virus infections, for example, encephalomyocarditis virus and dengue virus [14, 15]. However, it remains undetermined if RIG‐I plays a role in IL‐1β responses during HIV‐1 infection. Triggering of TLR8 during HIV‐1 infection by ssRNA results in IL1B transcription and, interestingly, also leads to inflammasome activation and subsequent pro‐IL‐1β processing [7], although the underlying mechanism is unresolved.
Other viral PAMPs are prematurely aborted HIV‐1 transcripts, which are produced during transcription initiation of the integrated provirus in the absence of transcription elongation [10, 11, 16], and which are also expressed in latent infected cells [17]. Abortive HIV‐1 RNAs are recognized by the RIG‐I‐related PRR Dead‐box RNA helicase DDX3, which leads to type I IFN responses [11, 18]. Dead‐box polypeptide 3 (DDX3) specifically recognizes abortive HIV‐1 transcripts by the 5’ cap in combination with the proximal TAR loop, a complex secondary structure of the first 58 nucleotides of any HIV‐1 transcript [11, 16, 18]. Another well‐described PRR to recognize abortive HIV‐1 RNAs is protein kinase RNA‐activated (PKR), which likely recognizes the double stranded (ds) structure of the TAR loop [19, 20], resulting in, among others, the phosphorylation of eukaryotic translation initiation factor 2 α. Thus, although different HIV‐1‐specific PAMPs trigger various PRRs either during acute or latent infection by HIV‐1, it is unclear how generation of mature, bioactive IL‐1β is achieved during HIV‐1 infection.
Here, we investigated the role of abortive HIV‐1 RNAs in the induction and maturation of IL‐1β upon HIV‐1 infection. Our data show that triggering of PKR by abortive HIV‐1 RNAs results in NLRP3 inflammasome activation and caspase‐1‐mediated processing of pro‐IL‐1β, via generation of ROS and MAP kinases. During HIV‐1 infection of PBMCs, the inhibition of PKR activity and NLRP3 oligomerization inhibited pro‐IL‐1β processing and attenuated the IL‐1β response. Thus, PKR serves as a crucial PRR in the induction of inflammation and might be a potential target to attenuate aberrant inflammation during HIV‐1 infection.
Results
Abortive HIV‐1 RNA induces pro‐IL‐1β processing
To assess the role of abortive HIV‐1 RNA in the induction of IL‐1β responses without the interference of other HIV‐1‐specific PAMPs during HIV‐1 infection, we transfected monocyte‐derived DCs with synthetic HIV‐1 Cap‐RNA58, similar to abortive HIV‐1 RNA [11, 18]. Stimulation of DCs with different TLR agonists, that is, TLR2/6 ligand lipoteichoic acid (LTA), TLR3 ligand polyinosinic:polycytidylic acid (poly‐I:C), TLR4 ligand lipopolysaccharide (LPS), or TLR7/8 ligand R848, induced robust IL1B transcription but only led to undetectable or low levels of secreted IL‐1β (Fig. 1A and B). Notably, costimulation of DCs with HIV‐1 Cap‐RNA58 with either LTA, poly‐I:C, or LPS significantly enhanced the levels of IL‐1β protein secretion, whereas it did not affect IL1B transcription (Fig. 1A and B). As a control, LPS‐primed DCs were stimulated with ATP, a well‐described activator of the NLRP3 inflammasome via P2X7 receptors [21, 22], which resulted in pro‐IL‐1β processing, similar to HIV‐1 Cap‐RNA58, compared to untreated or LPS‐treated DCs (Fig. 1C and D). Although this pro‐IL‐1β processing occurred in every donor, IL‐1β secretion was subject to donor variability (Fig. 1C, Supporting Information Fig. S1) and, therefore, we show the data as fold change of IL‐1β secretion to perform statistical analysis (Fig. 1D). These data suggest that abortive HIV‐1 RNA induces pro‐IL‐1β processing. We also observed that HIV‐1 Cap‐RNA58 enhanced R848‐mediated IL1B transcription as well as IL‐1β protein secretion, suggesting that abortive HIV‐1 RNA, aside from activating pro‐IL‐1β processing, can modulate IL1B transcription, depending on the PRR (Fig. 1B). These data strongly suggest that abortive HIV‐1 RNA induces inflammasome activation, leading to subsequent pro‐IL‐1β processing and bioactive IL‐1β secretion.
Figure 1.
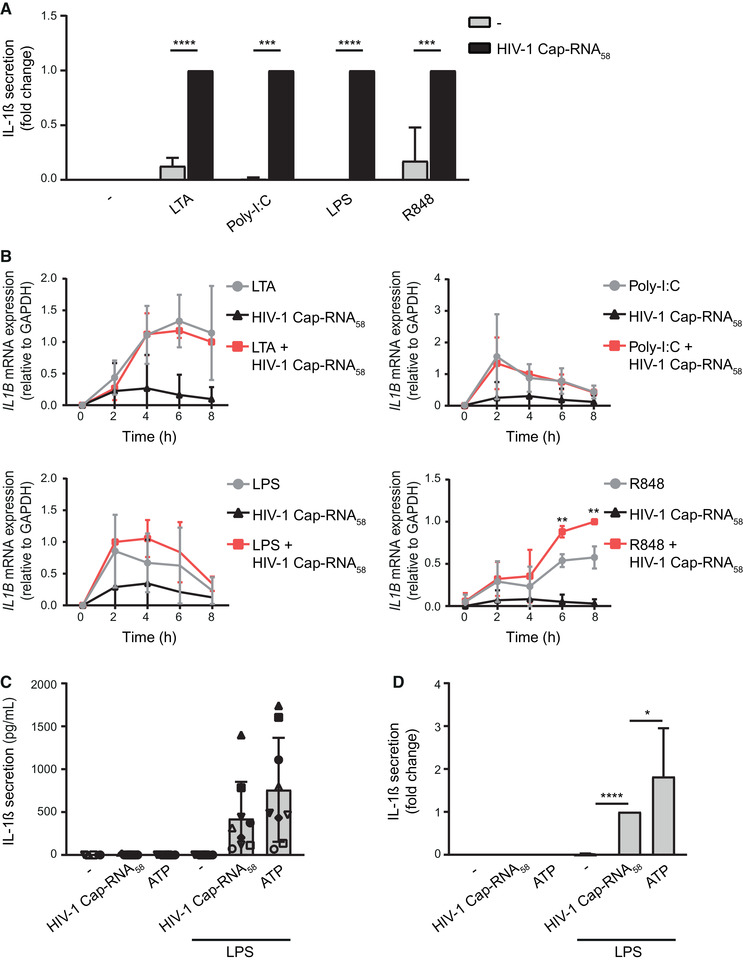
Abortive HIV‐1 RNA induces pro‐IL‐1β processing. (A) Monocyte‐derived DCs were (co)stimulated with different TLR ligands (LTA, Poly‐I:C, LPS, or R848) and/or HIV‐1 Cap‐RNA58. IL‐1β secretion in cell culture supernatant was measured after 24 h by ELISA. Responses induced by the combination of HIV‐1 Cap‐RNA58 and each TLR ligand were set at 1. See also Supporting information Fig. S1 for IL‐1β secretion (pg/mL). (B) Similarly, DCs were costimulated with HIV‐1 Cap‐RNA58 and TLR ligands, and mRNA was extracted every 2 h to analyze IL1B transcription by quantitative real‐time PCR. mRNA expression was relative to GAPDH. Responses induced by HIV‐1 Cap‐RNA58 with TLR ligand at 8 h (LTA, R848), 4 h (poly‐I:C), and 2 h (LPS) were set at 1. (C, D) DCs were stimulated with LPS, ATP, or costimulated with LPS and HIV‐1 Cap‐RNA58 for 24 h. ATP was added to the LPS alone condition for the final 4 h of stimulation. IL‐1β secretion in the supernatant was measured using ELISA with concentrations ranging from 50 to 1500 pg/mL, depending on donor variability (C). To statistically compare IL‐1β responses in different donors, IL‐1β secretion was shown as fold change compared to HIV‐1 Cap‐RNA58‐induced responses (D). Data are representative of collated data of six (A) and four (B) donors, or nine donors (C, D), with each symbol representing a different donor (C) (mean ± S.D.). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Student's t‐test.
Abortive HIV‐1 RNA activates the NLRP3 inflammasome
We next set out to elucidate the molecular mechanism through which abortive HIV‐1 RNA triggers pro‐IL‐1β processing. First, we silenced different components of known inflammasomes, that is, NLRP3 and NLRC4 [8, 23], in DCs by RNA interference and target gene expression was assessed (Supporting Information Fig. S2A and B). We observed a complete block of HIV‐1 Cap‐RNA58‐induced pro‐IL‐1β processing in LPS‐treated DCs upon silencing of NLRP3 protein expression, but not in control‐silenced or NLRC4‐silenced DCs (Fig. 2A). Silencing of the adaptor protein ASC, which is crucial for all known inflammasomes, significantly decreased HIV‐1 Cap‐RNA58‐induced pro‐IL‐1β processing, as shown by fold change of IL‐1β secretion due to high donor variability in IL‐1β levels (Fig. 2A). In addition, treatment with the NLRP3 inhibitor MCC950 abrogated IL‐1β secretion by both DCs and PBMCs stimulated with LPS and HIV‐1 Cap‐RNA58 (Fig. 2B and C, Supporting Information Fig. S2C). Caspase‐1 within the assembled NLRP3 inflammasome is the enzyme responsible for instigating pro‐IL‐1β processing [8, 23] and we, therefore, measured caspase‐1 activation. Treatment with HIV‐1 Cap‐RNA58 led to a significant increase in the percentage of DCs with active caspase‐1 (Fig. 2D). As a positive control, treatment of DCs with ATP strongly increased the percentage of DCs with activated caspase‐1 (Fig. 2D). Additionally, we observed that treatment of both DCs and PBMCs with caspase‐1 inhibitor Ac‐YVAD‐CMK abrogated IL‐1β secretion by cells costimulated with LPS and HIV‐1 Cap‐RNA58 (Fig. 2E and F, Supporting Information Fig. S2D). Stimulation of DCs with LPS and HIV‐1 Cap‐RNA58 did not lead to IL‐1β‐driven pyroptosis as no increase in LDH release was observed (Supporting Information Fig. S2E). These data strongly suggest that abortive HIV‐1 RNA activates the canonical NLRP3 inflammasome for caspase‐1‐dependent processing of pro‐IL‐1β.
Figure 2.
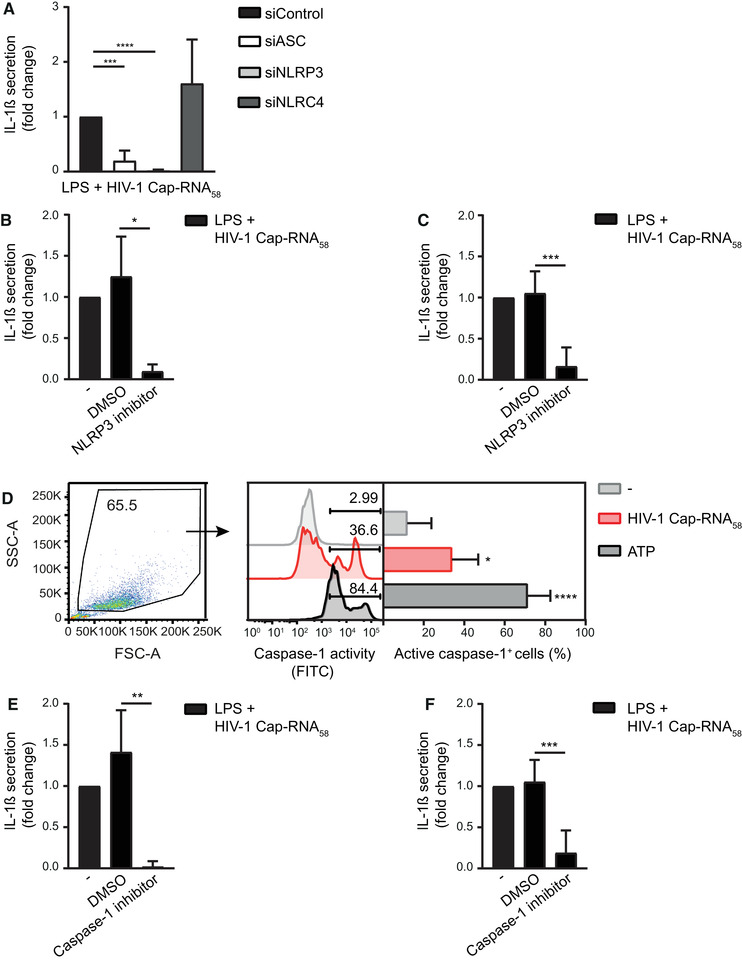
Abortive HIV‐1 RNA activates the NLRP3 inflammasome. (A) DCs were treated with LPS alone or in combination with HIV‐1 Cap‐RNA58 after silencing of ASC, NLRP3, and NLRC4 by RNA interference. IL‐1β secretion was measured by ELISA in the supernatant after 24 h. Responses induced by LPS with HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S2A and B. (B, C) DCs (B) or PBMCs (C) were incubated with DMSO control or with NLRP3 inhibitor MCC950 for 2 h, followed by costimulation with HIV‐1 Cap‐RNA58 and LPS. IL‐1β secretion in the supernatant was measured after 24 h by ELISA. Responses induced by LPS and HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S2C for IL‐1β secretion (pg/mL). (D) DCs were left untreated, or treated with either HIV‐1 Cap‐RNA58 or ATP. DCs with active caspase‐1 were detected after 22 h by flow cytometry using the FAM‐FLICA assay. (E, F) DCs (E) or PBMCs (F) were incubated with DMSO control or with caspase‐1 inhibitor Ac‐YVAD‐cmk for 2 h, followed by costimulation with HIV‐1 Cap‐RNA58 and LPS. IL‐1β secretion in the supernatant was measured after 24 h by ELISA. Responses induced by LPS and HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S2D for IL‐1β secretion (pg/mL). Data are representative of collated data of three (C, F) or four donors (A, B, D, E) (mean ± S.D.). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Student's t‐test.
Abortive HIV‐1 RNA induces pro‐IL‐1β processing via PKR
We next set out to identify the PRR through which abortive HIV‐1 RNA induces NLRP3 inflammasome activation. We have previously identified DDX3 as a sensor for HIV‐1 Cap‐RNA58 and shown that DDX3 triggering leads to type I IFN production [11, 18]. Notably, silencing of DDX3 protein expression did not interfere with HIV‐1 Cap‐RNA58‐induced pro‐IL‐1β processing in LPS‐treated DCs (Fig. 3A, Supporting Information Fig. S3C), suggesting that DDX3‐dependent sensing of HIV‐1 Cap‐RNA58 is dispensable for pro‐IL‐1β processing. Target gene and protein expression upon RNAi was assessed (Supporting Information Fig. S3A and B). It has been described that abortive HIV‐1 RNAs are also bound by ds RNA sensor PKR, a process that depends on the presence of the ds secondary structure of the TAR loop [19, 20]. We observed a strong reduction in pro‐IL‐1β‐processing after silencing of PKR expression in DCs costimulated with LPS and HIV‐1 Cap‐RNA58 (Fig. 3A), suggesting that PKR is the PRR that leads to inflammasome activation in response to the presence of abortive HIV‐1 RNA. Next, DCs were stimulated with a capless synthetic HIV‐1 RNA58, which still contains the double‐stranded TAR structure. We observed that capless HIV‐1 RNA58 resulted in PKR‐dependent pro‐IL‐1β processing in LPS‐treated DCs, similar to HIV‐1 Cap‐RNA58, which is also consistent with the sustained pro‐IL‐1β processing we observed in DDX3‐silenced cells (Fig. 3A and B, Supporting Information Fig. S3C and D). In order to investigate whether PKR kinase activity was needed for pro‐IL‐1β processing by abortive HIV‐1 RNA, we used the PKR inhibitor C16 that binds to the ATP site, thus, blocking its kinase activity. We observed that PKR inhibition abrogated IL‐1β production by DCs costimulated with not only TLR4 ligand LPS and HIV‐1 Cap‐RNA58, but also other TLR agonists (Fig. 3C, Supporting Information Fig. S3E). As treatment with LPS and HIV‐1 Cap‐RNA58 induced IL‐1β processing via caspase‐1, we next assessed the involvement of PKR in caspase‐1 cleavage. We found that treatment with LPS and HIV‐1 Cap‐RNA58 indeed induced caspase‐1 cleavage, which was blocked by PKR inhibition (Supporting Information Fig. S3F and G). Moreover, costimulation of PBMCs with either HIV‐1 Cap‐RNA58 or HIV‐1 RNA58 together with LPS, induced significant levels of IL‐1β secretion, which was significantly decreased upon inhibition of PKR (Fig. 3D, Supporting Information Fig. S3H). Next, we performed a PKR‐specific kinase activity assay. Both HIV‐1 Cap‐RNA58 and HIV‐1 RNA58 significantly increased the kinase activity of PKR in treated DCs compared to untreated DCs (Fig. 3E). These data strongly suggest that recognition of abortive HIV‐1 RNA by PKR results in PKR kinase activation and subsequent pro‐IL‐1β processing.
Figure 3.
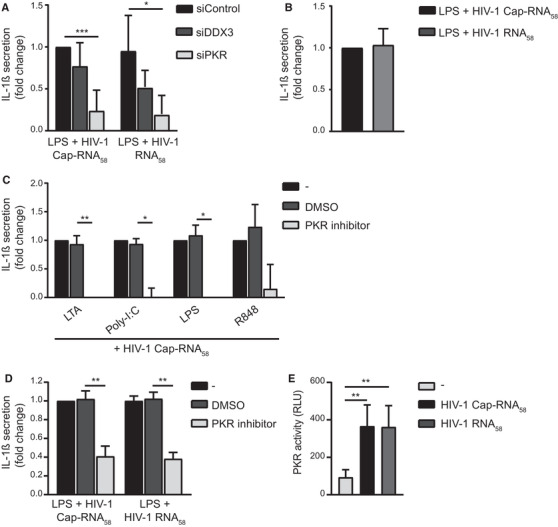
Abortive HIV‐1 RNA induces pro‐IL‐1β processing via PKR. (A) DDX3 and PKR‐silenced DCs (A) or unsilenced DCs (B) were costimulated with LPS and either HIV‐1 Cap‐RNA58 or HIV‐1 RNA58. IL‐1β secretion was measured in the supernatant by ELISA. Responses induced by LPS and HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S3A‐C. (B, C) DCs were incubated with DMSO or PKR inhibitor C16 for 2 h, followed by costimulation with either HIV‐1 Cap‐RNA58 and different TLR ligands (C) or with LPS and HIV‐1 Cap‐RNA58 or HIV‐1 RNA58 (B). IL‐1β secretion was measured in the supernatant after 24 h by ELISA. Responses induced by TLR ligand with HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S3D and E for IL‐1β secretion (pg/mL). (D) PBMCs were incubated with DMSO or PKR inhibitor C16 for 2 h, followed by costimulation with LPS and HIV‐1 Cap‐RNA58. IL‐1β secretion was measured in the supernatant after 24 h by ELISA. Responses induced by LPS and HIV‐1 Cap‐RNA58 were set at 1. See also Supporting information Fig. S3G for IL‐1β secretion (pg/mL). (E) Whole‐cell extracts were made from untreated DCs or DCs treated with HIV‐1 Cap‐RNA58 or HIV‐1 RNA58. After 2 h, PKR kinase activity was determined. Data are representative of collated data of three donors (C), four or more donors (A, B, D, E). (mean ± S.D.). *p < 0.05, **p < 0.01, ***p < 0.001, Student's t‐test.
PKR triggering by abortive HIV‐1 RNA induces inflammasome activation via ROS and MAP kinases
The precise role of PKR activation in inflammasome activation is unclear. It has been described that active PKR triggers NADPH oxidase (NOX)‐mediated generation of ROS [24, 25], and ROS has been previously linked to NLRP3 inflammasome activation [26]. To investigate whether abortive HIV‐1 RNA‐dependent pro‐IL‐1β processing is dependent on ROS generation, we treated DCs and PBMCs with Rac1 inhibitor NSC23766 or NOX inhibitor DPI, hereby interfering with NADPH oxidase assembly and activity, respectively. Treatment with either inhibitor significantly decreased pro‐IL‐1β processing after costimulation of DCs with LPS and HIV‐1 Cap‐RNA58, similar to the PKR inhibitor (Fig. 4A, Supporting Information Fig. S4A and B). Moreover, both Rac1 and NOX inhibitors blocked pro‐IL‐1β processing in PBMCs stimulated with LPS and HIV‐1 Cap‐RNA58, comparable to PKR inhibition (Fig. 4B, Supporting Information Fig. S4C and D). These data suggest that ROS generation is involved in abortive HIV‐1 RNA‐mediated pro‐IL‐1β processing. We also observed that NOX inhibitor DPI, but not Rac1 inhibitor NSC23766, strongly decreased IL1B transcription in DCs (Fig. 4C), which could account for the observed decreased IL‐1β levels. Therefore, we directly determined the role of the NADPH oxidase pathway on caspase‐1 activity. We observed that inhibition of both NOX and Rac1 significantly inhibited caspase‐1 activation by HIV‐1 Cap‐RNA58, to a similar level as PKR inhibition (Fig. 4D), further implying a role for ROS in inflammasome activation downstream of PKR.
Figure 4.
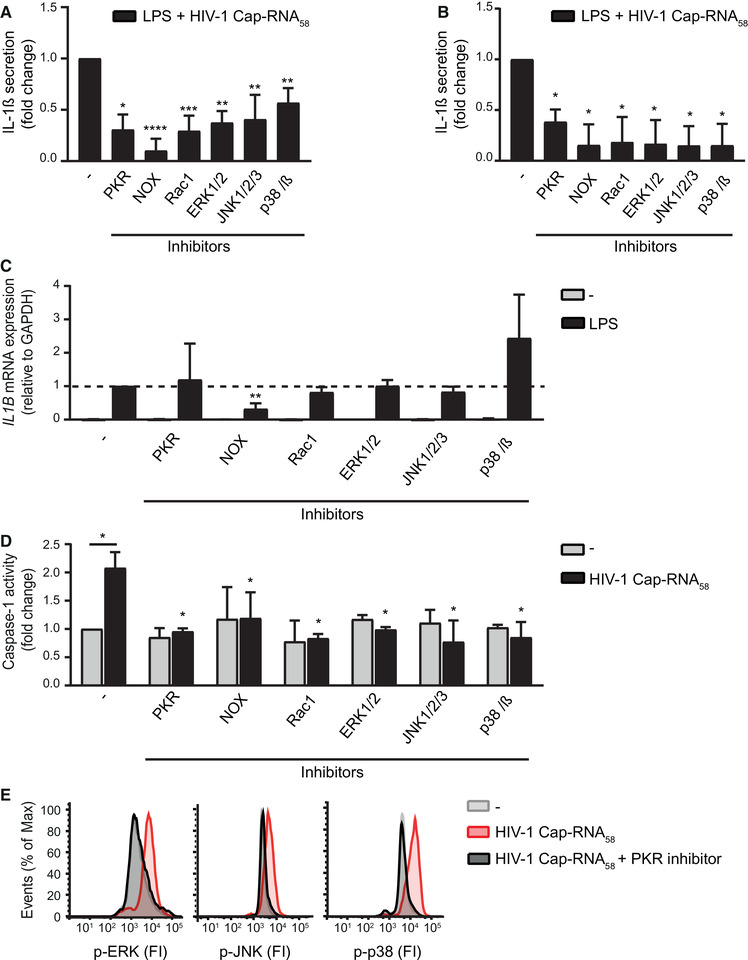
PKR triggering by abortive HIV‐1 RNA induces inflammasome activation via ROS and MAP kinases. (A‐C) DCs (A, C) or PBMCs (B) were incubated with inhibitors C16 (PKR), diphenyleneiodonium chloride (NAPDH oxidase (NOX)), NSC23766 (Rac1), FR180204 (ERK1/2), SP600125 (JNK1/2/3) and SB203580 (p38α/β) for 2 h, followed by costimulation with LPS and HIV‐1 Cap‐RNA58. IL‐1β was measured after 24 h in the supernatant, by ELISA (A, B), and IL1B transcription was determined after 6 h of stimulation by quantitative real‐time PCR with mRNA expression relative to GAPDH (C). Responses induced by LPS with HIV‐1 Cap‐RNA58 (A, B) or by LPS (C) were set at 1. See also Supporting information Fig. S4A and C for IL‐1β secretion (pg/mL) and Supporting information Fig S4B and D. (D) Similarly, DCs were treated with the different inhibitors and caspase‐1 activity was measured after stimulation with HIV‐1 Cap‐RNA58 by the caspase‐1‐Glo assay after 22 h. Background signal induced by unstimulated DCs was set at 1. (E) DCs were treated with PKR inhibitor C16 for 2 h, followed by stimulation with HIV‐1 Cap‐RNA58. After 3 h, cells were stained with phospho‐specific antibodies against ERK1/2, JNK, and p38 and analyzed using flow cytometry to determine fluorescence intensity (FI). Data are representative of collated data of four or more (A‐D) donors. Data are representative of at least three donors (E) from different experiments (mean ± S.D.). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Student's t‐test.
It also has been described that PKR activation leads to activation of the MAP kinases ERK1/2, JNK, and p38, which in turn have been linked to inflammasome activation [27, 28, 29, 30, 31]. We investigated whether either kinase is involved in inflammasome activation as a downstream target of PKR. Inhibition of either ERK1/2, JNK, or p38 significantly decreased IL‐1β secretion in DCs (Fig. 4A, Supporting Information Fig. S4A and B), while IL1B transcription remained unaffected (Fig. 4C). Inhibition of these MAP kinases significantly decreased IL‐1β secretion in PBMCs (Fig. 4B, Supporting Information Fig. S4C and D). Treatment of DCs with HIV‐1 Cap‐RNA58 induced phosphorylation of ERK1/2, JNK, and p38 in a PKR‐dependent manner as the PKR inhibitor C16 completely abrogated this phosphorylation (Fig. 4E). Moreover, inhibition of either ERK1/2, JNK, or p38 significantly inhibited caspase‐1 activation by HIV‐1 Cap‐RNA58 (Fig. 4D). Our results imply that PKR‐mediated canonical inflammasome activation by abortive HIV‐1 RNA is mediated by MAP kinases.
HIV‐1SF162 infection induces IL‐1β secretion via PKR and inflammasome activation
We next investigated whether PKR activation during HIV‐1 infection leads to IL‐1β maturation and secretion. HIV‐1 blocks cytosolic sensing in DCs by various mechanisms [11, 32], thereby suppressing innate immunity. Therefore, we investigated IL‐1β secretion upon HIV‐1 infection in PBMCs. We infected PBMCs with R5‐tropic HIV‐1SF162 in the absence or presence of the PKR inhibitor. PKR inhibition significantly reduced secreted IL‐1β levels (Fig. 5A, Supporting Information Fig. S5A), while HIV‐1 infection of total PBMC, CD3+ and CD3− cell populations was unaffected (Fig. 5B and C, Supporting Information Fig. S5C and D). Since PKR inhibition did not affect viral replication, these results suggest that PKR activation enhances IL‐1β production at the level of pro‐IL‐1β processing during HIV‐1 infection. Inhibition of NLRP3 in HIV‐1‐infected PBMCs showed decreased IL‐1β secretion (Fig. 5D, Supporting Information Fig. S5B). In contrast to the mock infection, caspase‐1 activity was increased upon HIV‐1 infection of PBMCs and PKR inhibition blocked caspase‐1 activation (Fig. 5E). To identify which cells in the PBMC pool secrete IL‐1β, we infected PBMCs and examined the intracellular expression of cleaved IL‐1β in CD3+ and CD11c+ cells, by flow cytometry. CD3+ T cells in infected PBMCs contained low levels of intracellular cleaved IL‐1β, at 24 and 48 h after infection (Fig. 5F). Furthermore, CD11c+ cells, such as DCs or monocytes, contained intracellular cleaved IL‐1β, which increased over time (Fig. 5F). These data strongly suggest that HIV‐1SF162‐infection of PBMCs results in IL‐1β secretion via PKR‐mediated inflammasome activation, similar as to what we observed for the synthetic treatment of DCs with abortive HIV‐1 RNA.
Figure 5.
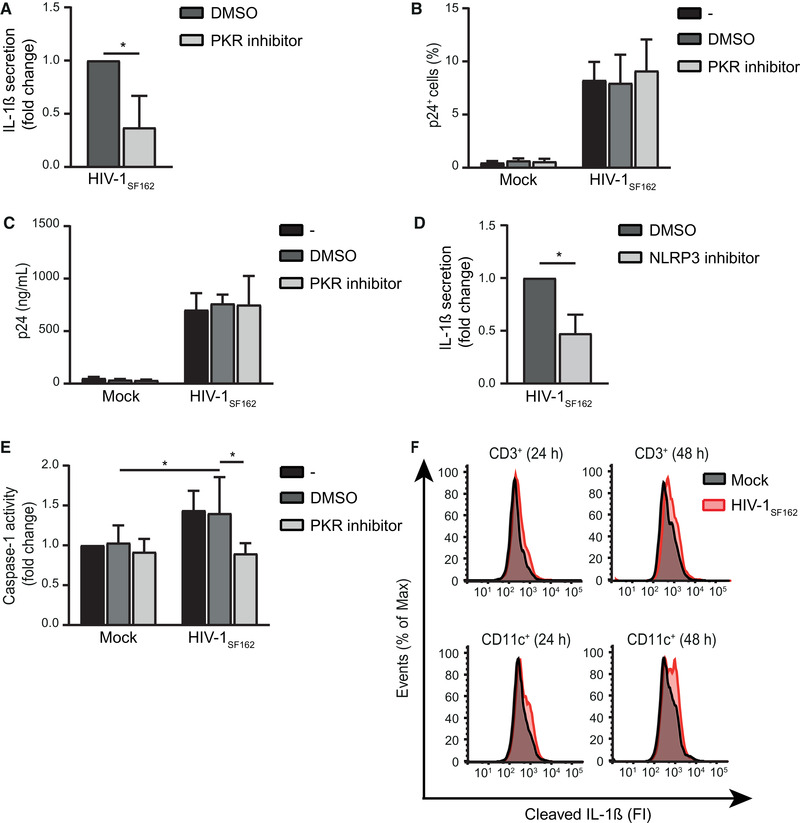
HIV‐1SF162 infection induces IL‐1β secretion via PKR and inflammasome activation. (A, D) PBMCs were incubated with IL‐2 for 3 days, treated with PKR inhibitor C16 (A) or NLRP3 inhibitor MCC950 (D) for 2 h and infected with CCR5 (R5)‐tropic virus HIV‐1SF162 with MOI 0.1. IL‐1β was measured in the supernatant after 48 h by ELISA. Responses induced by HIV‐1SF162 were set at 1. See also Supporting information Fig. S5A and B for IL‐1β secretion (pg/mL). (B,C) Similarly, IL‐2‐treated PBMCs were incubated with C16 for 2 h and infected with HIV‐1SF162. Intracellular p24 was measured after 4 days by flow cytometry (B) or in the supernatant after 5 days by ELISA (C). See also Supporting information Fig. S5C and D for intracellular p24 analysis in CD3+ and CD3− populations. (E) PBMCs were incubated with IL‐2 for 3 days, treated with PKR inhibitor C16 for 2 h and infected with HIV‐1SF162 with MOI 0.1. Caspase‐1 activity was assessed after 46 h, using a caspase‐1‐Glo assay. Responses induced by untreated mock controls were set at 1. (F) PBMCs were incubated with IL‐2 for 3 days, infected with HIV‐1SF162 with MOI 0.1, and treated with brefeldin A. Intracellular cleaved IL‐1β was assessed in CD3+ and CD11c+ cells of the PBMC pool and analyzed using flow cytometry to determine fluorescence intensity (FI). Data are representative of collated data of four (A) or three (B‐E) donors. Data are representative of at least two (F, 48 h timepoint) or three (F, 24 h timepoint) donors (mean ± S.D.). *p < 0.05, Student's t‐test.
Discussion
Chronic immune activation caused by proinflammatory cytokines, such as IL‐1β, accompanies HIV‐1 disease progression, leading to tissue damage and rapid cell turnover [1]. It has previously been described that HIV‐1 modulates the host pro‐IL‐1β levels [7, 33], but it remains unclear whether and how HIV‐1 directs pro‐IL‐1β processing. Our data strongly suggest that PKR detects abortive HIV‐1 RNA, which is produced during productive infection as well as in latent infected cells. Abortive HIV‐1 RNA‐dependent PKR activation leads to pro‐IL‐1β processing via activation of the canonical inflammasome consisting of NLRP3 and caspase‐1 in both DCs and PBMCs. Triggering of PKR mediates NLRP3 inflammasome activation via pathways involving ROS generation and activation of MAP kinases. Both transfection with abortive HIV‐1 RNAs as well as productive HIV‐1 infection of PBMCs induced PKR‐ and NLRP3‐mediated processing of pro‐IL‐1β. These data suggest that abortive HIV‐1 RNA produced during infection and latency contributes to immune inflammation by inducing IL‐1β via PKR‐mediated activation of the NLRP3 inflammasome.
During productive HIV‐1 infection, TLR8 detects genomic HIV‐1 ssRNA upon unveiling in the endosomal compartments [34], which induces expression of pro‐IL‐1β, while an unknown signal then proceeds to IL‐1β maturation via the activation of the NLRP3 inflammasome [7]. Our study reveals that the signal needed for NLRP3 inflammasome activation is provided by PKR when it detects the presence of abortive HIV‐1 transcripts. We show that abortive HIV‐1 RNA‐mediated PKR activation not only resulted in processing of TLR8‐induced pro‐IL‐1β, but that this mechanism also induced IL‐1β maturation after triggering of signaling pathways downstream of other TLRs.
Triggering of PKR by abortive HIV‐1 RNA resulted in the activation of caspase‐1 within the context of the canonical NLRP3 inflammasome during transfection of abortive HIV‐1 RNAs as well as during HIV‐1 infection. The role of PKR in inflammasome activation is controversial as some reports mention that PKR functions as a scaffold molecule during inflammasome assembly, a function that is independent of its kinase activity, while other reports state that this assembly brings the inflammasome components in proximity of PKR and their activation benefits from PKR kinase activity, while yet another study reports that PKR kinase activity dampens inflammasome activation [35, 36, 37, 38]. Here, we provide evidence that PKR‐mediated inflammasome activation required its kinase activity, as caspase‐1 activation and pro‐IL‐1β processing are abrogated after both PKR protein silencing as well as PKR kinase inhibition. Whether PKR is solely involved in inflammasome activation or whether it also affects inflammasome priming steps remains unclear. We have identified roles for both NADPH oxidase‐mediated ROS generation and the activation of the MAP kinases ERK1/2, JNK and p38, in PKR‐mediated NLRP3 inflammasome activation. The role of ROS in activation of the inflammasome has been studied extensively, identifying a crucial role for ROS‐mediated release of thioredoxin‐interacting protein from thioredoxin which then binds to NLRP3, resulting in NLRP3 inflammasome activation [22, 26]. PKR has previously been reported to induce the assembly of the NADPH oxidase complex that consists of several NOX proteins together with the small GTPase Rac1 via PKC [25]. Here, we show for the first time that PKR‐mediated ROS generation is linked to inflammasome activation. Interestingly, it has been described that during HIV‐1 infection, ROS formation is crucial in the production of IL‐1β [7].
Another link between PKR and inflammasome activation can be found in the three MAP kinases, ERK1/2, JNK, and p38: PKR activation has been shown to result in activation of all three MAP kinases in different settings [27–29, 39], while ERK1/2, JNK, and also p38 can target NLRP3 for phosphorylation, a priming event crucial for inflammasome activation [30, 31]. Activation of ERK1/2, JNK, and p38 as a result of ROS generation has been previously described [25, 40–42]. It will be interesting to investigate whether the two mechanisms we have identified downstream of PKR work sequentially or synergistically.
We show that not only transfection of DCs and PBMCs with abortive HIV‐1 RNA results in PKR activation and subsequent pro‐IL‐1β processing, but also HIV‐1 infection of PBMCs, a heterogeneous and, therefore, relevant population of cells, resulted in activation of caspase‐1 and subsequent IL‐1β production, which was attenuated upon inhibition of PKR activity and NLRP3 oligomerization. Transfection of PBMCs with abortive HIV‐1 RNA induced IL‐1β secretion. These data suggest that abortive HIV‐1 RNAs that are also observed in productively infected cells are sensed by PKR, leading to inflammasome activation. Both CD3+ T cells and CD11c+ cells in the PBMC pool expressed cleaved IL‐1β, indicating that these cells are responsible for IL‐1β secretion. CD11c is expressed by myeloid cells such as macrophages and DCs. These data suggest that both T cells and myeloid cells sense HIV‐1 infection via PKR to induce IL‐1β.
The role of PKR during HIV‐1 infection is versatile as activated PKR inhibits both cellular and viral translation of mRNAs via phosphorylation of eukaryotic translation initiation factor 2 α [28], hereby affecting viral replication. HIV‐1 escapes PKR‐dependent translational inhibition of viral mRNAs via binding of the HIV‐1 Tat protein to PKR, thus, repressing its activity or via protein‐dependent inhibition of PKR [43, 44]. We observed no differences in the levels of p24 in the PBMC population or in the supernatant, suggesting that the effect of the PKR inhibitor was on viral PAMP recognition and not on viral replication. Interestingly, it has recently been described that HIV‐1 protease induces caspase‐1 activation and IL‐1β secretion via sensing by the caspase recruitment domain‐containing protein 8 (CARD8) inflammasome [45]. In addition, NLRP1 activation has also been reported to induce IL‐1β secretion, via the detection of viral dsRNA [46]. Whether abortive HIV‐1 RNA is solely responsible for PKR‐dependent inflammasome activation during HIV‐1 infection or whether other types of viral dsRNA, viral PAMPs, or host sensors lead to pro‐IL‐1β processing remains to be determined.
Besides contributing to immune inflammation, HIV‐1‐associated IL‐1β production has also been linked to the induction of pyroptosis, IL‐1β‐mediated programmed cell‐death [47]. Pyroptosis occurs in nonpermissive CD4+ T cells that appear as dying uninfected “bystander” CD4+ T cells, but are in fact nonproductively infected cells [48]. In these nonproductively infected CD4+ T cells, impaired reverse transcription results in the accumulation of incomplete reverse transcription products, which trigger IL‐1β production through an unknown mechanism [49]. Importantly, abortive HIV‐1 RNA transcripts are generated during viral latency, as a product of incomplete transcription [16, 17]. Therefore, targeting PKR signaling might attenuate immune activation in chronic HIV‐1 infection.
In conclusion, HIV‐1 infection directs maturation of pro‐IL‐1β leading to excessive IL‐1β levels and as such continuous immune activation and inflammation. Triggering of PKR by detection of abortive HIV‐1 RNA, either synthetic or during HIV‐1 infection, leads to activation of the canonical NLRP3/caspase‐1 inflammasome that controls pro‐IL‐1β processing. Considering that abortive HIV‐1 transcripts are produced at different stages during the course of HIV‐1 infection, our data provide a rationale for targeting PKR to control continuous HIV‐1‐mediated immune activation.
Materials and methods
Cells, stimuli, and inhibitors
This study was performed according to the Amsterdam University Medical Centers, location AMC Medical Ethics Committee guidelines. All donors gave written informed consent in accordance with the Declaration of Helsinki. Human PBMCs from buffy coats of healthy individuals (Sanquin) were isolated using lymphoprep and cultured in RPMI medium supplemented with 10% fetal calf serum (FCS), penicillin (100 U/mL, Thermo Fisher), streptomycin (100 μg/mL, Thermo Fisher), and recombinant IL‐2 (20 U/mL, Novartis) at a cell density of 2 × 105 cells/mL at 37°C, 5% CO2. To obtain human monocytes, PBMC isolation was followed by percoll gradient steps. Monocytes were cultured in RPMI medium supplemented with 10% FCS, l‐glutamine (2 mM, Lonza), penicillin and streptomycin (100 U/mL and 100 μg/mL, respectively, Thermo Fisher) in the presence of GM‐CSF (800 U/mL, Invitrogen) and IL‐4 (500 U/mL Invitrogen) at 37°C, 5% CO2 to obtain monocyte‐derived DCs. On day 4, cells were plated at a cell density of 1 × 105 cells/mL and on day 6‐7 DCs were stimulated with 1 nM synthetic abortive HIV‐1 RNA complexed with transfection reagent lyovec (Invivogen), constituting the first 58 nucleotides of the HIV‐1 genome, including a 5’m7GTP cap incorporated using cocapping during in vitro transcription and lacking a polyA tail (HIV‐1 Cap‐RNA58, Biosynthesis) as previously described [11,18]. As a control, the same RNA sequence was synthesized, however, without the 5’m7GTP cap (HIV‐1 control RNA58, 1 nM). Furthermore, DCs were stimulated with lipopolysaccharide (LPS) Salmonella enterica serotype typhimurium (10 ng/mL, Sigma), LTA, poly‐I:C, R848 (all 10 μg/mL, Invivogen), recombinant adenosine triphosphate (5 mM, Promega), and staurosporin (1 μM, Merck). Cells were preincubated in the presence of various inhibitors as indicated for 2 h to target NLRP3 (MCC950, 5 μM, Invivogen), caspase‐1 (Ac‐YVAD‐cmk, 50 μM, Tocris), PKR (C16, 0.5 μM, Merck), NOX (diphenyleneiodonium chloride, 25 μM, Sigma), Rac1 (NSC23766, 100 mM, Tocris), ERK1/2 (FR180204, 10 μM, R&D systems), JNK1/2/3 (SP600125, 100 mM, Tocris), and p38α/β (SB203580, 25 mM, Invivogen). Upon treatment with the different inhibitors, cell viability was assessed using the CellTiter‐Glo 2.0 Viability Assay (Promega) according to manufacturer's instructions (Supporting information Fig. S4B and D).
IL‐1β secretion
DC and PBMC supernatants were harvested 24 or 48 h after stimulation as the most efficient inductions of IL‐1β secretion were observed during these time points. IL‐1β secretion levels were quantified using ELISA according to manufacturer's instructions (eBiosciences) and OD450 nm values were obtained using BioTek Synergy HT. The induction of IL‐1β‐mediated pyroptosis was assessed using an LDH cytotoxicity assay (Thermofisher) according to manufacturer's instructions of which the Positive Control of the kit was set at 100% LDH release.
Quantitative RT‐PCR
DCs were lysed to extract mRNA using the mRNA capture kit (Roche), followed by reverse transcription to generate cDNA using the Reverse transcriptase kit (Promega). Quantitative real‐time PCR was performed using SYBR Green (Thermo Fisher), with primer sets designed with Primer Express 2.0 (Applied Biosystems, Supporting information Table S1), on an ABI 7500 Fast Real‐Time PCR detection system (Applied Biosystems). Target gene expression levels were normalized to expression levels of household gene GAPDH, using the formula: Nt = 2 Ct (GAPDH)− Ct (target). Relative expression levels were calculated when Nt in HIV‐1 Cap‐RNA58 or LPS + HIV‐1 Cap‐RNA58‐treated cells was set at 1.
Caspase‐1 activity
Active caspase‐1 was detected using the FAM‐FLICA Caspase‐1 Assay kit (Immunochemistry Technologies) according to manufacturer's instructions. In brief, DCs were washed in IMDM medium lacking phenol red (Gibco), supplemented with 10% FCS, penicillin and streptomycin (100 U/mL and 100 μg/mL, respectively, Thermo Fisher) prior to stimulations. DCs were stimulated as previously described. After 22 h, DCs were treated with FAM FLICA reagent and incubated at 37°C, 5% CO2 for 1 h. Cells were washed three times in apoptotic wash buffer (Immunochemistry Technologies), immediately followed by flow cytometry analysis using the FACSCanto II (BD Bioscience) and FlowJo software version 10.7 and guidelines for the use of flow cytometry and cell sorting in immunological studies were followed. Live cells were gated based on FSC/SSC and the caspase‐1+ population within this live cell population was assessed. Processing of caspase‐1 was determined via detection of the processed 20 kDa unit (p20, 1:1000. 4199S, Cell Signaling) in DC cell lysates (40 μg) 8 h after stimulation, using immunoblot analysis. Primary antibody stainings were followed by HRP‐labeled anti‐rabbit (1:2000, P0217, Dako) and developed using an ECL detection system (Pierce). β‐Actin expression was used as a loading control (1:1000, sc‐81178, Santa Cruz) and primary antibody stainings were followed by HRP‐labeled anti‐mouse (1:2000, P0161, Dako). In addition, caspase‐1 activity was measured using the Caspase‐Glo 1 inflammasome assay (Promega) according to manufacturer's protocol. In short, DCs or PBMCs were treated as previously described and incubated with Caspase‐Glo 1 reagent containing the proteasome inhibitor MG‐132 to prevent nonspecific substrate cleavage. After 90 min, the cell/Caspase‐Glo 1 mixture was transferred to a white 96‐well opaque plate (Corning) and luminescence was measured using the TriStar² S LB 942 (Berthold).
RNA interference
On day 4 of monocyte‐derived DC cultures, cells were washed with PBS, resuspended in buffer R according to manufacturer's instructions (Thermo Fisher) and mixtures were divided for transfection with the different SMARTpool small interfering (si) RNAs (all from Dharmacon) to target DDX3 (siDDX3, M‐006874‐01), PKR (siEIF2AK2, L‐003527‐00), ASC (siASC, M‐004378‐01), NLRP3 (siNLRP3, M‐017367‐00), NLRC4 (siNLRC4, M‐004396‐00), or with a nontarget control (siControl, D‐001206‐13). Cells were transfected with 1500V for 20 ms using the Neon Transfection System (Thermo Fisher) and seeded in 24‐well plates in RPMI medium supplemented with 10% FCS and 2 mM l‐glutamine (Lonza) in the absence of antibiotics. Cells were collected, washed, seeded in a 96‐well round bottom plate, and incubated O/N at 37°C 5% CO2, 72 h after transfection, cells were stimulated as previously indicated and silencing efficiency was assessed using flow cytometry and/or quantitative RT‐PCR (Supporting information Figs. S2A, B and S3A, B). For flow cytometry analysis, DCs were fixed with 4% paraformaldehyde , permeabilized with 90% methanol at −20°C for at least 16 h and stained with anti‐ASC (1:50, 4628S, Cell Signaling), anti‐NLRP3 (1:50, 13158S, Cell Signaling), anti‐NLRC4 (1:50, 12421S, Cell Signaling), anti‐DDX3 (1:50, 2635S, Cell Signaling), and anti‐PKR (1:50, ab32052, Abcam), followed by PE‐conjugated donkey anti‐rabbit (1:200, Jackson Immuno Research), measured using FACSCanto II (BD Biosciences) and analyzed with FlowJo software version 10.7. Cells were gated on based on FSC/SSC and the ASC+, NLRP3+, NLRC4+, DDX3+, and PKR+ population within this live cell population was assessed.
PKR kinase assay
PKR kinase activity was assessed using a combination of an EIF2AK2 kinase enzyme kit (Promega) and ADP‐Glo kinase assay (Promega). DCs were stimulated as previously described to induce kinase activity. After 2 h, whole‐cell extracts were prepared using PKR lysis buffer (50 mM Tris‐HCl [pH 7.5], 1 mM EDTA, 150 mM NaCl, 0.5% Igepal, 12.5 mM beta‐glycerophosphate, 10% glycerol, 1 μg/mL leupeptin, 1 μg/mL pepstatin A, 0.4 mM PMSF). To capture PKR in whole‐cell extract, 96‐well high binding plates (Thermo Fisher) were coated with 10 μg/mL anti‐PKR (ab32052, Abcam) O/N at 4°C. Then, anti‐PKR‐coated wells were blocked with 1%BSA for 30 min and incubated with 10 μg whole‐cell extract for 2 h. Recombinant PKR was used to obtain a protein standard used for quantification (Promega). Wells were incubated with 0.1 mg/mL substrate (myelin basic protein) in 22.5 μL kinase activity buffer (40 mM Tris‐HCl [pH 7.5], 20 mM MgCl2, 50 μM DTT, 0.1 mg/mL BSA, 50 μM ATP) for 1 h at RT. Kinase activity termination and ATP depletion were achieved using the ADP‐Glo reagent (Promega) for 40 min at RT. Final PKR kinase activity was measured by incubation with kinase detection reagent for 30 min at RT, after which the mixture was transferred to a white opaque plate (Corning). Relative light units were detected as a measure for generated ADP and, thus, PKR activity, using the BioTek Synergy HT.
Protein phosphorylation
DCs were stimulated as previously described for 3 h (to detect p‐ERK, p‐p38, p‐JNK), after which cells were fixed with 4% paraformaldehyde and permeabilized with 90% methanol at −20°C for at least 16 h. Cells were stained with phospho‐specific antibodies against ERK1/2 Thr202/Tyr204 (1:50, 4377S, Cell Signaling) and p38 Thr180/Tyr182 (1:50, 9211S, Cell Signaling), followed by PE‐conjugated donkey anti‐rabbit (1:200, Jackson Immuno Research). Phospho‐specific antibodies against SAPK/JNK Thr183/Tyr185 (1:50, 9255S, Cell Signaling) were used, followed by Alexa647‐conjugated goat anti‐mouse (1:200, A21235, Invitrogen). Flow cytometry analysis was performed using the FACSCanto II (BD Biosciences) and analyzed with FlowJo software version 10.7.
Virus production and infection
R5‐tropic HIV‐1SF162 was obtained from Dr. Jay Levy and propagated on human phytohemagglutinin (PHA)‐stimulated PBMCs as previously described [50]. After 4 days, supernatant was harvested, filtered, and titrated using TZM‐bl cells (human cervical cancer cells). IL‐1β secretion was examined in infected PBMCs, treated as previously described. PBMCs were infected with HIV‐1SF162 at a multiplicity of infection (MOI) of 0.1 and cultured at 37°C 5% CO2, 46 h after infection, caspase‐1 activity was determined and after 48 h, supernatant was collected and IL‐1β secretion was assessed using ELISA. After 24 or 48 h of infection, cells were incubated with brefeldin A (10 μg/mL, Sigma) for the final 4 h, collected and CD3+ (APC‐Cy7‐conjugated anti‐CD3, 1:100, 300317, Biolegend) and CD11c+ (APC‐conjugated anti‐CD11c, 1:50, 33144, BD Biosciences) cells were intracellularly stained for cleaved IL‐1β (1:50, 83186S, Cell Signaling). Cells were gates based on FSC/SSC and assessed using flow cytometry. Four days after infection, cells were collected, stained, and gated based on FSC/SSC, followed by gating on single cells. Infection levels within the CD3+ and CD3− population of the PBMC pool were determined by staining PBMCs intracellularly using PE‐conjugated anti‐p24 (1:200, KC57‐RD1, Beckman Coulter) and APC‐Cy7‐conjugated anti‐CD3 (1:100, 300317, Biolegend). Flow cytometry analysis was performed using the FACSCanto II (BD Biosciences) and FlowJo software version 10.7. Alternatively, viral replication of HIV‐1SF162 was assessed by Gag p24 production in the PBMC culture supernatant 5 days after infection, using a quantitative in‐house p24 ELISA.
Data analysis
Statistical analysis was performed using Student's t‐test for paired observations using GraphPad version 8 and statistical significance was set at P < 0.05.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
M. S. designed, performed, and interpreted most experiments and wrote the manuscript; J. L. V. H. helped with silencing and performed HIV‐1 infection experiments; M. T. performed inhibitor and FLICA experiments; S. I. G. and T. B. H. G. supervised all aspects of this study.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149275
Abbreviations
- ASC
apoptosis‐associated speck‐like
- DDX3
Dead‐box polypeptide 3
- FCS
fetal calf serum
- HIV‐1
human immunodeficiency virus 1
- IL‐1β
interleukin‐1 beta
- LTA
lipoteichoic acid
- LPS
lipopolysaccharide
- MOI
multiplicity of infection
- NLRP3
NOD‐like receptor family pyrin domain containing 3
- NOX
NADPH oxidase
- poly‐I:C
polyinosinic:polycytidylic acid
- PKR
protein kinase RNA‐activated
- si
small interfering
- ssRNA
single‐stranded RNA
Supporting information
Supporting Information
Acknowledgments
This work was supported by Aidsfonds (P‐9906) and the European Research Council (Advanced Grant 670424).
Data availability statement
Data available on request from the authors.
References
- 1. Paiardini, M. and Müller‐Trutwin, M. , HIV‐associated chronic immune activation. Immunol. Rev. 2013. 254: 78–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leng, Q. , Borkow, G. , Weisman, Z. , Stein, M. , Kalinkovich, A. and Bentwich, Z. , Immune activation correlates better than HIV plasma viral load with CD4 T‐cell decline during HIV infection. 2001. 27: 389–397. [DOI] [PubMed] [Google Scholar]
- 3. Kamat, A. , Misra, V. , Cassol, E. , Ancuta, P. , Yan, Z. , Li, C. , Morgello, S. et al., A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PLoS One 2012. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dinarello, C. A. , Immunological and inflammatory functions of the interleukin‐1 family. Annu. Rev. Immunol. 2009. 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 5. Feria, M. G. , Taborda, N. A. , Hernandez, J. C. and Rugeles, M. T. , HIV replication is associated to inflammasomes activation, IL‐1β, IL‐18 and caspase‐1 expression in GALT and peripheral blood. PLoS One 2018. 13: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jalbert, E. , Crawford, T. Q. , D'Antoni, M. L. , Keating, S. M. , Norris, P. J. , Nakamoto, B. K. , Seto, T. et al., IL‐1Β enriched monocytes mount massive IL‐6 responses to common inflammatory triggers among chronically HIV‐1 infected adults on stable anti‐retroviral therapy at risk for cardiovascular disease. PLoS One 2013. 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo, H. , Gao, J. , Taxman, D. J. , Ting, J. P. Y. and Su, L. , HIV‐1 infection induces interleukin‐1β production via TLR8 protein‐dependent and NLRP3 inflammasome mechanisms in human monocytes. J. Biol. Chem. 2014. 289: 21716–21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schroder, K. and Tschopp, J. , The inflammasomes. Cell 2010. 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 9. Geijtenbeek, T. B. H. , Kwon, D. S. , Torensma, R. , van Vliet, S. J. , van Duijnhoven, G. C. , Middel, J. , Cornelissen, I. L. et al., DC‐SIGN, a dendritic cell‐specific HIV‐1‐binding protein that enhances trans‐infection of T cells. Cell 2000. 100: 587–597. [DOI] [PubMed] [Google Scholar]
- 10. Gringhuis, S. I. , van der Vlist, M. , van den Berg, L. M. , den Dunnen, J. , Litjens, M. and Geijtenbeek, T. B. H. , HIV‐1 exploits innate signaling by TLR8 and DC‐SIGN for productive infection of dendritic cells. Nat. Immunol. 2010. 11: 419–426. [DOI] [PubMed] [Google Scholar]
- 11. Gringhuis, S. I. , Hertoghs, N. , Kaptein, T. M. , Zijlstra‐Willems, E. M. , Sarrami‐Fooroshani, R. , Sprokholt, J. K. , van Teijlingen, N. H. et al., HIV‐1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV‐1 RNA by the host helicase DDX3. Nat. Immunol. 2017. 18: 225–235. [DOI] [PubMed] [Google Scholar]
- 12. Lahaye, X. , Satoh, T. , Gentili, M. , Cerboni, S. , Conrad, C. , Hurbain, I. , ElMarjou, A. et al., The capsids of HIV‐1 and HIV‐2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 2013. 39: 1132–1142. [DOI] [PubMed] [Google Scholar]
- 13. Unterholzner, L. , Keating, S. E. , Baran, M. , Horan, K. A. , Søren, B. , Sharma, S. , Sirois, C. M. et al., IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010. 11: 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poeck, H. , Bscheider, M. , Gross, O. , Finger, K. , Roth, S. , Rebsamen, M. , Hannesschläger, N. et al., Recognition of RNA virus by RIG‐I results in activation of CARD9 and inflammasome signaling for interleukin 1Β production. Nat. Immunol. 2010. 11: 63–69. [DOI] [PubMed] [Google Scholar]
- 15. Sprokholt, J. K. , Kaptein, T. M. , van Hamme, J. L. , Overmars, R. J. , Gringhuis, S. I. and Geijtenbeek, T. B. H. , RIG‐I–like receptor triggering by dengue virus drives dendritic cell immune activation and T H 1 differentiation. J. Immunol. 2017. 198: 4764–4771. [DOI] [PubMed] [Google Scholar]
- 16. Kao, S. , Calman, A. F. , Luciw, P. A. and Peterlin, B. M. , Anti‐termination of transcription within the long terminal repeat of HIV‐1 by TAT gene product. Nature 1987. 330: 489–493. [DOI] [PubMed] [Google Scholar]
- 17. Hladnik, A. , Ferdin, J. , Goričar, K. , Deeks, S. G. , Peterlin, B. M. , Plemenitaš, A. , Dolžan, V. et al., Trans‐activation response element RNA is detectable in the plasma of a subset of aviremic HIV‐1–infected patients. Acta Chim. Slov. 2017. 64: 530–536. [DOI] [PubMed] [Google Scholar]
- 18. Stunnenberg, M. , Sprokholt, J. K. , van Hamme, J. L. , Kaptein, T. M. , Zijlstra‐Willems, E. M. , Gringhuis, S. I. and Geijtenbeek, T. B. H. , Synthetic abortive HIV‐1 RNAs induce potent antiviral immunity. Front. Immunol. 2020. 11: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carpick, B. W. , Graziano, V. , Schneider, D. , Maitra, R. K. , Lee, X. and Williams, B. R. G. , Characterization of the solution complex between the interferon‐induced, double‐stranded RNA‐activated protein kinase and HIV‐I trans‐activating region RNA. J. Biol. Chem. 1997. 272: 9510–9516. [DOI] [PubMed] [Google Scholar]
- 20. Maitra, R. , McMillan, N. A.. , Desai, S. , McSwiggen, J. , Hivanessian, A. G. , Sen, G. , Williams, B. R. et al., HIV‐1 TAR RNA has an intrinsic ability to activate interferon‐inducible enzmes. Virology 1994. 204: 823–827. [DOI] [PubMed] [Google Scholar]
- 21. Kahlenberg, J. M. and Dubyak, G. R. , Mechanisms of caspase‐1 activation by P2X 7 receptor‐mediated K+ release. Am. J. cell physiol 2004. 286: 1100–1108. [DOI] [PubMed] [Google Scholar]
- 22. Schroder, K. , Zhou, R. and Tschopp, J. , The NLRP3 inflammasome: A sensor for metabolic danger? Science (80‐.). 2010. 327: 296–300. [DOI] [PubMed] [Google Scholar]
- 23. Franchi, L. , Muñoz‐planillo, R. and Núñez, G. , Sensing and reacting to microbes via the inflammasomes (NIH Public Access Author manuscript). Nat. Immunol. 2012. 13: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li, G. , Scull, C. , Ozcan, L. and Tabas, I. , NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010. 191: 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma, C. H. , Wu, C. H. , Jou, I. M. , Tu, Y. K. , Hung, C. H. , Hsieh, P. L. and Tsai, K. L. , PKR activation causes inflammation and MMP‐13 secretion in human degenerated articular chondrocytes. Redox Biol. 2018. 14: 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou, R. , Tardivel, A. , Thorens, B. , Choi, I. and Tschopp, J. , Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010. 11: 136–140. [DOI] [PubMed] [Google Scholar]
- 27. Goh, K. C. , DeVeer, M. J. and Williams, B. R. G. , The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000. 19: 4292–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. García, M. A. , Gil, J. , Ventoso, I. , Guerra, S. , Domingo, E. , Rivas, C. and Esteban, M. , Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006. 70: 1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gal‐Ben‐Ari, S. , Barrera, I. , Ehrlich, M. and Rosenblum, K. , PKR: A kinase to remember. Front. Mol. Neurosci. 2019. 11: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghonime, M. G. , Shamaa, O. R. , Das, S. , Eldomany, R. A. , Fernandes‐Alnemri, T. , Alnemri, E. S. , Gavrilin, M. A. et al., Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014. 192: 3881–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song, N. , Liu, Z. S. , Xue, W. , Bai, Z. F. , Wang, Q. Y. , Dai, J. , Liu, X. et al., NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017. 68: 185–197. [DOI] [PubMed] [Google Scholar]
- 32. Yin, X. , Langer, S. , Zhang, Z. , Herbert, K. M. , Yoh, S. , König, R. and Chanda, S. K. , Sensor sensibility‐HIV‐1 and the innate immune response. Cells. 2020. 9: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang, L. , Mosoian, A. , Schwartz, M. E. , Florman, S. S. , Schiano, T. , Fiel, M. I. , Jiang, W. et al., HIV infection modulates IL‐1β response to LPS stimulation through a TLR4‐NLRP3 pathway in human liver macrophages. J. Leukoc. Biol. 2019. 105: 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heil, F. , Hemmi, H. , Hochrein, H. , Ampenberger, F. , Kirschning, C. , Akira, S. , Lipford, G. et al., Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science (80‐.). 2004. 303: 1526–1529. [DOI] [PubMed] [Google Scholar]
- 35. He, Y. , Franchi, L. and Núñez, G. , The protein kinase PKR is critical for LPS‐induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013. 43: 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hett, E. C. , Slater, L. H. , Mark, K. G. , Kawate, T. , Monks, B. G. , Stutz, A. , Latz, E. et al., Chemical genetics reveals a kinase‐independent role for protein kinase R in pyroptosis. Nat. Chem. Biol. 2013. 9: 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu, B. , Nakamura, T. , Inouye, K. , Li, J. , Tang, Y. , Lundbäck, P. , Valdes‐Ferrer1, S. I. et al., Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012. 488: 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yim, H. C. H. , Wang, D. , Yu, L. , White, C. L. , Faber, P. W. , Williams, B. R. G. and Sadler, A. J. , The kinase activity of PKR represses inflammasome activity. Cell Res. 2016. 26: 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou, H. R. , Lau, A. S. and Pestka, J. J. , Role of double‐stranded RNA‐activated protein kinase R (PKR) in deoxynivalenol‐induced ribotoxic stress response. Toxicol. Sci. 2003. 74: 335–344. [DOI] [PubMed] [Google Scholar]
- 40. Zhang, J. , Wang, X. , Vikash, V. , Ye, Q. , Wu, D. , Liu, Y. and Dong, W. , ROS and ROS‐mediated cellular signaling. Oxid. Med. Cell. Longev. 2016. 2016: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Son, Y. , Kim, S. , Chung, H. T. and Pae, H. O. . 2013. Reactive oxygen species in the activation of MAP kinases, 1st ed. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 42. Sato, A. , Okada, M. , Shibuya, K. , Watanabe, E. , Seino, S. , Narita, Y. , Shibui, S. et al., Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor‐initiating capacity of glioma‐initiating cells. Stem Cell Res. 2014. 12: 119–131. [DOI] [PubMed] [Google Scholar]
- 43. Clerzius, G. , Gélinas, J. ‐ F. , Daher, A. , Bonnet, M. , Meurs, E. F. and Gatignol, A. , ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009. 83: 10119–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McMillan, N. A. J. , Chun, R. F. , Siderovski, D. P. , Galabru, J. , Toone, W. M. , Samuel, C. E. , Mak, T. W. et al., HIV‐1 TAT directly interacts with the interferon‐induced, double‐stranded rna‐dependent kinase, PKR. Virology 1995. 213: 413–424. [DOI] [PubMed] [Google Scholar]
- 45. Wang, Q. , Gao, H. , Clark, K. M. , Mugisha, C. S. , Davis, K. , Tang, J. P. , Harlan, G. H. et al., CARD8 is an inflammasome sensor for HIV‐1 protease activity. Science (80‐.). 2021. 371: eabe1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bauernfried, S. , Scherr, M. J. , Pichlmair, A. , Duderstadt, K. E. and Hornung, V. , Human NLRP1 is a sensor for double‐stranded RNA. Science (80‐.). 2021. 371: eabd0811. [DOI] [PubMed] [Google Scholar]
- 47. Bergsbaken, T. , Fink, S. L. and Cookson, B. T. , Pyroptosis: Host cell death. Nat. Rev. Microbiol. 2009. 7: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Doitsh, G. , Galloway, N. L. K. K. , Geng, X. , Yang, Z. , Monroe, K. M. , Zepeda, O. , Hunt, P. W. et al., Pyroptosis drives CD4 T‐cell depletion. Nature 2014. 505: 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doitsh, G. , Cavrois, M. , Lassen, K. G. , Zepeda, O. , Yang, Z. , Santiago, M. L. , Hebbeler, A. M. et al., Abortive HIV infection mediates CD4 T‐cell depletion and inflammation in human lymphoid tissue. Cell 2010. 143: 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarrami‐Forooshani R., Mesman A. W., Teijlingen N. H., Sprokholt J. K., Vlist M., Ribeiro C. M. S., Geijtenbeek T. B. H., Human immature Langerhans cells restrict CXCR4‐using HIV‐1 transmission. 2014;11: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Data available on request from the authors.