
Figure 4. The Aortic Valve in CAVD.
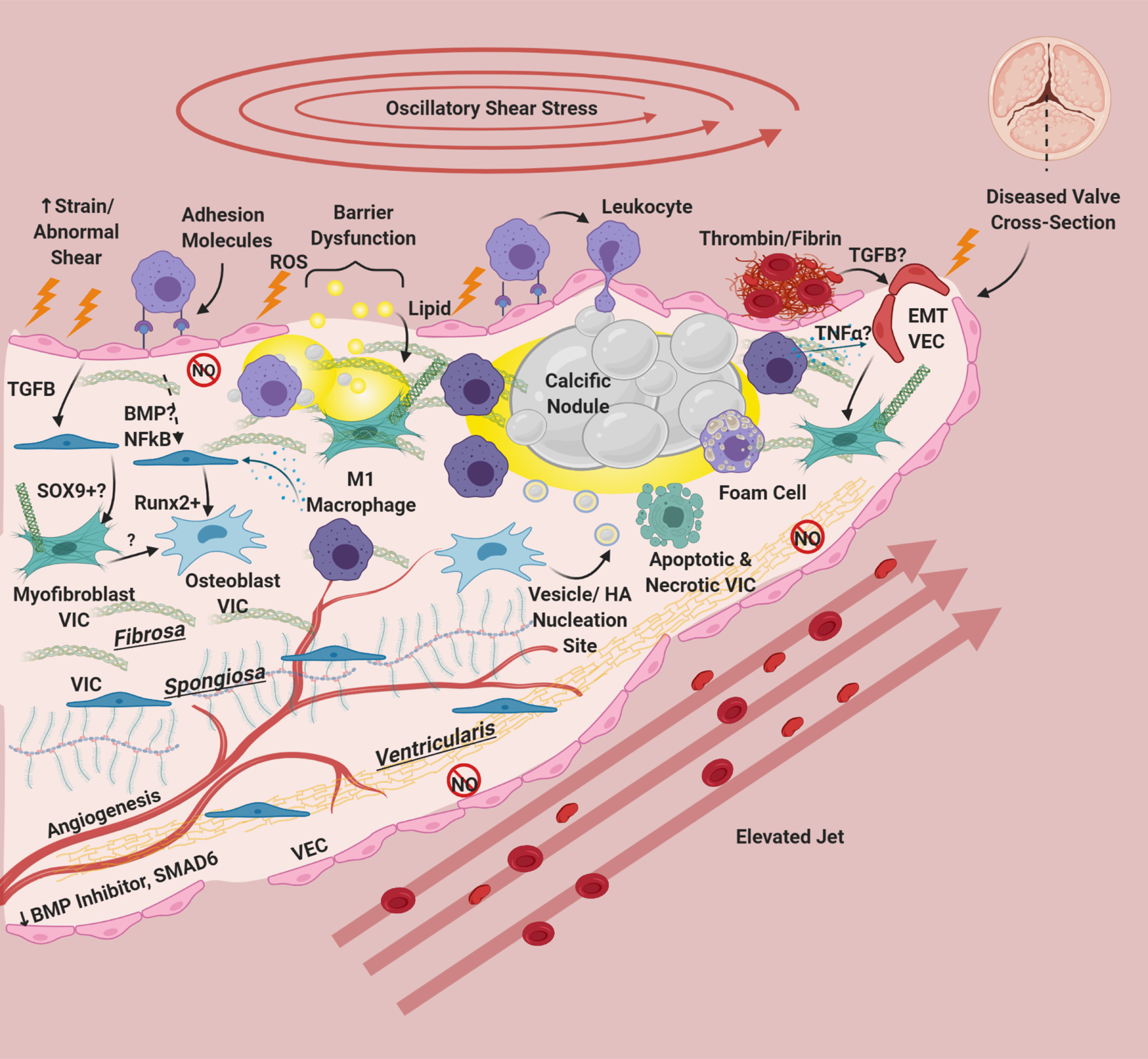
Risk factors cause damage, inflammatory adhesion expression, barrier disruption, and lipid deposition in/underneath the endothelium. Deposited lipids, specifically oxidized lipids can cause inflammation and damage in resident cells. Endothelial dysfunction results in eNOS down-regulation and dysfunction, abrogating protective eNOS signaling to VIC and in hemostasis. This is especially apparent on the ventricularis side of the valve. There is also a reduction in protective BMP inhibitors and SMAD6 on the ventricularis side of the valve. Leukocytes bind to VEC adhesion molecules and undergo extravasation into the tissue. Clotting factors including thrombin, tissue factor, and fibrin have been associated with calcified valves, and platelets recently have been implicated as sources of TGFB in valve disease. TGFB from VEC has been shown to induce Sox9 expression and to produce activated myofibroblast-like VIC. Paracrine signals from macrophages, specifically M1 macrophages has been shown to induce osteogenic differentiation of VIC. NFκB and BMP (possibly) from VEC can also modulate VIC osteoblastic differentiation. Osteoblastic VIC may excrete vesicles or undergo apoptosis/necrosis that deposit calcium and create HA nucleation sites, which grow over time to form large calcific nodules. Inflammatory infiltrate localizes around nodules, and areas of angiogenesis. Foam cells are observed in CAVD, but do not form dense necrotic cores. Inflammatory cytokines like TGFB and TNFα can cause VEC to undergo EndMT, creating fibrotic activated VIC-like cells. Thickening of the valve happens not only through lipid deposition/calcification but also fibrotic remodeling from activated VIC. CAVD induces angiogenesis in the normally avascular leaflet. For reasons unknown but hypothesized to be due to laminar shear stress, the ventricularis of the CAVD valve remains largely unaffected