
Figure 3.
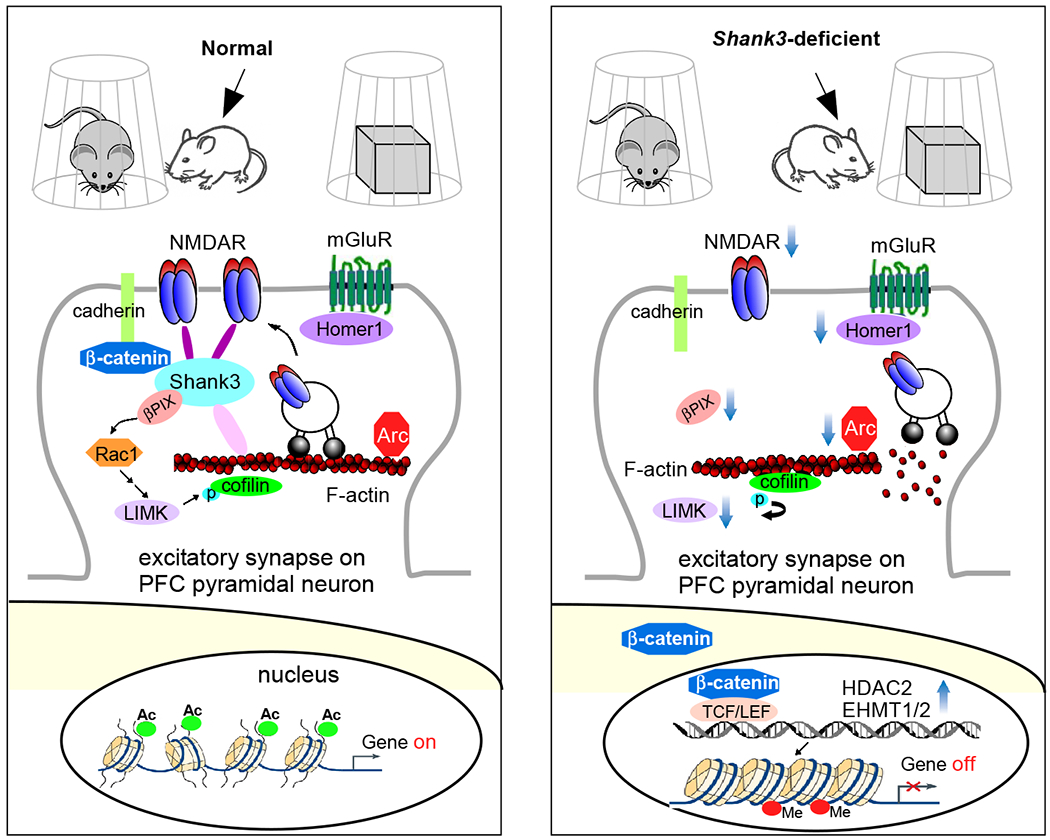
Schematic diagram illustrating a potential mechanism underlying the social deficits in Shank3 autism models. Normally, Shank3 crosslinks NMDARs to actin cytoskeleton, and binds to the adhesive junction-associated protein β-catenin. Loss of Shank3 leads to the translocation of β-catenin from synapses to nucleus, inducing the upregulation of histone modifiers HDAC2 and EHMT1/2. The ensuing transcriptional suppression of actin regulators, such as βPIX (Rac1 activator) and LIMK, and synaptic plasticity genes, such as Arc and Homer1 (mGluR anchor), results in the disruption of actin filaments (via cofilin-mediated depolymerization) and the diminished actin-based synaptic delivery of NMDARs in PFC pyramidal neurons. Consequently, the autism-like social preference deficits are manifested. Treatment with HDAC or EHMT inhibitors restores or elevates many target genes, which collectively leads to the normalization of NMDAR synaptic function in PFC and the rescue of social deficits127,145,146.