
Fig. 2.
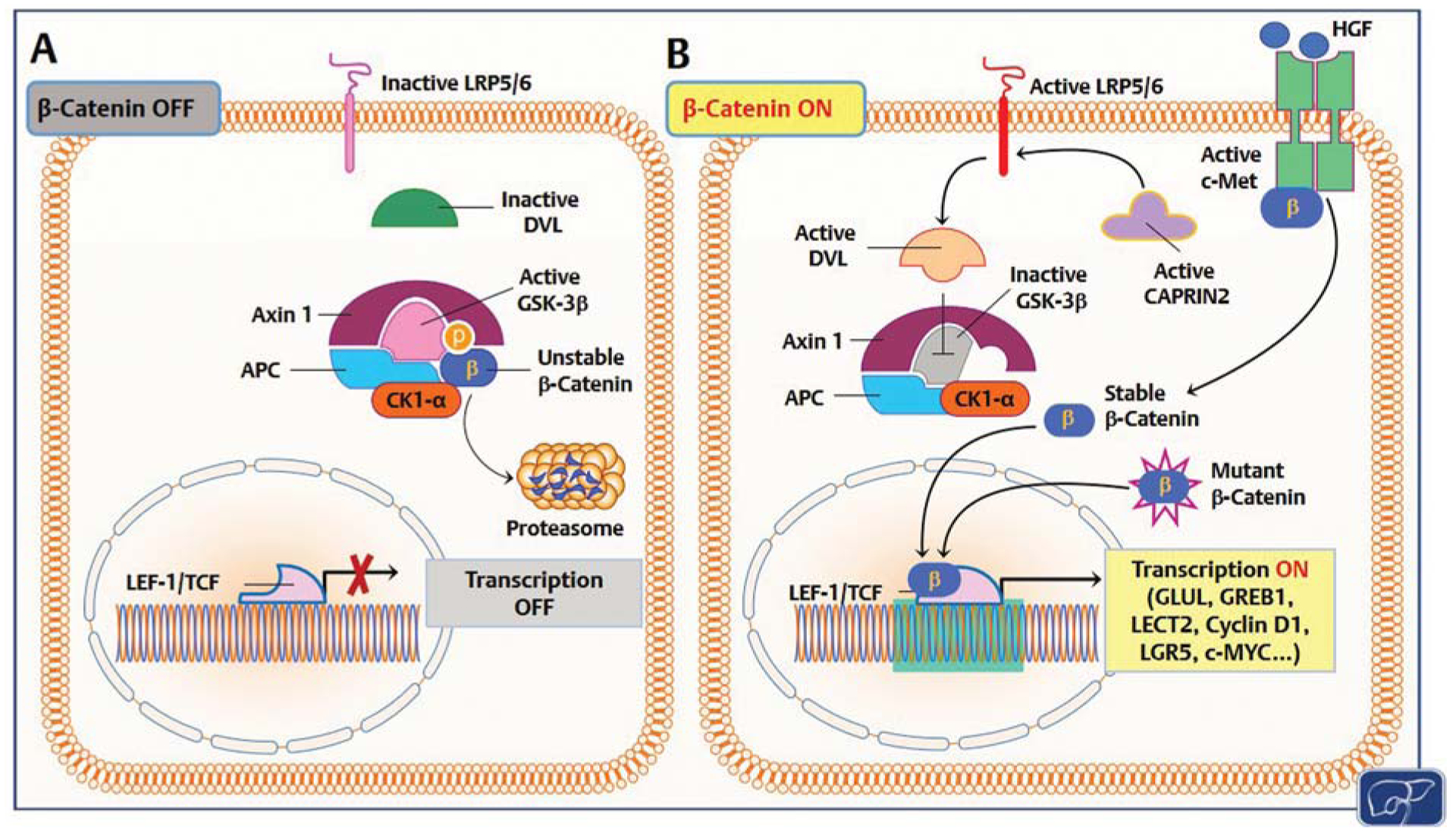
Mechanisms responsible for β-catenin activation in human hepatoblastoma (HB). (A) In quiescent hepatocytes, the β-catenin pathway is turned off (β-catenin OFF). Wild-type β-catenin (unstable β-catenin) is sequestered by a destruction complex consisting of adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK-3β), AXIN1, and casein kinase I α (CK1-α) and primed for proteolysis. (B) In HB, through somatic mutations (mutant β-catenin), β-catenin escapes proteasomal degradation and translocates into the nucleus, where it associates with T cell factor (TCF)/lymphoid enhancer 1 (LEF-1) transcription factors and induces the transcription (transcription ON) of target genes, including glutamine synthetase (GLUL), growth regulation by estrogen in breast cancer 1 (GREB1), leukocyte-cell-derived chemotaxin 2 (LECT2), cyclin D1, leucine-rich repeat containing G protein-coupled receptor 5 (LGR5), c-Myc, etc. Alternatively, β-catenin degradation is suppressed by HB tumor cells via CAPRIN2-activating mutations. Mutant CAPRIN2 activates LDL-receptor-related protein 5 and 6 (LRP5/6) than in turn activates the adaptor protein Disheveled (DVL). Consequently, DVL triggers GSK-3β inactivation and disrupts the β-catenin destruction complex. Furthermore, c-Met receptor activation by hepatocyte growth factor (HGF) induces nuclear translocation of β-catenin via tyrosine phosphorylation. In quiescence, c-Met and β-catenin physically interact at the inner surface of the hepatocyte plasma membrane.