

Abstract
Current treatments for unwanted antibody responses largely rely on immunosuppressive drugs compromising overall immunity. New approaches to achieve antigen-specific tolerance are desirable to avoid unwanted side effects. Several nanoparticle-based approaches that utilize different mechanisms to tolerize the B or T cell arms of the humoral immune response have shown promise for induction of antigen-specific tolerance, raising the possibility that they could work synergistically if combined. Earlier we showed that Siglec-engaging tolerance-inducing antigenic liposomes (STALs) that display both an antigen (Ag) and glycan ligands of the inhibitory co-receptor CD22 (CD22L) lead to robust antigen-specific B cell tolerance to protein antigens in naïve mice. In another approach, administration of free Ag with poly(lactic co-glycolic acid)-rapamycin nanoparticles (PLGA-R) induced robust antigen-specific tolerance through production of regulatory T cells. Here we illustrate that co-administration of STALs together with PLGA-R to naïve mice induced more robust tolerance to multiple antigen challenges than either nanoparticle alone. Moreover, in K/BxN mice that develop spontaneous autoimmune arthritis to the self-antigen glucose-6-phosphate-isomerase (GPI), co-delivery of GPI-LP-CD22L and PLGA-R delayed onset of disease, and in some mice prevented the disease indefinitely. The results show synergy between B cell-tolerizing STALs and T cell-tolerizing PLGA-R and the potential to induce tolerance in early stage autoimmune disease.
Keywords: Immune tolerance, autoimmune disease, arthritis, Siglec, nanoparticles, PLGA, liposome, K/BxN
Undesired immune responses are responsible for numerous human maladies including autoimmune diseases,1,2 allergies,3 transplantation rejection,4,5 and production of inhibitory antibodies to biotherapeutic medicines.6 Existing treatments for these conditions are not antigen-specific and therefore lead to broad immune suppression with numerous adverse effects.7 Thus, to suppress undesired responses to self-antigens that mediate autoimmune disease, antigen-specific tolerance approaches are needed that provide immune tolerance to the antigen of interest while leaving the rest of the immune system intact. Various approaches have been used to induce antigen-specific immune tolerance in the B cell and T cell arms of the humoral immune response.8–11 In principle, since some of the approaches used involve different mechanisms for tolerance induction, they may be combined for more robust tolerance.
A direct approach to induce antigen-specific B cell tolerance utilizes STALs (Siglec tolerizing antigenic liposomes) which co-present an antigen of interest with high affinity ligands of the B cell Siglec, CD22, on a liposomal nanoparticle platform. STALs target B cells that recognize the antigen and recruitment of the Siglecs to the immunological synapse formed when the B cell receptor (BCR) engages the antigen on an antigen presenting cell.12–17 This engagement leads to a potent apoptotic signal resulting in elimination of antigen-reactive B cells, leaving the rest of the B cell repertoire unaffected, and induction of B cell tolerance due to depletion of the antigen-specific B cells.18 The utility of STALs has been successfully demonstrated in several animal models of disease resulting from generation of antigen-specific antibodies including development of inhibitory antibodies to FVIII in hemophilia mice14 and development of anti-peanut IgE in a peanut antigen anaphylaxis model.19,20 Its potential for impacting rheumatoid arthritis has been highlighted by the demonstration that STALs induce apoptosis of memory B cells from RA patients that produce anti-citrullinated-protein antibodies (ACPA).21
STALs are particularly effective in naïve animals since there is minimal activation of CD4+ T cells that are required for B cells to differentiate into cells that produce high affinity antibodies of the IgG class, and are less suitable for inducing tolerance in animals with an ongoing immune response with antigen-specific CD4+ helper or memory T cells. Several approaches have been used to induce antigen-specific tolerance in the T cell arm of the immune response. Currently available to allergic patients is allergen immunotherapy (AIT) where gradually increasing doses of antigen are administered by injection or orally to patients over several years to build up tolerance. While AIT has been helpful in desensitizing patients to antigen, there is limited long-lasting desensitization when treatment ends.20,22,23 Several approaches in development seek to create antigen-specific T regulatory cells (Tregs) or delete/reprogram antigen-specific T cells.10,11,24 These include use of peptide-MHC bound nanoparticles,24,25 antigen coupled to syngeneic cells,26,27 or antigen and/or immune-modulators comprised in synthetic nanoparticles.28–35
An attractive option for inducing T cell tolerance currently in clinical development involves administering antigen with poly(lactic co-glycolic acid) (PLGA) particles containing the immunomodulator rapamycin. Initially, Maldonado et al. showed that PLGA nanoparticles encapsulated with both antigen and rapamycin could induce robust tolerance to a variety of antigens in naïve animals.29 Subsequently, it was shown that PLGA nanoparticles with rapamycin (PLGA-R) co-injected with soluble antigen produced equally robust tolerance.30,31,34–36 Mechanistic studies support the idea that the non-targeted rapamycin-PLGA particles efficiently deliver the drug to APCs to create a tolerogenic cytokine environment, which in turn induces Tregs that prevent activation of T helper cells needed by B cells to produce inhibitory antibodies.29–31,36
Because the tolerogenic nanoparticle technologies (STALs and PLGA-R) impact immune responses of the B cell and T cell arms of the immune system by different mechanisms, we reasoned that they might be synergistic, providing more robust tolerance than either alone. Initial attempts to incorporate rapamycin into the lipid bilayer of STALs showed potential for improving tolerance in naïve mice, but were limited by the dose of rapamycin that could be incorporated and inability to induce tolerance in previously sensitized mice.37 Here we have compared use of STALs and PLGA-R for suppressing antibody production in naïve mice for preventing inflammatory disease in the antigen-specific K/BxN arthritis model. Administering STALs and PLGA-R in combination significantly suppressed antibody production to liposomal-antigen over either nanoparticle alone. In K/BxN mice that develop autoimmune rheumatoid arthritis (RA) dependent on production of antibodies to the self-protein, glucose-6-phosphate isomerase (GPI),38–40 neither GPI-STALs or PLGA-R alone had significant impact on the onset of disease. When administered together weekly for up to 5 weeks, the onset of disease was delayed, and some mice remained disease free for up to 150 days. Some benefit could also be achieved by delaying treatment until ankle swelling was apparent. The results suggest that combining these B and T cell tolerance strategies hold promise for suppressing unwanted immune reactions.
RESULTS AND DISCUSSION
Nanoparticle synthesis and characterization.
Unless otherwise noted, STALs and PLGA-R nanoparticles were prepared as previously described with few modifications.14,29 For incorporation into liposomes (LP), the mouse CD22 ligand (CD22L) was coupled to pegylated-lipid (Figure 1A), and the protein antigen (Ag) was also coupled to lipid as described.14 Schematic illustrations of STALs (Ag-LP-CD22L) and PLGA-R nanoparticles are shown in Figure 1B.
Figure 1.
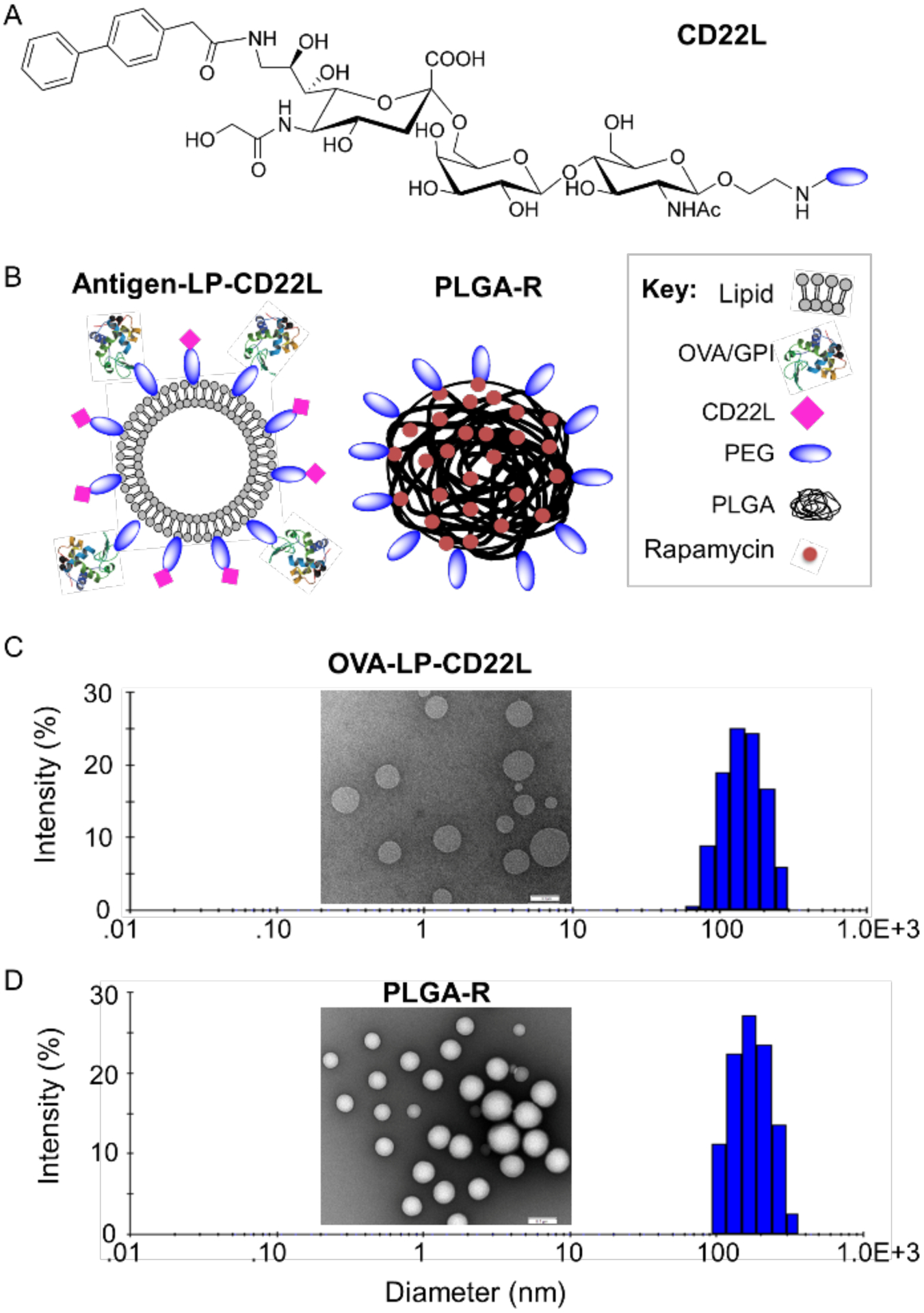
Nanoparticle formulation and characterization. (A) Chemical structure of the murine CD22 ligand linked to PEG. (B) Schematic illustration of targeted OVA/GPI-STALs (OVA/GPI-LP-CD22L) and PLGA rapamycin (PLGA-R) nanoparticles. (C-D) Representative transmission electron microscopic images of OVALP-CD22L (inset C) and PLGA-R (inset D), and hydrodynamic size distribution of OVA-LP-CD22L (156±32; plot C) and PLGA-R (182±31; plot D) as determined by dynamic light scattering.
STALs with ovalbumin (OVA) as antigen (OVA-STALs; OVA-LP-CD22L) and biodegradable PLGA-R nanoparticles were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Figure 1C–D). The hydrodynamic diameter of OVA-LP-CD22L and PLGA-R was found to be 156±32 and 182±31, respectively, as measured by DLS. TEM characterization confirmed that both OVA-LP-CD22L and biodegradable PLGA-R showed uniform spherical morphology.
STALs and PLGA-R synergize to produce enhanced tolerance.
Initial reports showed that PLGA-R nanoparticles together with antigen induced tolerance to the antigen of interest by inducing Tregs without inducing broad immune suppression to other antigens.29 Induction of tolerance in B cells by recruitment of the inhibitory receptor CD22 using STALs was also shown to be antigen-specific.14,16 Accordingly, we set out to evaluate the potential for these two approaches to more robustly induce tolerance if used together. Since STALs are formulated with protein antigen, STALs and PLGA-R were tested alone and in combination for their ability to suppress antibody production against a multivalent antigen, liposomal ovalbumin (OVA-LP or OVA-LP-CD22L), in C57BL/6J mice. The concentration of OVA (0.1 mol %) and CD22 ligand (1.5 mol %) used in the STALs was selected based on previously optimized formulations.14,37 PLGA-R nanoparticles contained 50–100 μg of rapamycin since earlier studies indicated this as an optimal dose for inducing tolerance.34,35 Similar formulations of STALs and PLGA nanoparticles have been reported to have serum half-lives in mouse blood of 11 and 16 hours, respectively.41,42
Mice were treated (i.v.) once (day 0) or twice (days 0 and 14) with OVA-LP, OVA-LP + PLGA-R, OVA-STALs (OVA-LP-CD22L), or OVA-STALs + PLGA-R, followed by challenge (i.p.) 14 and 35 days later with OVA/Alum and free OVA (fOVA), respectively. With one treatment, analysis of anti-OVA serum titers showed that treatment with OVA-STALs significantly reduced serum titers of OVA relative to mice treated with OVA-LP. Moreover, OVA-LP-CD22L and PLGA-R together further reduced anti-OVA IgG1 titers relative to mice treated with STALs alone or OVA-LP + PLGA-R, demonstrating synergy between the two tolerogenic nanoparticle systems (Figure 2A).
Figure 2.
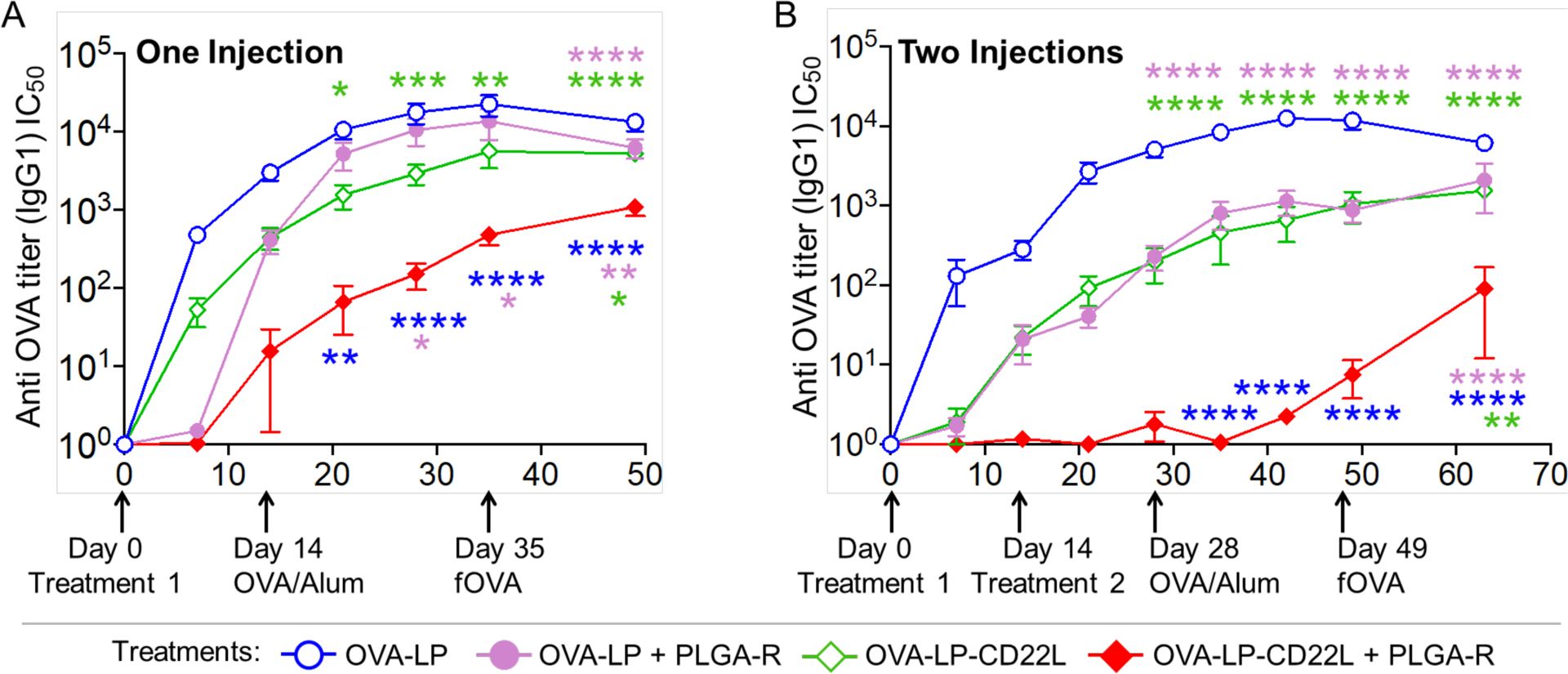
Co-delivery of OVA-STALs (OVA-LP-CD22L) and PLGA-R nanoparticles induce greater tolerance to liposomal OVA. (A) C57BL/6J mice (n = 16) were immunized on day 0 with the indicated nanoparticle treatments i.v. and then challenged i.p. with OVA/Alum on day 14 and fOVA on day 35. Data were pooled from three independent experiments. (B) C57BL/6J mice (n = 5) were immunized on days 0 and 14 with the indicated nanoparticle treatments i.v. and then challenged i.p. with OVA/Alum on day 28 and fOVA on day 49. Results are representative of two independent experiments. PLGA-R nanoparticles contained 100 μg of rapamycin.35 Mice were bled weekly after the treatment, and IgG1 titers were assessed for OVA. Blood from naïve mice was used as a control to normalize the data. Normalized titers were obtained by dividing the observed titers with the mean titer from naïve mice. All data represent the mean ± SEM. All statistical analyses were performed on raw data using two-way ANOVA with Tukey’s post-test (**** P ≤ 0.0001; ***P ≤ 0.001; ** P ≤ 0.01; *P ≤ 0.05).
With two treatments at 0 and 14 days, greater tolerance to OVA challenge was evident with the combined treatment with OVA-LP-CD22L + PLGA-R showing significantly greater suppression than other treatment groups (Figure 2B). Notably, while OVA-LP + PLGA-R induced weak suppression of anti-OVA production, two treatments with fOVA + PLGA-R induced strong tolerance equivalent to OVA-STALs + PLGA-R (Supplementary figure S1). Thus, we suggest that PLGA-R is more effective at inducing tolerance with monovalent antigen than the multivalent antigen exemplified by OVA-LP.
Since the strategy for combining STALs and PLGA-R envisioned the induction of CD4+ antigen-specific regulatory T cells that would synergize with STALs for suppression of B cell activation,29 we investigated production of Tregs in naïve mice. OVA-specific CD4+ T cells (OTII cells) were adoptively transferred into naïve mice, and the next day mice were treated with OVA-STALs (OVA-LP-CD22L) alone or with one or two doses of OVA-STALs + PLGA-R. On day 28 post-treatment, the number of Foxp3+CD25+OTII T cells in the spleen were evaluated (Supplementary figure S2). While no Treg induction was observed with OVA-STALs alone, Tregs were induced when OVA-STALs were administered together with PLGA-R nanoparticles.
Treatment of K/BxN arthritis mice with tolerogenic nanoparticles delays onset of disease symptoms.
To evaluate the potential for the combined STALs + PLGA-R nanoparticle treatments to modulate ongoing autoimmune disease, we chose the K/BxN autoimmune rheumatoid arthritis (RA) model.38–40 The K/BxN arthritis model recapitulates many histological features of human rheumatoid arthritis, including cartilage and bone destruction, leukocyte invasion, synovitis, and pannus formation. This is an ideal model since it is caused by development of autoimmune antibodies to the self-antigen glucose-6-phosphate isomerase (GPI), and both B and T cells are required for disease onset and pathology. K/BxN mice express the TCR transgene KRN and MHC class II molecule Ag7, spontaneously produce GPI reactive antibodies, and develop chronic RA symptoms as they age.38–40 Since the relevant antigen for the K/BxN model is GPI, an abundant self-antigen made by every cell, K/BxN mice are not “naïve” to the antigen, even at birth. All mice develop symptoms of RA and begin to exhibit ankle swelling between 25–30 days of age, allowing treatments to begin prior to disease onset. For these experiments, GPI-STALs were prepared in a manner analogous to OVA-STALs.14
K/BxN mice were injected i.v. with 1, 2, or 5 doses of nanoparticles every 6–7 days, starting at 21–25 days of age, and monitored for disease progression by measuring front and hind paw thickness three times per week (Figure 3A). Mice were considered to be at the study endpoint when one or more joint(s) measured 4 mm. GPI-STALs (GPI-LP-CD22L) and PLGA-R nanoparticles administered separately in two doses failed to alter disease progression since the median survival of mice (percentage of mice without disease) treated with either nanoparticles was not significantly different from untreated mice (37, 39, and 36 days for untreated, GPI-LP-CD22L-treated, and PLGA-R-treated mice, respectively; Figure 3B). However, when GPI-LP-CD22L and PLGA-R were co-delivered to K/BxN mice using the 1, 2, or 5-time dosing scheme (Figure 3C), disease progression was significantly delayed, demonstrating synergy of combining the two nanoparticles. The delay in disease progression was dose-dependent, as the median survival of mice without disease was 47.5, 56, and 80 days for mice treated with 1, 2, or 5 doses of GPI-LP-CD22L + PLGA-R, respectively. Astonishingly, none of the mice treated 5 times with GPI-LP-CD22L + PLGA-R had disease by 70 days of age, twenty days after dosing ended, and a few mice remained disease free to the end of the 150 day study. It will be important to determine if more prolonged administration will further delay in onset of disease and/or result in a greater proportion of animal subjects developing tolerance long term.
Figure 3.
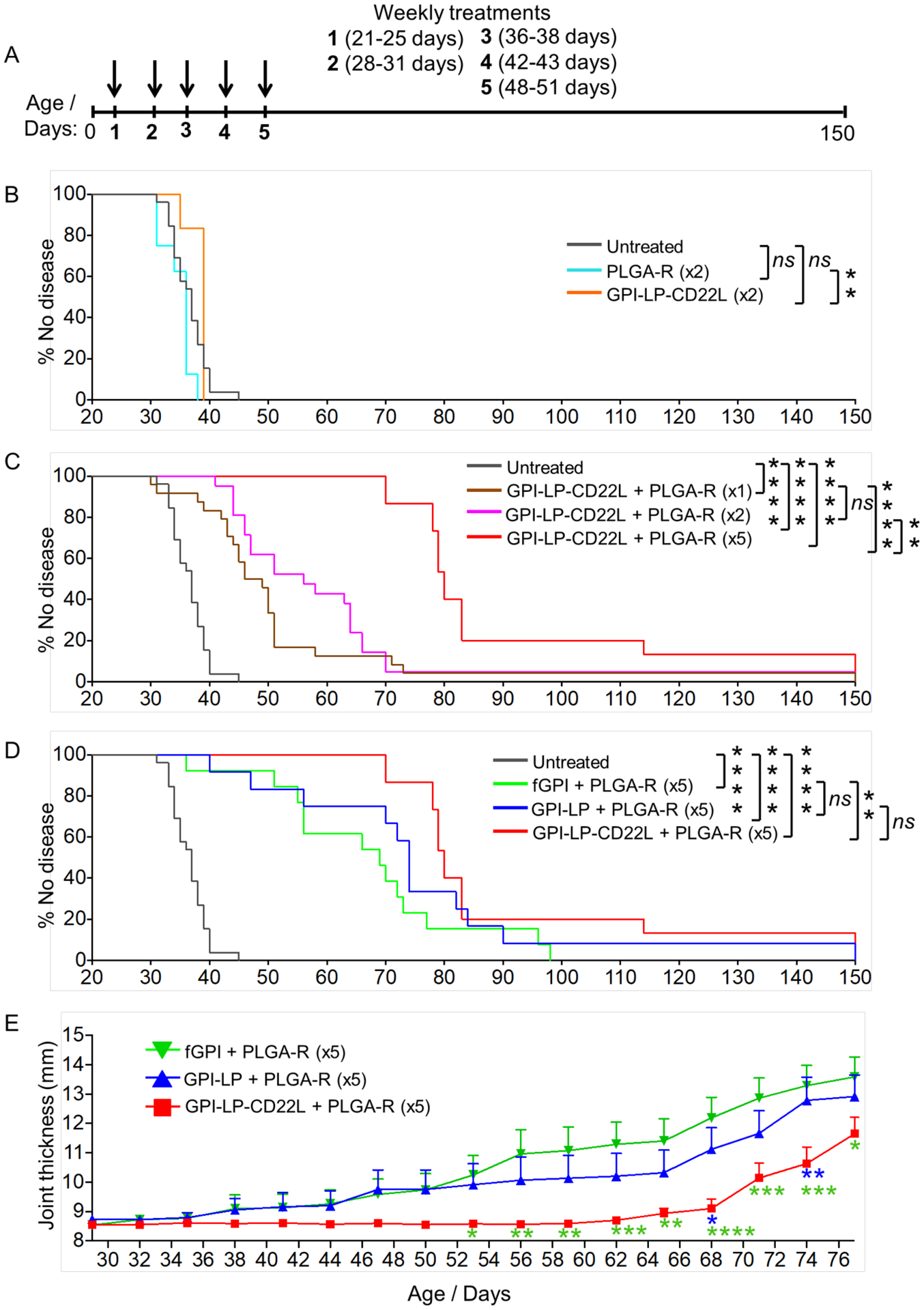
Administration of nanoparticles to K/BxN mice prior to onset of arthritis delayed disease. (A) Strategy for treating K/BxN mice with one, two, or five doses of nanoparticles prophylactically. PLGA-R nanoparticles contained 100 μg of rapamycin. (B-D) K/BxN mice, 21–25 days of age, were left untreated or treated once (x1) or weekly (every 6–7 days) for 2 (x2) or 5 (x5) consecutive weeks. Joint measurements were performed three times a week, and mice were considered to be at the study endpoint when one or more joint(s) measured 4 mm. Results over time for each group (n = 6–26), are shown as percent of mice with no joint swelling over 4 mm. Results are representative of two experiments. To facilitate comparisons, results for untreated mice and GPI-LP-CD22L (GPI-STALs) + PLGA-R are shown in panels B-D and C-D, respectively. (E) Disease severity assessed as total joint thickness for all four paws in treated mice out to 77 days. Statistical analyses were performed using a log-rank test in B-D and by two-way ANOVA with Tukey’s post-test in E (**** P ≤ 0.0001; *** P ≤ 0.001; ** P ≤ 0.01; *P ≤ 0.05 and ns indicates not significant).
Although we assumed that mice had abundant endogenous free antigen, we also investigated treatment of mice with PLGA-R with exogenous free soluble GPI (fGPI) or GPI liposomes (GPI-LP) without CD22L. Accordingly, mice were injected with fGPI (25 μg) + PLGA-R or GPI-LP + PLGA-R using the 5-dose injection scheme. Co-injection of PLGA-R with exogenous fGPI or GPI-LP also delayed onset of disease (Figure 3D). However, in contrast to GPI-STALs + PLGA-R both of these groups began to get disease during the treatment period, with 25–40% of the mice developing disease before the end of dosing at 50 days. GPI-STALs + PLGA-R was the only treatment that delayed the onset of disease for all mice during treatment and for nearly 20 days after treatment ended. Notably, while the survival curve of GPI-STALs + PLGA-R showed a statistically significant benefit with fGPI + PLGA-R, the difference did not reach statistical significance when compared to GPI-LP + PLGA-R (Figure 3D). However, when mean disease severity was based on the sum of ankle thickness for all four paws over time (Figure 3E), GPI-STALs + PLGA-R showed statistically significant benefit over both groups. Overall, by several measures GPI-STALs + PLGA-R exhibited enhanced suppression of disease over STALs or antigen + PLGA-R alone.
To assess disease progression histologically, ankle sections from untreated wild-type C57BL/6J mice (Figure 4A–D) and K/BxN mice at 55 days that were untreated (Figure 4E–H) or treated with 5 weekly doses of GPI-LP-CD22L+PLGA-R (Figure 4I–L) were stained with Safranine O/Fast Green to assess protection of the joints under the different treatment conditions. Normal cartilage in wild-type mice is characterized by the maintenance of Safranine O cartilage staining (Figure 4B), synovial tissue (Figure 4C), and the bone matrix (Figure 4D). This is in contrast to the untreated K/BxN mice that exhibit severe loss of cartilage, inflamed synovium with infiltration of immune cells into the joint space, and severe bone resorption at the cartilage/bone margin (Figure 4F, G, H, respectively). Conversely, joints in the treated K/BxN mice were nearly normal, with healthy cartilage, synovium, and minimal bone resorption (Figure 4J, K, L, respectively). Together these histochemical data suggest treatment with the combined nanoparticles can prevent damage to the cartilage, bone, and joint space.
Figure 4.
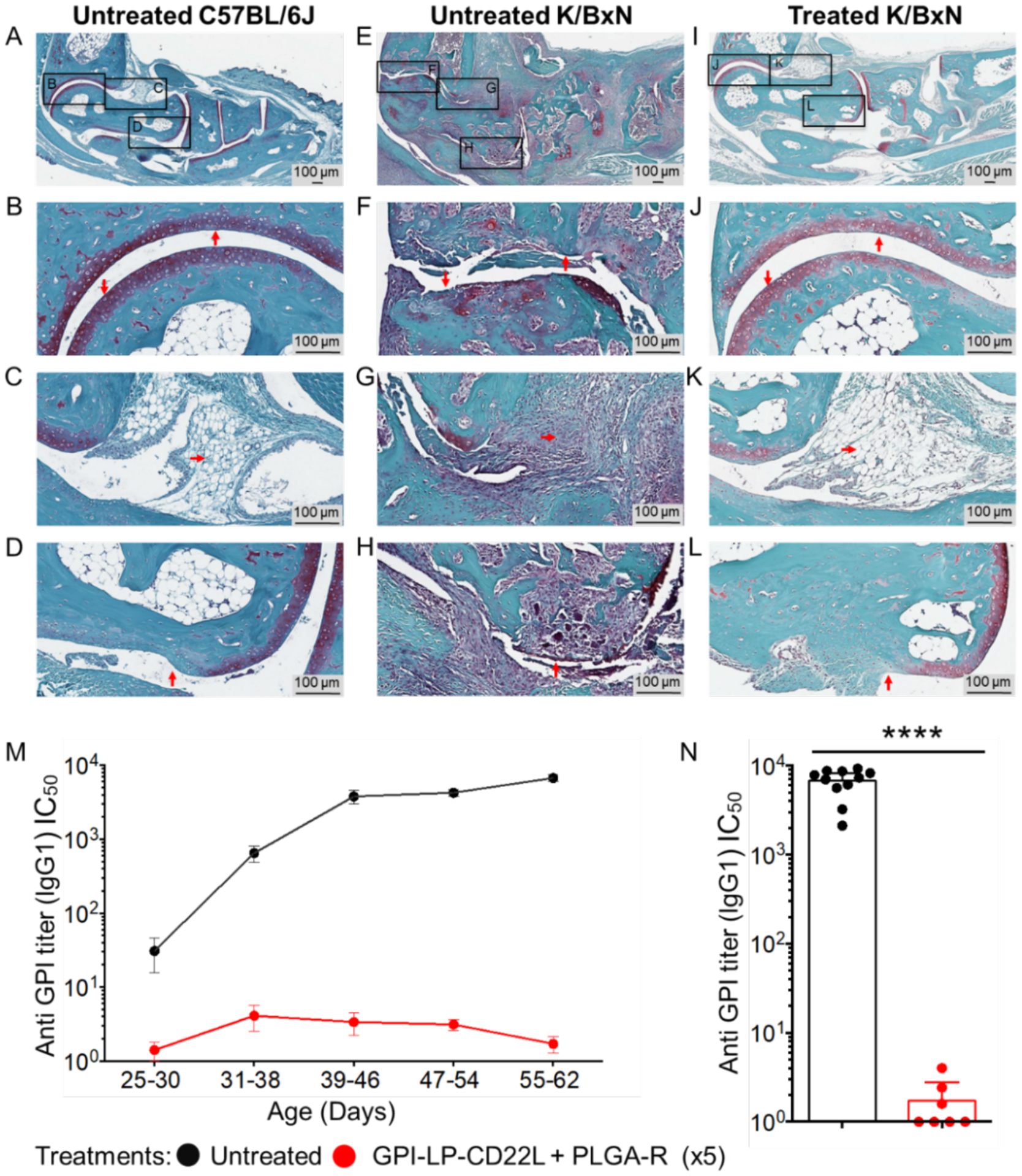
Co-administration of GPI-LP-CD22L and PLGA-R protects K/BxN mice from joint damage. Histochemical staining of day 55 paraffin-embedded ankle sections with Safranine O/Fast Green and hematoxylin counterstain reveals gross differences in the joint health of control wild-type mice left untreated (A), K/BxN diseased mice left untreated (E), and K/BxN diseased mice treated 5 times with GPI-LP-CD22L and PLGA-R (I). Higher magnification reveals distinct maintenance of cartilage (J), preservation of synovial tissue (K), and minimal bone resorption (L) in treated K/BxN mice, comparable to healthy controls (B, C, and D), which is in stark contrast to the loss of cartilage (F), severe infiltration of immune cells into the synovium (G), and severe bone resorption (H) seen in diseased K/BxN mice left untreated. Images are representative of two mice for each condition. (M) Co-administration of GPI-LP-CD22L and PLGA-R to K/BxN mice suppresses production of anti-GPI serum titers. Anti-GPI IgG titers from untreated K/BxN mice (black, n = 11) versus mice co-administered GPI-LP-CD22L and PLGA-R (red, n = 7) for 5 consecutive weeks as determined by ELISA. (N) Anti-GPI IgG1 titers plotted for days 55–62. All data represent the mean ± SEM. Statistical analysis was performed using the Mann-Whitney test (**** P ≤ 0.0001).
We also assessed the serum anti-GPI IgG titers from treated and untreated K/BxN mice by ELISA (Figure 4M–N). While untreated K/BxN mice developed high anti-GPI titers, mice treated with GPI-STALs + PLGA-R for 5 consecutive weeks, maintained very low anti-GPI titers, providing direct evidence that the treatments suppressed antibody production to GPI.
Therapeutic treatment of K/BxN arthritis mice delays progression of disease.
Although K/BxN mice are born with the propensity to develop autoimmune joint disease mediated by antibodies to GPI, we considered that once the disease symptoms had started, progression may be resistant to treatment with GPI-STALs + PLGA-R. However, motivated by the delayed onset of arthritis when treatment was initiated soon after weaning, we sought to test for efficacy when the first treatment was initiated after disease onset when at least one paw was swollen to a thickness of 2.8–3.05 mm. Mice were then either left untreated or given 5 weekly doses of GPI-LP-CD22L + PLGA-R containing either 50 or 100 μg of rapamycin (Figure 5A). For this study, we used total joint thickness, the sum of the thickness of all four paws, as a measure of disease severity. For untreated mice (n = 17), disease progressed rapidly with all mice achieving a score of >12 mm by day 40. Of the mice administered with GPI-LP-CD22L + PLGA-R containing 50 μg of rapamycin per treatment (Figure 5B, middle panel, n = 17), all but one mouse showed a delay in achieving a score of 12, and 9 out of 17 mice showed a more significant delay. Remarkably, 3 mice either had no progression, or progressed and then reversed disease severity. Moreover, 2 out of 5 mice administered with GPI-LP-CD22L + PLGA-R containing 100 μg of rapamycin per treatment showed no progression (Figure 5B, right panel). These results suggest that the combination of GPI-STALs + PLGA-R is able to significantly delay or prevent disease in a prophylactic modality and even reverse the disease progression in a therapeutic treatment modality. The majority of treated mice have a delay in disease onset relative to untreated mice (Supplementary figure S3) and have shown tolerance and suppression of ongoing disease. We further analyzed the anti-GPI titers for the mice left untreated versus mice treated with GPI-STALs + PLGA-R on days 30–35 and 50–55 (Figure 5C). While anti-GPI titers for the mice left untreated or treated with 50 μg/dose increased significantly over this period, there was no significant increase in titers for mice treated with 100 μg/dose rapamycin. Disease severity in the untreated and treated groups correlates with anti-GPI IgG titers in these mice (Supplementary figure S3).
Figure 5.
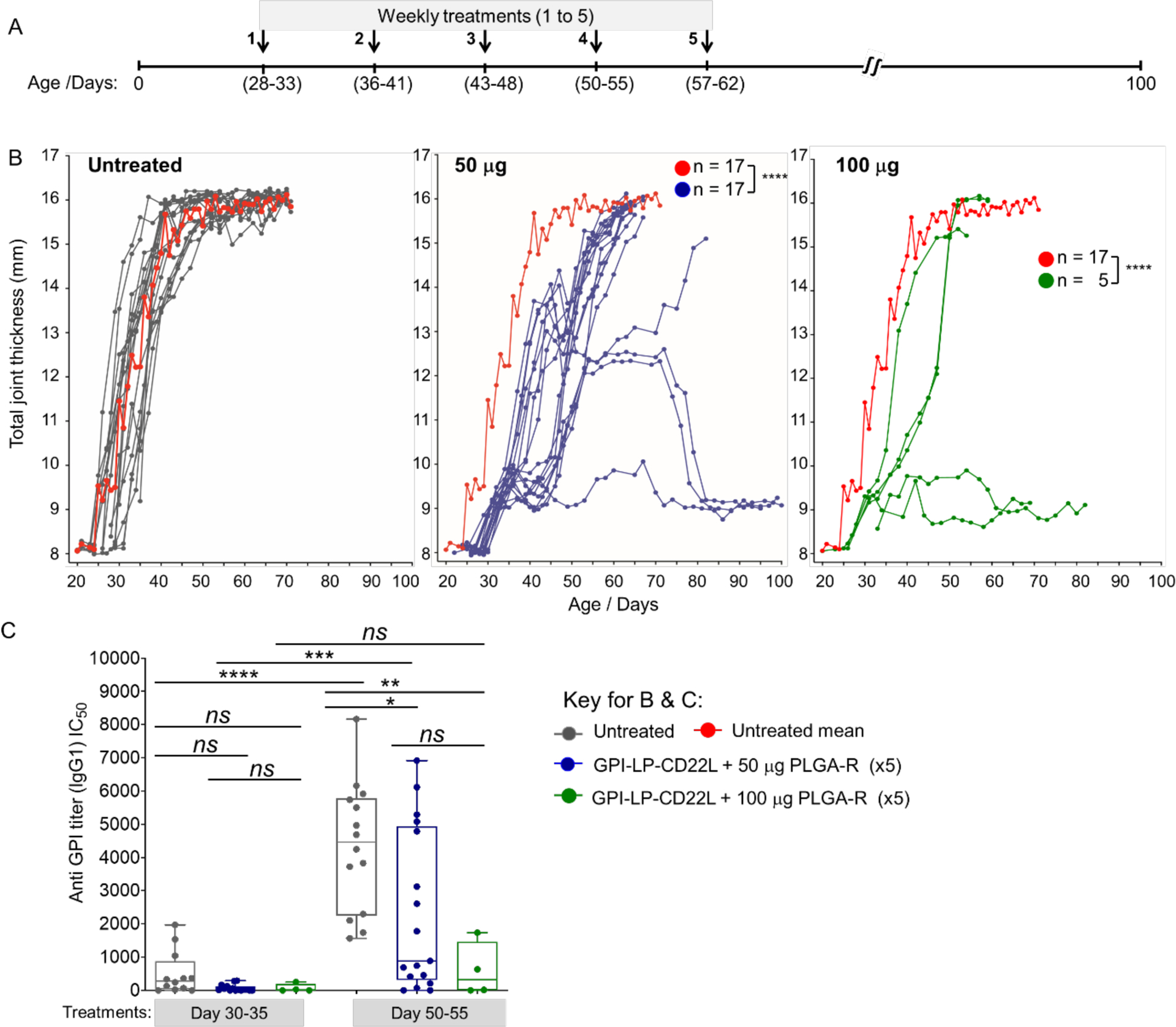
Treatment of K/BxN mice after disease onset reduces disease progression. (A) K/BxN mice were treated after disease onset, measured as >3mm in one or more joints. Mice were either left untreated or treated with 5 weekly injections of both GPI-STALs (GPI-LP-CD22L) and PLGA-R nanoparticles. (B) Progression of disease severity measured by total joint thickness for untreated mice (left panel), and mice treated with GPI-STALs (GPI-LP-CD22L) and PLGA-R nanoparticles containing either 50 μg (middle panel) or 100 μg (right panel) of rapamycin. The mean progression for untreated mice (red curve left panel) is repeated in the middle and right panels for comparison. (C) Anti-GPI antibody titers from untreated and GPI-STALs + PLGA-R treated K/BxN mice on days 30–35 and day 50–55. Statistical analyses were performed using mixed-effect analysis with a Sidak post-test in 5B and two-way ANOVA followed by Tukey’s test in 5C (**** P ≤ 0.0001; *** P ≤ 0.001; ** P ≤ 0.01; *P ≤ 0.05 and ns indicates not significant).
CONCLUSION
In conclusion, we found that co-administration of STALs targeting B cells that recognize GPI and PLGA-R that induces antigen-specific T cell tolerance act synergistically to induce more robust antigen-specific tolerance compared to either nanoparticle alone. Repeated dosing was beneficial, which was particularly evident in the delay of disease onset and progression in the K/BxN arthritis model. Co-administration of GPI-LP-CD22L and PLGA-R with 5 weekly doses delayed disease onset for 20 days after the final dose, with some mice remaining disease-free for the duration of the 150–day study. Significant delay in the onset of disease was also observed in animals treated with PLGA-R with fGPI or LP-GPI alone, but in both cases disease onset occurred in some mice during treatment. Therapeutic treatment of K/BxN mice with GPI-LP-CD22L and PLGA-R, after disease onset, when anti-GPI IgG1 titers had increased, delayed disease progression in most mice, and in some cases prevented and even reversed the disease. Taken together the results suggest the potential of antigen-specific therapy that simultaneously targets the B cell and T cell arms of the immune system to treat early stage autoimmune diseases where the antigen is known. Indeed, while the broadly immunosuppressive B cell depleting therapy with rituximab (anti-CD20) is therapeutically effective43,44 in a rheumatoid arthritis setting, there is also potential for an antigen-specific approach preventing production of anti-citrulline antibodies.21 We believe that further development is needed for combining the STAL and PLGA-R nanoparticle platforms for clinical application, in particular to determine the extent to which repeated dosing can suppress immune responses and induce long term tolerance.
Supplementary Material
ACKNOWLEDGMENTS
This manuscript has been released as a pre-print at [BioRxiv], ([Srivastava] et al.).45
We thank the following individuals at The Scripps Research Institute: K. Worrell for synthesis of the mCD22 ligand, M. Olmer for the histochemical staining protocol, T. Fassel for microscopy, and A. Tran-Crie for help with preparation of the manuscript.
Funding
This work was funded in part by NIH grants R01AI099141 & R01AI050143.
Footnotes
Supporting Information
Complete description of all experimental methods with associated figures and supporting references (PDF)
Complete contact information is available at: https://pubs.acs.org/
Author Kei Kishimoto was employed by the company Selecta Biosciences. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- [1].Naparstek Y, and Plotz PH (1993) The role of autoantibodies in autoimmune disease, Annu Rev Immunol 11, 79–104. [DOI] [PubMed] [Google Scholar]
- [2].Lleo A, Invernizzi P, Gao B, Podda M, and Gershwin ME (2010) Definition of human autoimmunity-autoantibodies versus autoimmune disease, Autoimmun Rev 9, A259–266. [DOI] [PubMed] [Google Scholar]
- [3].Gould HJ, and Sutton BJ (2008) IgE in allergy and asthma today, Nat Rev Immunol 8, 205–217. [DOI] [PubMed] [Google Scholar]
- [4].Kwun J, Bulut P, Kim E, Dar W, Oh B, Ruhil R, Iwakoshi N, and Knechtle SJ (2012) The role of B cells in solid organ transplantation, Semin Immunol 24, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Clatworthy MR (2011) Targeting B cells and antibody in transplantation, Am J Transplant 11, 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Singh SK (2011) Impact of product-related factors on immunogenicity of biotherapeutics, J Pharm Sci 100, 354–387. [DOI] [PubMed] [Google Scholar]
- [7].Vassilopoulos D, and Calabrese LH (2007) Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and co-existing chronic viral infections, Curr Opin Rheumatol 19, 619–625. [DOI] [PubMed] [Google Scholar]
- [8].Miller SD, Turley DM, and Podojil JR (2007) Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease, Nat Rev Immunol 7, 665–677. [DOI] [PubMed] [Google Scholar]
- [9].Sabatos-Peyton CA, Verhagen J, and Wraith DC (2010) Antigen-specific immunotherapy of autoimmune and allergic diseases, Curr Opin Immunol 22, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pearson RM, Casey LM, Hughes KR, Miller SD, and Shea LD (2017) In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance, Adv Drug Deliv Rev 114, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kishimoto TK, and Maldonado RA (2018) Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance, Front Immunol 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crocker PR, Paulson JC, and Varki A (2007) Siglecs and their roles in the immune system, Nat Rev Immunol 7, 255–266. [DOI] [PubMed] [Google Scholar]
- [13].Jellusova J, and Nitschke L (2011) Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22, Front Immunol 2, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, and Paulson JC (2013) Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis, J Clin Invest 123, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pfrengle F, Macauley MS, Kawasaki N, and Paulson JC (2013) Copresentation of antigen and ligands of Siglec-G induces B cell tolerance independent of CD22, J Immunol 191, 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Macauley MS, and Paulson JC (2014) Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells, J Immunol 193, 4312–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Macauley MS, Crocker PR, and Paulson JC (2014) Siglec-mediated regulation of immune cell function in disease, Nat Rev Immunol 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson JC, and Nemazee D (2010) Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo, J Exp Med 207, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Orgel KA, Duan S, Wright BL, Maleki SJ, Wolf JC, Vickery BP, Burks AW, Paulson JC, Kulis MD, and Macauley MS (2017) Exploiting CD22 on antigen-specific B cells to prevent allergy to the major peanut allergen Ara h 2, J Allergy Clin Immunol 139, 366–369 e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Burks AW, Calderon MA, Casale T, Cox L, Demoly P, Jutel M, Nelson H, and Akdis CA (2013) Update on allergy immunotherapy: American Academy of Allergy, Asthma & Immunology/European Academy of Allergy and Clinical Immunology/PRACTALL consensus report, J Allergy Clin Immunol 131, 1288–1296 e1283. [DOI] [PubMed] [Google Scholar]
- [21].Bednar KJ, Nycholat CM, Rao TS, Paulson JC, Fung-Leung WP, and Macauley MS (2019) Exploiting CD22 To Selectively Tolerize Autoantibody Producing B-Cells in Rheumatoid Arthritis, ACS Chem Biol 14, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Investigators, P. G. o. C., Vickery BP, Vereda A, Casale TB, Beyer K, du Toit G, Hourihane JO, Jones SM, Shreffler WG, Marcantonio A, Zawadzki R, Sher L, Carr WW, Fineman S, Greos L, Rachid R, Ibanez MD, Tilles S, Assa’ad AH, Nilsson C, Rupp N, Welch MJ, Sussman G, Chinthrajah S, Blumchen K, Sher E, Spergel JM, Leickly FE, Zielen S, Wang J, Sanders GM, Wood RA, Cheema A, Bindslev-Jensen C, Leonard S, Kachru R, Johnston DT, Hampel FC Jr., Kim EH, Anagnostou A, Pongracic JA, Ben-Shoshan M, Sharma HP, Stillerman A, Windom HH, Yang WH, Muraro A, Zubeldia JM, Sharma V, Dorsey MJ, Chong HJ, Ohayon J, Bird JA, Carr TF, Siri D, Fernandez-Rivas M, Jeong DK, Fleischer DM, Lieberman JA, Dubois AEJ, Tsoumani M, Ciaccio CE, Portnoy JM, Mansfield LE, Fritz SB, Lanser BJ, Matz J, Oude Elberink HNG, Varshney P, Dilly SG, Adelman DC, and Burks AW (2018) AR101 Oral Immunotherapy for Peanut Allergy, N Engl J Med 379, 1991–2001. [DOI] [PubMed] [Google Scholar]
- [23].MacGinnitie AJ, Rachid R, Gragg H, Little SV, Lakin P, Cianferoni A, Heimall J, Makhija M, Robison R, Chinthrajah RS, Lee J, Lebovidge J, Dominguez T, Rooney C, Lewis MO, Koss J, Burke-Roberts E, Chin K, Logvinenko T, Pongracic JA, Umetsu DT, Spergel J, Nadeau KC, and Schneider LC (2017) Omalizumab facilitates rapid oral desensitization for peanut allergy, J Allergy Clin Immunol 139, 873–881 e878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Irvine DJ, Swartz MA, and Szeto GL (2013) Engineering synthetic vaccines using cues from natural immunity, Nat Mater 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, and Santamaria P (2010) Reversal of autoimmunity by boosting memory-like autoregulatory T cells, Immunity 32, 568–580. [DOI] [PubMed] [Google Scholar]
- [26].Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJ, and Miller SD (2011) Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells, J Immunol 187, 2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, and Hubbell JA (2015) Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens, Sci Rep 5, 15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, and Miller SD (2012) Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis, Nat Biotechnol 30, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, and Kishimoto TK (2015) Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance, Proc Natl Acad Sci U S A 112, E156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, Chan VT, Ahmed S, Srinivasan A, Deitemeyer P, Maldonado RA, and Kishimoto TK (2018) Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis, Front Immunol 9, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mazor R, King EM, Onda M, Cuburu N, Addissie S, Crown D, Liu XF, Kishimoto TK, and Pastan I (2018) Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity, Proc Natl Acad Sci U S A 115, E733–E742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hlavaty KA, Luo X, Shea LD, and Miller SD (2015) Cellular and molecular targeting for nanotherapeutics in transplantation tolerance, Clin Immunol 160, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, and Miller SD (2014) A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease, ACS Nano 8, 2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang AH, Rossi RJ, Yoon J, Wang H, and Scott DW (2016) Tolerogenic nanoparticles to induce immunologic tolerance: Prevention and reversal of FVIII inhibitor formation, Cell Immunol 301, 74–81. [DOI] [PubMed] [Google Scholar]
- [35].Kishimoto TK, Ferrari JD, LaMothe RA, Kolte PN, Griset AP, O’Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, and Maldonado RA (2016) Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles, Nat Nanotechnol 11, 890–899. [DOI] [PubMed] [Google Scholar]
- [36].Lim HH, Yi H, Kishimoto TK, Gao F, Sun B, and Kishnani PS (2017) A pilot study on using rapamycin-carrying synthetic vaccine particles (SVP) in conjunction with enzyme replacement therapy to induce immune tolerance in Pompe disease, Mol Genet Metab Rep 13, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pang L, Macauley MS, Arlian BM, Nycholat CM, and Paulson JC (2017) Encapsulating an Immunosuppressant Enhances Tolerance Induction by Siglec-Engaging Tolerogenic Liposomes, Chembiochem 18, 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Monach PA, Mathis D, and Benoist C (2008) The K/BxN arthritis model, Curr Protoc Immunol Chapter 15, Unit 15 22. [DOI] [PubMed] [Google Scholar]
- [39].Ditzel HJ (2004) The K/BxN mouse: a model of human inflammatory arthritis, Trends Mol Med 10, 40–45. [DOI] [PubMed] [Google Scholar]
- [40].Monach P, Hattori K, Huang H, Hyatt E, Morse J, Nguyen L, Ortiz-Lopez A, Wu HJ, Mathis D, and Benoist C (2007) The K/BxN mouse model of inflammatory arthritis: theory and practice, Methods Mol Med 136, 269–282. [DOI] [PubMed] [Google Scholar]
- [41].Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, and Paulson JC (2010) In vivo targeting of B-cell lymphoma with glycan ligands of CD22, Blood 115, 4778–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rafiei P, and Haddadi A (2017) Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile, Int J Nanomedicine 12, 935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Finckh A, Ciurea A, Brulhart L, Kyburz D, Moller B, Dehler S, Revaz S, Dudler J, Gabay C, and Physicians of the Swiss Clinical Quality Management Program for Rheumatoid, A. (2007) B cell depletion may be more effective than switching to an alternative anti-tumor necrosis factor agent in rheumatoid arthritis patients with inadequate response to anti-tumor necrosis factor agents, Arthritis Rheum 56, 1417–1423. [DOI] [PubMed] [Google Scholar]
- [44].Brulhart L, Ciurea A, Finckh A, Notter A, Waldburger JM, Kyburz D, and Gabay C (2006) Efficacy of B cell depletion in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor alpha agents: an open-label observational study, Ann Rheum Dis 65, 1255–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Srivastava A, Arlian BM, Pang L, Kishimoto TK, and Paulson JC (2020) Tolerogenic Nanoparticles Impacting B and T Lymphocyte Responses Delay Autoimmune Arthritis in K/BxN Mice, 2020.2007.2002.185140 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.