

Abstract
Background and Objectives
Costimulation by CD40 and its ligand CD40L (CD154) is important for the functional differentiation of T cells. Preclinical studies have recognized the importance of this costimulatory interaction in the pathogenesis of experimental models of multiple sclerosis (MS). To determine safety, pharmacokinetics, and immune effect of a humanized monoclonal antibody (mAb) against CD40 ligand (toralizumab/IDEC-131) in patients with relapsing-remitting MS (RRMS).
Methods
This single-institution open-label dose-escalation study (phase I) enrolled 12 patients with RRMS to receive 4 doses of 1, 5, 10, or 15 mg/kg of humanized αCD40L (toralizumab) IV infusion every other week. Patients were followed up to 18 weeks, annually, and finally at 5 years. In addition to safety and pharmacokinetics, other secondary and exploratory measurements are immune effects, clinical, MRI, laboratory, and neuropsychological evaluations.
Results
Fifteen adverse events, all of mild to moderate severity, were considered to be of possible or of unknown relationship to treatment. No serious adverse events, including thromboembolic events, occurred during the 18-week defined study period. Annual and long-term follow-up at 5 years revealed no delayed toxicity. Pharmacokinetics were nonlinear between the 5 and 10 mg/kg dose groups. The serum half-life of toralizumab was consistent between the dose groups with a mean of 15.3 days (SD = 1.9). Flow cytometry revealed no depletion of lymphocyte subsets. An increase in the CD25+/CD3+ and CD25+/CD4+ ratio and a shift toward an anti-inflammatory cytokine response were seen after treatment.
Discussion
Our study suggests that blocking CD40L is safe and well tolerated in patients with RRMS while increasing CD25 + T cells and anti-inflammatory cytokine profile. These findings support further studies to assess the efficacy of blocking CD40L as a potential treatment of RRMS.
Classification of Evidence
This study provides Class IV evidence on the safety, pharmacokinetics, and immune effects of an mAb to CD40L in patients with RRMS.
CD40 and its ligand, CD40L (CD154), have a central role in the regulation of both humoral and cell-mediated immunity.1 Blockade of CD40L is effective in ameliorating experimental autoimmune conditions and thus is an attractive therapeutic intervention.2-6 A functional role for CD40L-CD40 in peptide-induced experimental autoimmune encephalomyelitis (EAE) is suggested by the observations that brief, prophylactic treatment with an antibody against CD40L (αCD40L) blocks disease in several animal models.5,7-9 CD40-CD40L interactions within the CNS are critical determinants of disease development and progression.10 The lack of expression of CD40 by CNS-resident cells diminishes the intensity and duration of myelin oligodendrocyte glycoprotein –induced EAE and reduces the degree of inflammatory cell infiltrates into the CNS. Encephalitogenic T cells that enter a CNS environment in which CD40 is absent from parenchymal microglia cannot elicit the expression of chemokines within the CNS.
In humans, both clinical and pathologic observations suggest the involvement of CD40L and CD40 in multiple sclerosis (MS). CD40L+-expressing cells (CD4+) are present in the perivascular infiltrate in human MS brain sections but not in tissue from control individuals without MS. In brain tissue from patients with MS, cells expressing CD40 are juxtaposed to cells expressing CD40L, suggesting that CD40-CD40L interactions are ongoing in human MS plaques. A study showed an increase in CD4+ T cells expressing CD40L in the peripheral blood of patients with MS.11 Of particular interest is the possible association between the expression of CD40L and clinical status.11-13 Increased concentrations of IFNγ found in patients with MS may be due to higher production of interleukin (IL)12 or perhaps IL23 that is induced by CD40L.12,14,15 These data support the concept that therapeutic blocking of the CD40-CD40L interaction may be an effective approach in the management of MS. Toralizumab is a humanized monoclonal antibody (mAb) against CD40L comprised of human gamma 1 heavy chains and human kappa light chains with murine complementarity determining regions.
Toralizumab binds specifically to human CD40L on T cells, thereby preventing CD40 signaling. Herein, we present the safety profile and immune effects after the administration of a humanized αCD40L antibody in patients with clinically defined relapsing-remitting MS (RRMS).
Methods
Study Design
This was a phase I dose-escalation, single-center open-label trial, designed to determine the safety, feasibility, and immune effects of administering multiple IV doses of a humanized mAb against CD40L to patients with RRMS. Patients in 4 separate cohorts (n = 3 patients/group) were enrolled to assess the maximum tolerated dose (MTD) of humanized αCD40L. The initial cohort received a dose of 1 mg/kg, and if no severe toxicity was found, the dose was escalated in each successive cohort: 5, 10, and 15 mg/kg. We performed pharmacokinetic and immunologic assays in blood of all patients enrolled. Other parameters measured included neuropsychological testing, MRI, and disease clinical activities (relapse and disability) Figure 1. Patients who enrolled in the treatment phase but dropped out early were evaluated in the data pool and included in the outcome analysis.
Figure 1. Phase I Clinical Trial Design.

Initial cohort received anti-CD40L mAb 1 mg/kg IV; dose was escalated in each successive cohort: 5, 10, and 15 mg/kg. The primary outcome was safety. The secondary outcome was pharmacokinetics. Exploratory measures included immunologic analysis, clinical assessment of relapse and EDSS score, neuropsychological test, and MRI. mAb = monoclonal antibody.
Patient Selection
Patients were included if they had a clinically definite or laboratory-supported diagnosis of RRMS16, 1 clinical relapse within the previous year, and an Expanded Disability Status Scale (EDSS) score of 0–3.5. All patients were aged between 18 and 54 years, had baseline laboratory tests, chest x-ray and ECG within established parameters, and had the ability to give informed consent. Patients of childbearing potential were required to use an approved form of birth control.
Patients were excluded if they had major medical or psychiatric illness that would interfere either with administration of therapy, compliance with scheduled follow-up appointments, or prevention from giving informed consent. Patients who were pregnant or breast feeding or attempting to become pregnant were excluded. Other exclusion criteria consisted of treatment with interferon-β (IFN−β), glatiramer acetate, or steroids within the past 30 days or exposure to cytotoxic chemotherapy within 6 months before entry. No patients had received any mAb-mediated therapy such as anti-CD20, anti-CD52, anti-integrin, or S1P modulators.
Intervention
Toralizumab has human gamma 1 heavy chains and human kappa light chains with murine complementarity determining regions and binds specifically to human CD40L. The humanized mAb was produced under good manufacturing practice conditions and supplied by IDEC pharmaceuticals (San Diego, CA). The mAb was given IV over a 1-hour period, followed by a 2-hour observation period. Patients were treated every other week for 4 doses per patient. The frequency of administration was based on the preliminary data in the EAE model and pharmacokinetic studies in humans in which it was observed that the half-life for this antibody in circulation is approximately 12–13 days.17 The first group received a dose of 1 mg/kg, with subsequent dose escalation for each additional group to 5, 10, and 15 mg/kg. Initiation of the next treatment group started after the previous group completed the first follow-up visit at week 8 (2 weeks after the final treatment). MRI scans and neuropsychological assessment were performed at baseline, 8 weeks (2 weeks post–final treatment), and 18 weeks (12 weeks post–final treatment) postbaseline. MRI (number of enhancing lesions and lesion volume) and clinical activities (relapse and EDSS score) were exploratory outcome measurements.
Safety Assessments
Safety of the drug in patients with MS was assessed by a variety of clinical and laboratory evaluations including a brief standardized neuropsychological test battery eTable 3 (links.lww.com/NXI/A639). Toxicity was scored according to version 2.0 of the National Cancer Institute Common Toxicity Criteria. Before each treatment, adverse events were obtained by patient interview and by performing physical/neurologic examinations. Additional safety parameters were monitored by blood chemistries including liver and kidney function tests and complete blood counts performed before each treatment. All adverse drug reactions were recorded, and their frequency and severity were compared between dose levels. Adverse events resulting from concurrent illnesses, reactions to concurrent medications, or progression of disease activity were also recorded.
Pharmacokinetic Analysis and Measurement of Anti-CD40L Antibody
Serum samples were collected for pharmacokinetic assessment of mAb concentrations before (zero time) and immediately (within 10 minutes) on completion of each study infusion and at weeks 8 (2 weeks after the final treatment) and 18 (12 weeks after the final treatment). An ELISA using goat anti-toralizumab determined the serum concentration of anti-CD40L antibody. The lower limit of detection was 125 ng/mL, and the lower limit of quantification was 500 ng/mL. Posttreatment sera, obtained at week 18 after first treatment, were analyzed for the development of anti-toralizumab antibody response using a 96-well microtiter plate coated with toralizumab F(ab)'2. For this assay, the lower limit of detection was 125 ng/mL, and the lower limit of quantification was 250 ng/mL.
Immunologic Analysis
Lymphocyte proliferation, flow cytometry, and ELISA assays were performed on freshly isolated peripheral blood mononuclear cells (PBMCs) from each patient before and after antibody treatments. Flow cytometry analysis was performed using a FACSCalibur flow cytometer (Becton Dickinson) on blood samples obtained before each study infusion and at weeks 8 (2 weeks after the final treatment) and 18 (12 weeks after the final treatment). Peripheral blood flow cytometry determined the total, CD45+, CD3+/CD4+, CD3+/CD8+, CD3+, CD3-/CD56/CD16+, CD19+, and CD25+ lymphocyte populations. Patients in the high-dose cohort (15 mg/kg) were selected for further immunologic studies. PBMCs were isolated at baseline and weeks 2, 4, 6, and 8 and analyzed for phenotypic shift by flow cytometry and proliferative response in vitro. PBMC were stimulated for 4 hours with 1 μg/ml phorbol myristate acetate and 50 ng/ml Ionomycin at 37°C and the supernatant was assayed for cytokine/chemokine production using a Milliplex human 17-plex kit (Millipore). Delayed type hypersensitivity (DTH) reactions to tetanus toxoid and mumps were performed.
Safety Measures on Clinical Activity
The relapse rate during the study period was monitored. Relapses were defined as the appearance of a new symptom or worsening of an old symptom lasting more than 48 hours, attributable to MS activity, accompanied by a documented neurologic abnormality, and preceded by a period of stability or improvement for at least 30 days. Individual's raw scores on neuropsychological testing were converted to standard (z) scores using demographically appropriate normative data. Domain-based z-scores were obtained by averaging across selected subscales within each domain. Negative z values reflect greater levels of impairment or symptoms in all cases. The EDSS score was assessed at the initiation of therapy and before each treatment. A follow-up EDSS score was performed at weeks 8 and 18 (2 and 12 weeks after the final treatment).
MRI Acquisition and Analysis Protocol
Only brain MRI images were obtained with a single 1.5 T MR unit (General Electric, Milwaukee, WI) following a standardized protocol. To ensure uniformity of angulation, the axial sections were aligned with the anterior and posterior inferior margins of the corpus callosum. Axial T2-weighted scans were obtained with fast spin echo technique (the repetition time [TR] 3,000, the echo time [TE] 100, 3 mm thickness, and 0 gap). Fluid-attenuated inversion recovery (FLAIR) axial scans were obtained (TR 10,000, TE 156, TI 2200, and 3 mm/0 gap).
Images were reviewed by a certified neuroradiologist. Data were sent for quantification of lesion burden to the Brain Imaging Laboratory at our institution where an experienced image analyst (interrater and intrarater intraclass correlation coefficient reliabilities >.95) defined lesion boundaries using a semiautomated segmentation program (Alice, Hayden Imaging Processing, Boulder, CO). This technique allowed calculation of total lesion volume after the processed lesion margins were reviewed by the neuroradiologist.
MRI evidence of lesion activity was determined by gadolinium diethylenetriamine pentaacetate–enhanced T1- weighted images and was used as a secondary outcome for this study. The number of lesions at the time of therapy induction was compared with the number of lesions at 2 and 12 weeks posttherapy. The number of lesions in the pretreatment and 2-week postfinal treatment at each dose level was evaluated before dose escalation. Areas of MS lesions identified on the FLAIR sequence were multiplied by slice thickness and summed across slices to yield the total lesion load in mm3. All scans were performed on the same machine and reviewed by the same neuroradiologist.
Dose Escalation and Statistical Consideration
For the purpose of this study, the MTD was defined as the maximum αCD40L dose at which 33% or less of the patients experienced a dose-limiting toxicity. A dose-escalation protocol was followed based on the doses as described above. The first 3 patients were assigned the 0 tier dose. If no patients experienced severe toxicity (greater than grade 3), the dose was incremented to the next higher dose tier for the second group of 3 patients. If a single patient experienced greater than grade 3 toxicity, the second group received the initial dose level; if more than 1 patient experienced greater than grade 3 toxicity, the second group received the next lower dose tier. This procedure continued until a maximum accrual of 12 patients had been accomplished. In presenting data from this trial, continuous data were summarized in tables listing the mean, SD or standard error, median, minimum and maximum values, and number of subjects in a group. Categorical data were summarized in tables listing the frequency and the percentage of subjects in a group. These summaries were presented separately for subjects on different treatment arms.
Data and Safety Monitoring Committee Review
The Data and Safety Monitoring Committee was comprised of 4 independent investigators and charged with reviewing all the data after each cohort had completed treatment. If there were no safety or data management concerns, permission was granted to escalate the dose in the next group. The study was to be on hold if any of the members requested a halt of the study because of safety issues or if there were any serious toxicities that required Food and Drug Administration (FDA) report.
Standard Protocol Approvals, Registrations, and Patient Consents
Ethical approval was obtained from the Committee for the Protection of Human Subjects at Dartmouth College (CPHS Approval #14070 Date of approval 10/2/98) and Investigational New Drug status granted by the FDA (IND# 8265). Written informed consent was obtained from all patients participating in the study.
Data Availability
Data will be shared with qualified investigators on request; please contact CEF3W@virginia.edu.
Results
Patient Characteristics
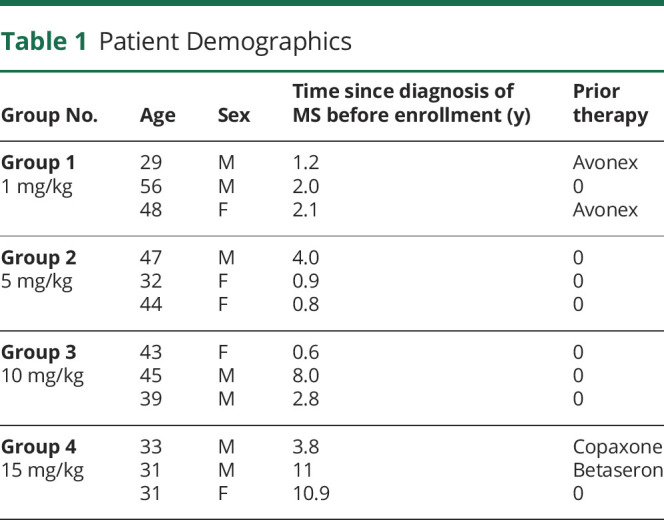
Twelve patients with definite RRMS and a relapse in the previous 12 months were included in the study (Table 1). The first patient received the initial dose on June 1999, and the 12th patient received the last dose on April 2001. The median age was 41 years (range of 29–56 years), and 58% were men. At enrollment, the median time since diagnosis of MS was 2.1 years, with a range of 6 months–11 years. The median EDSS score was 2. Four patients had previously received either an interferon (IFN)-β preparation or glatiramer acetate but had disease activity as determined by > 1 clinically confirmed relapse in the year preceding enrollment.
Table 1.
Patient Demographics
Safety and Tolerability
All patients completed the planned treatment schedule without any serious adverse events. An MTD was not found following a dose-escalation protocol based on the doses of 1, 5, 10, and 15 mg/kg of antibody. No significant abnormalities were detected in vital signs or clinical laboratory studies. There was a protocol deviation in 1 of the patients at the 1 mg/kg dose, who started IFN-β therapy 4 weeks after completing mAb treatment, without consulting with the study treating physicians.
Of 49 adverse events reported, 15 were considered to be of possible relationship to treatment (Table 2). All the events were either grade 1 (mild) or grade 2 (moderate). The most frequent adverse events of possible relationship to treatment occurring on the day of infusion included gastric disturbance, fatigue, and headache and associated with the higher dose of toralizumab (10 and 15 mg/kg cohorts). Six infections were reported in 5 patients; all events were grade 1 or 2, and all patients recovered. One case of nondisseminated herpes zoster and 1 case of upper respiratory infection were the only infections considered to be possibly related to treatment. Of the 15 adverse events possibly related to treatment, 1 appeared in the second group, 4 in the third group, and 10 in the fourth group. No serious adverse events including thromboembolic events occurred during the 18-week study period. There were no significant abnormalities noted in laboratory tests that could be related to the treatment under study.
Table 2.
Reported Adverse Events

Pharmacokinetic Studies
The results of the pharmacokinetic studies for each group can be observed in Table 3. Serum concentrations of mAb increased proportionally with dose, and the mean AUC was proportional to the dose administered between the 1 and 5 mg/kg groups. Patients in the 10 and 15 mg/kg dose group exhibited a similar mean Cmax. The pharmacokinetic studies between the 5 and 10 mg/kg dose groups were nonlinear, suggesting that saturation of clearance mechanisms takes place after multiple IV mAb administration between these doses. The serum half-life of toralizumab was consistent between the dose groups with a mean of 15.3 days (SD = 1.9).
Table 3.
Pharmacokinetic Parameters

Immunologic Parameters
Peripheral blood cell counts obtained at baseline and various intervals posttherapy revealed no evidence of infusion-related T-cell (CD3+, CD4+, or CD8+) depletion (Figure 2A). There was no change in peripheral white blood cell counts or absolute lymphocytes counts following treatment (Figure 2B). Immunologic analysis of T-cell markers in the high dose cohort revealed a trend toward increased CD25/CD3 phenotype expression at week 8 (Figure 3A). A similar trend in the CD25/CD4 population was further noted in this cohort beginning at week 6 and extending into week 8. Analysis of specific cytokines demonstrated a modest rise in the interleukin 10 (IL10): monocyte chemoattractant protein-1 (MCP1) ratio and a more robust change in the IL10:IL17 ratio at weeks 4 and 8 (Figure 3B). In addition to CD25 being recognized as a T-cell activation marker, it is also considered to be an important hallmark of regulatory T cells. At the time this study was performed, it was considered the primary phenotypic marker along with enhanced IL10 of T-cell immune regulation. Although the number of samples available for analysis was limited, there is a trend for both enhanced CD25+ expression and increased IL10:IL17 ratio consistent with the induction of immune regulation following treatment with anti-CD40L.
Figure 2. Leukocyte and Lymphocyte Changes After Anti-CD40L.

(A) Percentage changes of total CD3+, CD3+CD4+, and CD3+CD8+ over time for each patient according to dose tier. (B) Leukocyte and lymphocyte count for all patients.
Figure 3. Heat Maps of CD25 and Anti-inflammatory Cytokine Shift After Anti-CD40L.

(A) Increased CD25/CD3 and CD25/CD4 ratios were seen following treatment. Flow cytometry measured CD25+ vs CD3+ and CD4+ shift after treatment as Z-score heat map. The values on the heat map indicate the ratio of CD25+ cells compared with CD3+ (left) and CD4+ (right). The heat map indicates the change over time in individual patients in the high-dose group (15 mg/kg). (B) IL10/MCP1 and IL10/IL17 were increased following treatment in the same patients. Cytokines in PBMC culture supernatant were measured. Quantity of anti-inflammatory IL10 and proinflammatory cytokine MCP1 and IL17 was measured, and their ratios are presented as Z-score heat map. The ratios are derived by dividing the concentration of IL10 by the concentration of MCP1 (left) or the concentration of IL10 by the concentration of IL17 (right). Across all patients and time points, IL10 ranged between 197 and 565 pg/mL, MCP1 ranged between 5,280 and 15,627 pg/mL, and IL17 ranged between 54 and 1,138 pg/mL. The heat maps indicate the mean change over time in the high-dose group (15 mg/kg). MCP1 = monocyte chemoattractant protein-1; PBMC = peripheral blood mononuclear cell.
There was no change in skin DTH response to tetanus or mumps antigens posttherapy. There was no demonstrable immunoglobulin isotype switching. There was no significant difference in patient responses before or 4 and 8 weeks after treatment to lymphoproliferative responses to phytohemagglutinin, anti-CD3, and tetanus toxoid (data not shown). No patient developed detectable antibodies against toralizumab measured at week 18 after the first infusion (10 weeks after finishing treatment). There was no evidence of CD40 expression on CD3+ T cells or CD40L expression of the CD3− population postinfusion.
Clinical Parameters
Patients were selected based on 1 clinical relapse within the previous year with a baseline annualized relapse rate (ARR) of 1. The relapse rate was monitored throughout the study, and there were no relapses during the 18-week study period. With a follow-up of 5 years, the average ARR of all 4 groups was 0.125. Higher dose 10 and 15 mg/kg group had ARR of 0.08, and lower dose 1 and 5 mg/kg group ARR was 0.17 (eTable 1, links.lww.com/NXI/A637). The EDSS score was performed at each assessment timepoint as shown in Figure 1. The comparison of baseline and week 18 can be observed in eTable 2 (links.lww.com/NXI/A638). In 6 of the 12 patients, there was an improvement in the disability scores, whereas in the other 6 patients, the scores remained stable. Neuropsychological testing, performed as a safety parameter, revealed no evidence of decline in any of the measured domains during the trial (eFigure 1, links.lww.com/NXI/A636, and eTable 3, links.lww.com/NXI/A639).
MRI Parameters
Active inflammation, as determined by standard dose gadolinium-enhanced T1-weighted MRI, was used as the secondary outcome for this study. Toxicity and response were determined by measuring the number of lesions at the time of therapy induction and compared with the number of lesions in MRI performed at 18 weeks (end of the study). At baseline MRI, 3 patients demonstrated evidence of gadolinium-enhancing lesions. Two of these 3 patients continued to demonstrate MRI lesion enhancement at the end of the study, although a reduction in gadolinium lesion burden was observed. On follow-up MRI studies, no enhancing lesions were observed in any of the other 9 patients without baseline enhancing lesions. There was no increase in the number of gadolinium-enhancing lesions in any of the MRI scans. The T1 gadolinium-enhanced and FLAIR lesion load was determined in all studies by semiautomated volumetric analysis. The mean volume of enhanced lesions decreased from 1,114 mm3 at baseline to 470 mm3 (n = 3) at week 18. Mean FLAIR lesion volume was 10,902 (±12,764) mm3 at baseline and 12,117 (±15,303) mm3 at week 18 (n = 12). Qualitative examination revealed no differences in FLAIR lesion volume over time when the patients were stratified according to their toralizumab dose.
Five-Year Follow-up
The patients were evaluated every 6 months after the initial 18-week follow-up for adverse event assessment and changes in disease activity (relapse rate and EDSS score). Patients with acute clinical relapses were treated with steroids if indicated. Nine of the 12 patients elected to be on either IFN-β or glatiramer acetate during the first year after anti-CD40L treatment, whereas the other 3 patients opted for no treatment.
The mean EDSS score at baseline was 2.3 ± 0.8 and 2.5 ± 1.6 at 5 years, and the change over the follow-up period can be observed on eTable 2 (links.lww.com/NXI/A638). On follow-up, 6 patients experienced a clinical relapse over the preceding 5 years, whereas 6 had no relapses. One patient in the lowest dose cohort (1 mg/kg dose) developed a secondary progressive course 3 years after treatment. The ARR was reduced in all treatment groups when compared with baseline (eTable 1, links.lww.com/NXI/A637). The effect of treatment appeared to be dose dependent in that the cohorts treated with 10 and 15 mg/kg had a reduction in the relapse rate of 0.08 compared with the lower dose 0.17.
Discussion
In this study, blocking CD40L with escalating doses of toralizumab was safe and well tolerated in patients with RRMS. The safety profile showed mild or moderate adverse events without a dose-limiting toxicity at doses up to 15 mg/kg. Specifically, we observed no thromboembolic events in any of the treated patients. Treatment with this anti-CD40L mAb did not result in exacerbation of the disease, as measured by clinical (EDSS, neuropsychological evaluation, and number of relapses) and MRI parameters. The pharmacokinetic studies confirm that saturation of clearance mechanisms takes place after multiple IV mAb doses between 5 and 10 mg/kg.
A major safety concern associated with blocking CD40L is that patients would have increased susceptibility to infection. Previous clinical trials examining dose escalation of this antibody for treatment of other autoimmune diseases showed no definite increased risk of infection; when infection occurred, the relationship to the drug was unclear, and all patients recovered without complication at doses up to 15 mg/kg.17,18,19 In our study, the 2 patients who developed infections were in the highest treatment group (15 mg/kg). One patient developed a grade 1 reactivation of herpes zoster that was treated without complication. Although this may have been an opportunistic infection, there was no evidence of immune compromise in this cohort after treatment as determined by lymphocyte subset analysis, proliferation assays and antigen recall. Including this study, reports on 120 patients treated with toralizumab revealed no severe infectious complications related to treatment.17,18
As a component of the safety outcomes for treatment with anti-CD40L, we assessed exploratory clinical and MS relevant MRI outcomes. The absence of relapses and the stable EDSS scores demonstrated that there was no increased clinical activity secondary to the treatment during the first 18 weeks. A similar MRI outcome further supports the lack of significant neurologic toxicity. It is noteworthy that the neuropsychological test performance results did not reveal decline in cognition, mood, or fatigue over the course of the trial. Furthermore, although not inclusive in the trial design, we found a reduced ARR in the higher dose group compared with the lower treatment group, and we did not observe any long-term toxicity at 5 years posttreatment.
In addition to T cells, a number of cellular compartments including platelets and endothelium express CD40L. Thromboembolic complications were observed in humans and monkeys with other mAb preparations that target CD40L.20 The antibody we used in this study, toralizumab (IDEC-131), was withdrawn from further development because of these potential thromboembolic complications. No thromboembolic events were reported in 85 patients with systemic lupus erythematosus (SLE) receiving toralizumab in clinical trials.17,18,21 We found no significant systemic toxicity, including thromboembolic events, following treatment with this specific anti-CD40L mAb in 12 patients with MS.
There are several potential explanations to account for the difference in thromboembolic complications between the antibody preparations including potency, manufacturing techniques or binding to dissimilar ligand epitopes. It has also been suggested that the antibody forms immune complexes with platelet constant fragment (Fc) receptors and soluble CD40L that results in platelet activation and hypercoagulability.22 Therefore, different molecules targeting CD40 ligand have been developed to circumvent the Fc-mediated effect and potentially decrease the risk of this complication.20,23 There are ongoing studies examining these new approaches to target the CD40-CD40L axis with promising results in a variety of autoimmune diseases.20,24 More recent therapeutic agents targeting CD40L invoke the benefits of earlier antibody studies through the use of the anti-CD40L variable fragment Fab region (or an analogous protein region) in the absence of the Fc domain. There are several completed and ongoing studies using these new approaches to block CD40L as a treatment for SLE (NCT02804763), primary Sjogren syndrome (NCT04572841), immune thrombocytopenic purpura (NCT02273960), and adult-onset rheumatoid arthritis (NCT02780388).
Our results on the immune effects of CD40 blockade in MS gain relevance in light of these new therapeutic opportunities and a better understanding of the immune biology of the interaction of CD40L with antigen-presenting cells. Membrane-bound CD40L is a costimulatory molecule located on T cells; it interacts with CD40 on the surface of antigen-presenting cells and has an integral function in the T-B cell interaction that occurs during autoimmune disease (Figure 4). Our data showed that CD40L blockade increased the ratios of CD25/CD3 and CD25/CD4, which may be essential for the induction of tolerance. The increased anti-inflammatory to proinflammatory cytokine ratios IL10/IL17 and IL10/MCP1 further implicate potential tolerance mechanisms.
Figure 4. Potential Mechanism of Action of Anti-CD40L mAb in Treating MS.

Activation of T cells by B cell/antigen-presenting cells (APC) is dependent on engagement of costimulatory molecules and the successful activation of T cell receptor (TCR) signal. TCR signal is upregulated when the major histocompatibility complex class II (MHCII) containing the appropriate peptide on the B cell/APC is recognized by the TCR. This interaction leads to the upregulation of CD40L (CD154) on the T cell that engages the constitutively expressed CD40 on the APC. Monoclonal antibodies against CD40L (anti-CD40L mAb) block further activation of downstream signaling and prevent T- and B-cell activation; which can potentially induce tolerance in antigen-specific manner. mAb = monoclonal antibody; TCR = T-cell receptor.
Antigen-presenting capacity of B cells is enhanced on ligation of CD40 by CD40L.25,26 Soluble CD40L (sCD40L) in plasma correlated with EDSS score changes and was significantly upregulated in actively progressive secondary progressive MS.27 Plasma sCD40L concentration has the greatest capacity to discriminate actively progressive MS from nonprogressive benign and RRMS.27 These studies in combination with our results suggest that CD40L plays a major role in disease pathogenesis and that blockade may have a beneficial effect on MS disease progression. Because strategies to manipulate the CD40-CD40L axis remain a promising therapeutic approach28, a recent phase II double-blind randomized placebo-controlled study assessing the efficacy and safety of a newer CD40L mAb (SAR441344) in MS was started on May 2021 (NCT04879628).
Our study is limited by the small number of patients and may have missed significant toxicity in a larger cohort. However, when combining the 88 patients with SLE17,18, 20 patients with immune thrombocytopenic purpura19 and our 12 patients with MS treated with toralizumab, there were no serious adverse effects. Seven (58%) of the 12 patients in our study were male, but 104 (86%) of the 120 patients with autoimmune disease treated with toralizumab were female. Therefore, it is unlikely that there would be a higher risk of side effects, like thromboembolic complications, according to sex. Furthermore, a phase II double-blind, placebo-controlled trial showed similar type and frequency of adverse events between the toralizumab and placebo groups.18 Another concern was that patients with more severe MS would have higher likelihood of side effects. However, the 3 patients in the 15 mg/kg dose cohort with the highest baseline EDSS score did not have increased short- or long-term adverse effects. Finally, we measured clinical and imaging outcomes with the goal to establish safety but not to determine benefit of this therapeutic approach.
Preclinical studies suggest that blocking CD40L has the potential to be an effective treatment for patients with MS. Our study showed that blocking CD40Lwith a targeted antibody had short- and long-term safety in patients with RRMS and that treatment has the potential to elicit a beneficial immune response. As dysfunction of T-cell regulation has been appreciated in those with MS, our findings offer encouraging preliminary data that an important mechanism of anti-CD40L would be via the induction of an effective T regulatory response in the absence of inducing circulating CD3+/CD4+/CD8+ depletion (Figure 2 and 3). Although future studies with toralizumab are no longer feasible, our study supports and paves the way for future phase II MS clinical trials using newer anti-CD40L mAb such as the ongoing study with SAR441344 (Sanofi). Future studies targeting CD40L alone or in combination with other disease modifying treatments will determine their efficacy and safety in RRMS and actively progressive secondary MS.
Acknowledgment
The authors acknowledge the contributions of Alexander Mamourian, Mark Totoritis, Tor Tosteson, John Baron, and the members of the Data and Safety Committee (Lionel Lewis, MD, Alexander Reeves, MD, and David Roberts, MD). They thank all their patients for participating in the study.
Glossary
- ARR
annualized relapse rate
- DTH
delayed type hypersensitivity
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Status Scale
- FDA
Food and Drug Administration
- IFN
interferon
- IL
interleukin
- IND
investigational new drug
- mAb
monoclonal antibody
- MCP
monocyte chemoattractant protein
- MS
multiple sclerosis
- MTD
maximum tolerated dose
- PBMC
peripheral blood mononuclear cell
- RRMS
relapsing-remitting ms
- SLE
systemic lupus erythematosus
- sCD40L
soluble CD40L
Footnotes
Class of Evidence: NPub.org/coe
Contributor Information
Camilo E. Fadul, Email: cef3w@virginia.edu.
Kathleen A. Ryan, Email: kthleenryan@gmail.com.
Randolph J. Noelle, Email: rjn@dartmouth.edu.
Heather A. Wishart, Email: heather.a.wishart@dartmouth.edu.
Jacqueline Y. Channon, Email: jacqueline.smith@dartmouth.edu.
Isaac R. Kasper, Email: ikasper@melrosewakefield.org.
Brant Oliver, Email: brant.j.oliver@dartmouth.edu.
Daniel W. Mielcarz, Email: daniel.w.mielcarz@dartmouth.edu.
Lloyd H. Kasper, Email: lloydkasper@gmail.com.
Study Funding
No targeted funding reported.
Disclosure
Y. Mao-Draayer has served as a consultant and/or received grant support from Acorda, Bayer Pharmaceutical, Biogen Idec, Celgene/Bristol Myers Squibb, EMD Serono, Sanofi-Genzyme, Genentech-Roche, Novartis, Questor, Janssen, and Teva Neuroscience. Y. Mao-Draayer was supported by grants from NIH NIAID Autoimmune Center of Excellence: UM1-AI110557-05, UM1 AI144298-01, PCORI, Novartis, Genentech-Roche, Sanofi-Genzyme, and Chugai. L.H. Kasper was supported by National Institutes of Health AI061938 and a grant from IDEC Pharmaceuticals.Go to Neurology.org/NN for full disclosures.
References
- 1.Noelle RJ. CD40 and its ligand in cell-mediated immunity. Agents Actions Suppl. 1998;49:17-22. [DOI] [PubMed] [Google Scholar]
- 2.Carayanniotis G, Masters SR, Noelle RJ. Suppression of murine thyroiditis via blockade of the CD40-CD40L interaction. Immunology. 1997;90(3):421-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howard LM, Miga AJ, Vanderlugt CL, et al. . Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest. 1999;103(2):281-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadlack B, Lohler J, Schorle H, et al. . Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25(11):3053-3059. [DOI] [PubMed] [Google Scholar]
- 5.Gerritse K, Laman JD, Noelle RJ, et al. . CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93(6):2499-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balasa B, Krahl T, Patstone G, et al. . CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol. 1997;159(9):4620-4627. [PubMed] [Google Scholar]
- 7.Constantinescu CS, Hilliard B, Wysocka M, et al. . IL-12 reverses the suppressive effect of the CD40 ligand blockade on experimental autoimmune encephalomyelitis (EAE). J Neurol Sci. 1999;171(1):60-64. [DOI] [PubMed] [Google Scholar]
- 8.Laman JD, Maassen CB, Schellekens MM, et al. . Therapy with antibodies against CD40L (CD154) and CD44-variant isoforms reduces experimental autoimmune encephalomyelitis induced by a proteolipid protein peptide. Mult Scler. 1998;4(3):147-153. [DOI] [PubMed] [Google Scholar]
- 9.Schaub M, Issazadeh S, Stadlbauer TH, Peach R, Sayegh MH, Khoury SJ. Costimulatory signal blockade in murine relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 1999;96(2):158-166. [DOI] [PubMed] [Google Scholar]
- 10.Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193(8):967-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen J, Krakauer M, Sellebjerg F. Increased T cell expression of CD154 (CD40-ligand) in multiple sclerosis. Eur J Neurol. 2001;8(4):321-328. [DOI] [PubMed] [Google Scholar]
- 12.Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci U S A. 1997;94(2):599-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teleshova N, Bao W, Kivisakk P, Ozenci V, Mustafa M, Link H. Elevated CD40 ligand expressing blood T-cell levels in multiple sclerosis are reversed by interferon-beta treatment. Scand J Immunol. 2000;51(3):312-320. [DOI] [PubMed] [Google Scholar]
- 14.Filion LG, Matusevicius D, Graziani-Bowering GM, Kumar A, Freedman MS. Monocyte-derived IL12, CD86 (B7-2) and CD40L expression in relapsing and progressive multiple sclerosis. Clin Immunol. 2003;106(2):127-138. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Chu N, Hu A, Gran B, Rostami A, Zhang GX. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain. 2007;130(Pt 2):490-501. [DOI] [PubMed] [Google Scholar]
- 16.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National multiple sclerosis society (USA) advisory committee on clinical trials of new agents in multiple sclerosis. Neurology. 1996;46(4):907-911. [DOI] [PubMed] [Google Scholar]
- 17.Davis JC Jr., Totoritis MC, Rosenberg J, Sklenar TA, Wofsy D. Phase I clinical trial of a monoclonal antibody against CD40-ligand ( Toralizumab) in patients with systemic lupus erythematosus. J Rheumatol. 2001;28(1):95-101. [PubMed] [Google Scholar]
- 18.Kalunian KC, Davis JC Jr., Merrill JT, Totoritis MC, Wofsy D. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(12):3251-3258. [DOI] [PubMed] [Google Scholar]
- 19.Kuwana M, Nomura S, Fujimura K, et al. . Effect of a single injection of humanized anti-CD154 monoclonal antibody on the platelet-specific autoimmune response in patients with immune thrombocytopenic purpura. Blood. 2004;103(4):1229-1236. [DOI] [PubMed] [Google Scholar]
- 20.Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv Drug Deliv Rev. 2019;141:92-103. [DOI] [PubMed] [Google Scholar]
- 21.Davis JC Jr., Totoritis MC, Sklenar TA, Wofsy D. Results of a phase I, single-dose, dose-escalating trial of a humanized anti-CD40l monoclonal antibody (toralizumab) in patients with systemic lupus erythematosus (SLE). Arthritis Rheum. 1999;42(9):S281. [Google Scholar]
- 22.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6(2):114. [DOI] [PubMed] [Google Scholar]
- 23.Karnell JL, Albulescu M, Drabic S, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med. 2019;11(489):eaar6584. [DOI] [PubMed] [Google Scholar]
- 24.Pucino V, Gardner DH, Fisher BA. Rationale for CD40 pathway blockade in autoimmune rheumatic disorders. Lancet Rheumatol. 2020;2(5):e292-e301. [DOI] [PubMed] [Google Scholar]
- 25.Harp CT, Lovett-Racke AE, Racke MK, Frohman EM, Monson NL. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clin Immunol. 2008;128(3):382-391. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Ireland SJ, Remington G, et al. . CD40-Mediated NF-κB activation in B cells is increased in multiple sclerosis and modulated by therapeutics. J Immunol. 2016;Vol 197(11):4257-4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Q, Wang Q, Yang J, et al. Elevated sCD40L in secondary progressive multiple sclerosis in comparison to non-progressive benign and relapsing remitting MS. J Cent Nerv Syst Dis. 2021:14;1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aarts S, Seijkens TTP, van Dorst KJF, Dijkstra CD, Kooij G, Lutgens E. The CD40-CD40L dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol. 2017:8;1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be shared with qualified investigators on request; please contact CEF3W@virginia.edu.