

Abstract
Platinum-based chemotherapy remains widely used in advanced non-small cell lung cancer (NSCLC) despite experimental evidence of its potential to induce long-term detrimental effects, including the promotion of pro-metastatic microenvironments. In this study, we investigated the interconnected pathways underlying the promotion of cisplatin-induced metastases.
In tumor-free mice, cisplatin treatment resulted in an expansion in the bone marrow of CCR2+CXCR4+Ly6Chigh inflammatory monocytes (IMs) and an increase in lung levels of stromal SDF-1, the CXCR4 ligand. In experimental lung metastasis assays, cisplatin-induced IMs promoted the extravasation of tumor cells and the expansion of CD133+CXCR4+ metastasis-initiating cells (MICs). Peptide R, a novel CXCR4 inhibitor designed as an SDF-1 mimetic peptide, prevented cisplatin-induced IM expansion, the recruitment of IMs into the lungs, and the promotion of metastasis. At the primary tumor site, cisplatin treatment reduced tumor size while simultaneously inducing tumor release of SDF-1, MIC expansion, and recruitment of pro-invasive CXCR4+ macrophages. Co-recruitment of MICs and CCR2+CXCR4+ IMs to distant SDF-1-enriched sites also promoted spontaneous metastases that were prevented by CXCR4 blockade. In clinical specimens from NSCLC patients SDF-1 levels were found to be higher in platinum-treated samples and related to a worse clinical outcome. Our findings reveal that activation of the CXCR4/SDF-1 axis specifically mediates the pro-metastatic effects of cisplatin and suggest CXCR4 blockade as a possible novel combination strategy to control metastatic disease.
Keywords: lung cancer stem cells, metastasis, SDF-1/CXCR4 axis, chemotherapy, peptide anti-CXCR4, inflammatory monocytes, combination therapy
Graphical abstract
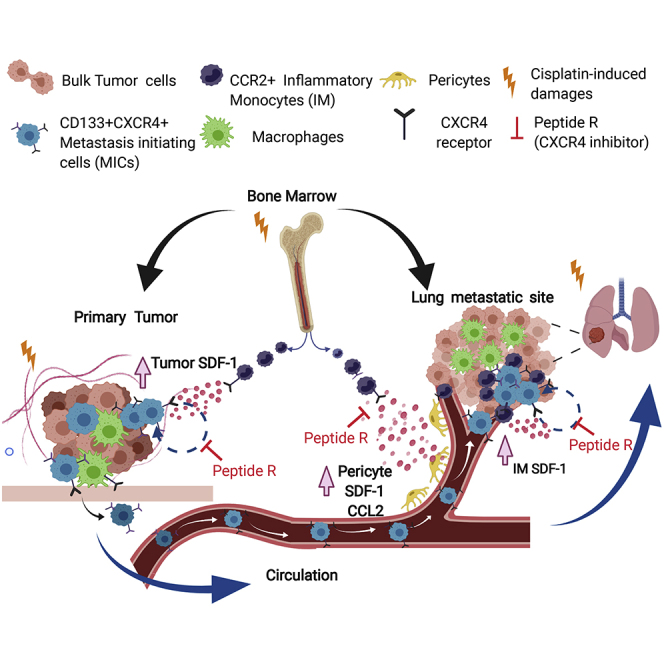
This study shows that cisplatin treatment of NSCLC uniquely activates the SDF-1-CXCR4 pathway in stroma and tumor cells, ultimately boosting metastasis formation. Combination strategies with CXCR4 inhibitors can revert this effect and improve long-term efficacy of chemotherapy.
Introduction
Lung cancer represents the first cause of cancer-related mortality worldwide.1 Platinum-based chemotherapy, alone or in combination with immunotherapy, is the standard of care in neo-adjuvant/adjuvant treatment of patients with non-small cell lung cancer (NSCLC).2, 3, 4, 5, 6 However, the long-term efficacy of standard chemotherapy is undermined by potential paradoxical detrimental effects such as the promotion of tumor relapse and metastasis spread.7,8
In preclinical settings, chemotherapy treatments, while demonstrating an effective control of primary tumors, can in fact stimulate pro-metastatic effects mediated by the host’s inflammatory responses to cytotoxic damage.9,10 Chemokines released by damaged stromal/tumor cells after chemotherapy can promote tumor cell dissemination, survival, and growth at distant sites.7,11,12 However, how platinum compounds can orchestrate multiple pro-metastatic activities is not fully understood.
The myeloid subset of C-C motif chemokine receptor type 2 (CCR2)high inflammatory monocytes (IMs) promotes the early phases of metastasis development by fostering extravasation of tumor cells at distant sites.13,14 Chemotherapy can exacerbate recruitment of CCR2+ IMs and the consequent promotion of metastasis by inducing tumor/stromal release of the CCR2 ligand, CCL2.15,16 Furthermore, chemotherapy can induce the release of other chemokines such as SDF-1, the ligand for the chemokine receptor CXCR4, resulting in the recruitment of a subset of tumor-associated macrophages (TAMs) (CD206+TIE2highCXCR4high) with pro-angiogenic activity,17,18 thus favoring primary tumor re-growth and dissemination.19
CXCR4, a G protein-coupled receptor expressed on multiple cell types, including lymphocytes, hematopoietic stem cells, endothelial cells, fibroblasts, and cancer cells,20,21 has also been implicated in several mechanisms that promote tumor progression and metastasis.20,22,23 CXCR4 inhibitors are being studied for the treatment of advanced refractory solid tumors with promising results.20,24,25 In pre-clinical models of colorectal cancer and melanoma, a novel CXCR4 peptide inhibitor (peptide R), designed as an SDF-1 mimetic peptide,26 has been shown to increase the efficacy of chemotherapy or immunotherapy in controlling the growth of primary tumors and the formation of metastases.27,28 Notably, the CXCR4/SDF-1 axis can also modulate immunosuppressive tumor microenvironments, and its inhibition can enhance the efficacy of immunotherapy by preventing tumor infiltration by myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs).29,30
We previously reported that in lung cancer the fraction of CD133+ cancer stem cells (CSCs) that co-express the chemokine receptor CXCR4 can be spared by chemotherapy and possess the highest ability to disseminate and initiate distant metastasis (metastasis-initiating cells [MICs]).31, 32, 33 In particular, cisplatin treatment of lung cancer patient-derived xenografts (PDXs), despite the effective decrease in the size of primary tumors, results in an enrichment of MICs and enhancement of metastasis that can be counteracted by CXCR4 inhibition.31
In this study, we hypothesized a central role for the CXCR4/SDF-1 axis in orchestrating multiple chemotherapy-induced pro-metastasis effects in NSCLC and tested the effectiveness of a combination therapy with peptide R to counteract the promotion by cisplatin of pro-metastatic microenvironments and improve treatment efficacy.
Results
Cisplatin promotes expansion of bone marrow (BM)-derived IMs and their lung recruitment through CXCR4/SDF-1 axis activation
In light of pre-clinical evidence on the functional relevance of systemic pro-metastatic changes induced by chemotherapy, we initially investigated the direct effect of cisplatin in shaping the microenvironment and promoting the generation of a pre-metastatic niche in healthy tissues. We focused in particular on the CXCR4/SDF-1 axis, which we previously identified as a crucial modulator of CSC dynamics in cisplatin-treated PDX models31 and that has been shown to modulate immune microenvironments composition.25,27 Tumor-free naive severe combined immunodeficiency (SCID) mice were treated with cisplatin alone or in combination with the peptide R, a cyclic peptide antagonist able to inhibit CXCR4 function,26 and reactive changes were evaluated in the BM and lungs. After 72 h, cisplatin caused the expansion of BM IMs (CD11b+Ly6C+Ly6G−), without substantial changes in the frequency of other myeloid cells or natural killer (NK) cells (Figure 1A; Figure S1A). Notably, IMs in cisplatin-treated group were 4-fold enriched in the subset co-expressing CXCR4 and CCR2 (Figure 1B). CXCR4 inhibition prevented IM expansion caused by cisplatin and the enrichment of the CXCR4+/CCR2+ subset (Figures 1A and 1B). In the lungs, cisplatin-induced damage resulted in the release of several inflammatory cytokines, including CCL2 and SDF-1. This was partially impaired by CXCR4 inhibition (Figures S1B and S1C; Figure 1C). Notably, SDF-1 levels in the BM remained substantially unaffected by the treatments (Figure S1D). Immunofluorescence analysis of lung tissues showed that SDF-1 production was predominantly contributed by interstitial lung pericytes, defined by the expression of platelet-derived growth factor receptor β (PDGFRβ)/CD146/α-smooth muscle actin (α-SMA)/nestin markers, while it was only minimally associated to CD45+ elements, data that were also confirmed by in vitro combination treatments of both human and murine endothelial cells (Figure 1D; Figures S1E and S1F).
Figure 1.

Cisplatin promotes lung recruitment of BM inflammatory monocytes through SDF-1/CXCR4 activation
(A) FACS analysis of DX5+ NKs and CD11b+ myeloid cell subsets in bone marrow (BM) of SCID mice 72 h after treatment with cisplatin alone or in combination with peptide R (CXCR4 inhibitor). n = 5 mice/group, two independent experiments. Bars are the mean value ± SE. ∗p ≤ 0.05. Nϕ, neutrophil; DC, dendritic cells; IM, inflammatory monocyte; RM, resident monocyte. (B) Frequency of CXCR4+ and CCR2+ inflammatory monocytes, detected in (A). ∗∗∗p ≤ 0.001. (C) IHC for SDF-1 in lung tissue of SCID control and treated mice, 72 h after administration of cisplatin alone and in combination with peptide R. Quantification of SDF-1+ cells, counted in five random areas of n = 5 lung sections/group, is shown on the right. ∗∗∗p ≤ 0.0001. (D) Double immunofluorescence for SDF-1 and PDGFRβ+; CD146 pericytes cells and CD45+ immune cells (on the bottom) performed on lung tissue from the same experimental groups as in (C). (E) Median percentage of inflammatory monocytes, gated total lung live cells, detected by FACS in lungs of mice 72 h after treatment with cisplatin and combination therapy compared to untreated control. n = 5 mice/group in two independent experiments. ∗p ≤ 0.05. (F) Frequency of CXCR4+ and CCR2+ inflammatory monocytes, detected in (E). (G) Percentage of inflammatory monocytes, relative to the gated live lung cells, evaluated by FACS in BM and lungs of BALB/c mice, 72 h after treatments with cisplatin and combination therapy compared to untreated control. n = 5 mice/group. Bars are the mean value ± SD. ∗p ≤ 0.05. (H) Median frequency of CXCR4+CCR2+ cells within gated inflammatory monocytes, detected in (G). ∗p ≤ 0.05. (I) Analysis for Cxcl12 gene expression performed in lung tissue of n = 5 BALB/c mice treated with cisplatin or combination. Untreated mouse lung tissue was used as a calibrator.
To shed light on the mechanistic links between cisplatin-induced damage and upregulation of the CXCR4/SDF-1 pathway, we investigated the potential role of two factors previously involved in regulation of cisplatin activity at the intracellular level, activating transcription factor 3 (Atf3)34 and high mobility group box 1 (Hmgb1).35,36 Gene expression analysis of cisplatin-treated lungs revealed that Hmgb1 was significantly increased by cisplatin and that peptide R counteracted this effect (Figure S1G).
Consistently with the release of SDF-1 and CCL2, cisplatin increased the relative abundance of CCR2+ IMs (2.3-fold change) expressing high levels of CXCR4; the combination with peptide R significantly prevented the increase in IMs induced by cisplatin in the lungs, even when a similar relative percentage of CXCR4+CCR2+ IMs was detected (Figures 1E and 1F; Figure S1H).
Since IMs share a similar phenotype with monocytic MDSCs (M-MDSCs),37 we also assessed the expression of a series of immunosuppressive (Interleukin [IL]-10, Nos1, Nox2) and MDSC-related genes (CD49d and Arginase), but no modulation (or even detection) was observed in treated lung tissue (Figure S1I), suggesting that the cisplatin-recruited CD11b+Ly6C+Ly6G− myeloid subset is likely represented by IMs rather than M-MDSCs.
Inflammatory status promoted by cisplatin and the resulting increase of IMs at BM and lung sites were mainly observed in an acute response (72 h after treatment) and were alleviated during prolonged treatments (2–4 weeks) (Figures S2A–S2C).
Acute treatment (72 h) with cisplatin of tumor-free immunocompetent mice also confirmed the expansion of BM-derived IMs and their recruitment to the lungs through a CXCR4-dependent mechanism (Figures 1G–1I). Cisplatin treatment did not significantly affect other myeloid or lymphoid immune cell compartments (Figure S2D). Interestingly, no modulations in the expression of M-MDSC-associated genes or in Treg frequency were observed in lungs after acute treatment with cisplatin (Figures S2D and S2E).
Treatments of naive SCID mice with other drugs commonly used for lung cancer treatment (gemcitabine, paclitaxel, pemetrexed, etoposide) failed to cause the expansion of BM-derived IMs (Figure S3A) and to significantly increase stromal expression of SDF-1 and CCL2 cytokines in the lungs that abundantly chemoattracted CXCR4+CCR2+ IMs (Figures S3B–S3E).
Overall, these data indicate the potential of CXCR4/SDF-1 axis inhibition to prevent cisplatin-induced inflammation.
IMs recruited by cisplatin support lung metastasis outgrowth
Next, to elucidate the impact of cisplatin-induced host inflammatory responses in metastasis promotion, the human metastatic lung cancer cell line H460 (large cell carcinoma) was inoculated in the tail vein of SCID mice 72 h after administration of cisplatin, alone or in combination with peptide R. One week after injection, the number of H460 cells seeding the lungs was only slightly increased in cisplatin pre-treated animals but significantly enriched for the MIC subset compared to untreated controls (Figures 2A and 2B). Three weeks after injection, the cisplatin pre-treated group showed a massive increase in the number and size of metastatic foci that also maintained MIC enrichment compared to untreated controls (Figures 2C–2E). Pre-treatment with peptide R prevented MIC survival and expansion caused by cisplatin (Figures 2A–2E).
Figure 2.

Cisplatin-activated microenvironment induces MIC selection and expansion via the SDF-1/CXCR4 axis
(A) Percentage of H460 cells surviving in lungs of SCID mice pre-treated with cisplatin or combination with CXCR4, 1 week after i.v. injection. Tumor cells were detected by FACS as live (7-AAD−) and murine HLA− cells. n = 3 mice/group for two independent experiments. (B) Frequency of CD133+CXCR4+ MICs within gated H460 cells in murine lung (as detected in A). n = 3 mice/group for two independent experiments. ∗p ≤ 0.05. (C) Median percentage of metastatic H460 cells detected by FACS in murine lung, 3 weeks after i.v. injection in SCID mice pre-treated with cisplatin or combination with CXCR4. n = 6 lungs/group, for three independent experiments. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001. (D) Frequency of CD133+CXCR4+ MICs within H460 lung metastases, detected in (C). Bars are the mean value ± SD of n = 6 lungs/group for three independent experiments. ∗p ≤ 0.05. (E) H&E of murine lungs, 3 weeks after H460 i.v. injection in SCID mice pre-treated with cisplatin or combination with CXCR4 (on the left) and histological quantification of the area of lung invasion and number of metastatic foci (on the right). n = 3 mice/group, two independent experiments. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. (F) Percentage of metastasis-associated macrophages (MAMs) detected by FACS in murine lungs 72 h after treatment with cisplatin and 1 and 3 weeks after H460 cell i.v. injection. Bars are the value ± SD. n = 3 mice/group; two independent experiments were performed for the 72 h time point. ∗p ≤ 0.05. (G) Frequency of CXCR4+ cells in MAMs detected in (F). Bars are the value ± SD. n = 3 mice/group; two independent experiments were performed for the 72 h time point. ∗p ≤ 0.05. (H) IHC staining for CKs of lungs of SCID mice pre-treated with cisplatin/combination therapy and injected i.v. with A549 cells (on the left). Quantification of CK+ human metastatic positive cells relative to total lung cells is reported on the right. n = 5 mice/group. ∗p < 0.05, ∗∗p < 0.01. (I) Percentage of MAMs detected by FACS in murine lungs 1 month after A549 cell i.v. injection. n = 5 mice/group. ∗p ≤ 0.05. (J) H&E of murine lungs 4 weeks after LCC i.v. injection in C57BL/6 mice pre-treated with cisplatin or combination with CXCR4 (on the left) and histological quantification of the area of lung invasion (on the right). n = 5 mice/group. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. (K) Percentage of MAMs detected by FACS in C57BL/6 lungs 4 weeks after injection of LCC cells. n = 5 mice/group. ∗p ≤ 0.05.
During lung metastasis development, we also observed an increasing number of metastasis-associated macrophages (MAMs) (CD11b+CD11c−F480+GR1−),38 highly expressing CXCR4, that remained more abundant in lungs of cisplatin pre-treated mice (Figures 2F and 2G; Figures S4A and S4B). Accordingly, in metastatic lungs of cisplatin pre-treated mice, we found an increased expression of genes typically associated with immunosuppressive MAMs (Arginase, Csf1r, Mdr1, Nos1, Ros1) (Figure S4C), possibly derived from differentiation of initially cisplatin-recruited IMs.39
We repeated the same experimental metastasis assay by injecting in pre-treated SCID mice the NSCLC adenocarcinoma A549 cell line. After 1 month, mice pre-treated with cisplatin showed a significant increase in the number of metastases along with expansion of CD133+CXCR4+ MICs and MAMs, as well as upregulation of stromal SDF-1 and of genes associated with immunosuppressive MAMs (Arginase, Mrc1, Csf1r) (Figures 2H and 2I; Figures S4D and S4E). Peptide R treatment was able to revert such effects (Figures 2H and 2I; Figures S4D and S4E), confirming its effectiveness in counteracting metastatic growth promoted by cisplatin-damaged microenvironments in different NSCLC histological subtypes through repression of the CXCR4/SDF-1 axis.
We then assessed cisplatin-induced pro-metastatic effects in a syngeneic mouse model of lung cancer to investigate the mechanism in fully immunocompetent mice. First, we confirmed also in the C57BL/6 mouse strain the increase of CCR2+CXCR4+ IMs at lung sites induced by cisplatin in tumor-free mice, without any significant modulation in the lymphoid compartment (Figures S4F, S4G, and S4I). Then, murine Lewis lung carcinoma (LLC) cells were injected in the tail vein of C57BL/6 mice that had received a single dose treatment of cisplatin, alone or in combination with peptide R. Four weeks after injection, a massive increase in metastasis formation was observed in the lungs of mice pre-treated with cisplatin, together within MAM expansion and Cxcl12 and Ccl2 upregulation (Figures 2J and 2K; Figure S4H). In these mice we also observed an increase in the subsets of exhausted tumor-infiltrating PD1+ and TIM3+ CD8 T cells and of Tregs, along with an immunosuppressive molecule IL-10 increase (Figures S4I and S4J). CXCR4 inhibition was able to partially prevent an SDF-1 increase and MAM and Treg expansion, overall counteracting metastasis formation fostered by cisplatin.
Finally, pre-treatment of tumor-free SCID mice with cisplatin and a neutralizing antibody (nAb) against CCL2 prevented BM expansion and lung recruitment of CCR2+CXCR4+ IMs (Figures 3A and 3B). Tail vein injection of H460 cells 72 h after treatments revealed that anti-CCL2 nAb significantly reduced cisplatin-induced metastasis and MIC expansion and partially decreased the number of MAMs (Figures 3C–3F), pointing to CCR2+CXCR4+ IMs as the primary mediators of cisplatin pro-metastatic effects.
Figure 3.

CCL2 inhibition counteracts cisplatin pro-metastatic effects
(A) Percentage of CCR2+ inflammatory monocytes detected by FACS in SCID mouse BM and lungs, 72 h after treatment with cisplatin alone or with anti-CCL2 neutralizing antibody (nAb) CCL2, 150 μg/mice). n = 3 mice/group. ∗p ≤ 0.05. (B) Median frequency of CXCR4+ cells detected in lung CCR2+ IMs, analyzed in (A). n = 3 mice/group. ∗p ≤ 0.05. (C) Median percentage of lung metastatic H460 cells detected by FACS 3 weeks after i.v. injection in SCID mice pre-treated with cisplatin or a combination with nAb CCL2. n = 6 lungs/group. ∗∗p ≤ 0.001, ∗∗∗p ≤ 0.0001. (D) H&E of H460 lung metastases in treatment groups described in (C) and histological quantification of the area of lung invasion by tumor cells (on the right; n = 6 lungs/group). ∗p ≤ 0.05. (E) Median percentage of CD133+CXCR4+ MICs detected by FACS in H460 lung metastasis, analyzed in (C). n = 6 lungs/group. ∗p ≤ 0.05. (F) Percentage of MAMs detected in metastatic murine lungs, analyzed in (C) and (D).
IMs increase tumor cell extravasation and expansion of MICs through the CXCR4/SDF-1 axis
Chemotherapy can increase vascular permeability,16 which might be implicated in the early retention of tumor cells at the lungs observed in cisplatin-treated mice. Indeed, cisplatin treatment upregulated in lung tissue several adhesion molecules able to mediate vascular adhesion and extravasation of leukocytes and tumor cells (Icam-1, Vcam1, P-selectin) (Figure 4A), and data were also confirmed in vitro in murine and human endothelial cells treated with cisplatin (Figure S5A). This modulation was associated with the deconstruction of α-SMA+ endothelial layers, suggestive of an increased vascular leakiness (Figure S5B).
Figure 4.

IMs promote tumor cell extravasation and MIC expansion
(A) Gene expression analysis of adhesion molecules performed for n = 4 SCID mice lungs, 72 h after treatment with cisplatin. Untreated lung tissue was used as a calibrator. (B) Transendothelial migration assay: murine RAW 264.7 macrophages, treated with cisplatin (5 μM) or a combination with anti-CXCR4 (10 μM), were used to chemoattract PKH-labeled H460 cells through the endothelial cell layer, treated or not with cisplatin (5 μM). Serum-free medium was used to assess the basal transmigration ability of tumor cells. Migrated PKH-labeled H460 cells were counted by fluorescent microscopy in four random fields of each insert, in duplicate. Bars are the mean ± SD of three independent experiments. ∗p ≤ 0.05, ∗∗∗p ≤ 0.0001. (C) H460 cells were chemoattracted by conditioned medium (CM) from sorted BM-derived CCR2+ or CCR2− cells through the endothelial cell layer, treated or not with cisplatin, and analyzed as in (B). Bars are the mean ± SD of n = 2 independent experiments. ∗∗p ≤ 0.001. (D) Transendothelial migration assay as in (C), using CM from BM CCR2+ and CCR2− cells with or without nAb against VEGF (150 ng/mL). Bars are the mean ± SD of n = 2 independent experiments. ∗p ≤ 0.05, ∗∗p ≤ 0.001. (E) Percentage of CD133+CXCR4+ MICs evaluated by FACS in H460 cells co-cultured for 48 h with BM-sorted CCR2+ and CCR2− cells. Bars are the mean ± SD of n = 5 independent experiments. ∗p ≤ 0.05, ∗∗p ≤ 0.001. (F) Percentage of CD133+CXCR4+ MICs evaluated by FACS in H460 cells co-cultured with BM sorted CCR2+ and CCR2− cells with and without nAb SDF-1 (25 μM). Bars are the mean ± SD of n = 3 independent experiments, each in technical duplicate. ∗p ≤ 0.05, ∗∗p ≤ 0.001. (G) Mean percentage of lung disseminated tumor cells (DTCs) from subcutaneous xenografts ensuing from parental H460 and SDF-1 selected H460 cell lines. n = 4 mice/group. ∗p ≤ 0.05.
In vitro transendothelial migration of H460 cells demonstrated that cisplatin-treated human umbilical vein endothelial cells (HUVECs) allowed an enhanced transmigration of tumor cells chemoattracted by a murine macrophage cell line (RAW 264.7) (Figure 4B). Moreover, RAW 264.7 macrophages treated with cisplatin showed a 10-fold enrichment in the CCR2+CXCR4+ cell subset (Figure S5C), which was associated with increased H460 transmigration. This phenomenon was inhibited by peptide R, able to prevent CCR2+CXCR4+ subset expansion (Figure 4B).
We also confirmed that conditioned medium (CM) from BM-sorted CCR2+ IMs increased the transmigration ability of H460 cells, especially through cisplatin-treated endothelial cells, compared to CM from CCR2− cells (comprising neutrophils, NK cells, resident monocytes, and dendritic cells) (Figure 4C).
We verified that compared to CCR2− myeloid cells, CCR2+ IMs expressed higher levels of vascular endothelial growth factor (Vegf) (2.7-fold change ± SD 1.2), known to mediate tumor cell extravasation at metastatic sites.13,14 Coherently, cisplatin-treated RAW 264.7 cells and murine lung tissues, both enriched in CCR2+CXCR4+ cells, showed that upregulation of Vegf expression and neutralization of murine VEGF-A in CM from CCR2+ cells prevented tumor cell extravasation (Figures S5D and S5E; Figure 4D).
Next, we investigated the link between lung IM increase and MIC expansion caused by cisplatin treatment. Co-cultures of H460 cells with BM-sorted CCR2+ IMs determined a 20-fold increase of CD133+CXCR4+ MICs compared to co-cultures with CCR2− myeloid cells (Figure 4E). Interestingly, we verified that Cxcl12 expression was 12.7-fold (±SD 7.5) increased in CCR2+ IMs compared to CCR2− BM cells. Neutralization of SDF-1 specifically prevented the CCR2+ IM-induced MIC increase in H460 cells and also in lung adenocarcinoma cell lines (A549 and LT73), proving the ability of CCR2+ IMs to foster MIC expansion via CXCR4/SDF-1 axis activation (Figure 4F; Figure S5F). Similarly, cisplatin-treated HUVECs, which also showed upregulation of CXCL12 levels, promoted the expansion of CD133+ cells, even if at a lesser extent than CCR2+ IMs (Figure S5G).
Finally, the key role of SDF-1 in mediating CD133+CXCR4+ expansion was functionally demonstrated by the high content of MICs (4.3-fold increase) (Figure S5H) in a sub-line induced by the chronic exposure of H460 cells to SDF-1 that was associated with enhanced in vivo ability to colonize murine lungs compared to the parental cell line (Figure 4G).
Cisplatin treatment of lung cancer xenografts expands MICs and recruits CXCR4+ TAMs through the CXCR4/SDF-1 axis, promoting MIC intravasation
Cisplatin treatment of H460 subcutaneous xenografts indicated that combination with peptide R did not enhance cisplatin efficacy in controlling tumor growth, as expected by the low percentage of CXCR4+ tumor cells within the tumor bulk (Figure S6A). However, CXCR4 inhibitor prevented the enrichment in MIC content and the increase of tumor SDF-1 induced by cisplatin (Figures 5A and 5B). Differently from the SDF-1 stromal modulation observed at distant sites (Figure 1D; Figure S1E), we verified by immunofluorescence analysis in xenografts that the SDF-1 increase was mostly provided by tumor cells rather than stromal cells (Figure S6B).
Figure 5.

Cisplatin-induced SDF-1 in tumor cells favors MIC expansion and intravasation
(A) Content of CD133+CXCR4+ MICs in H460 xenografts at the end of treatments. n = 4 mice/group, two independent experiments. ∗p ≤ 0.05. (B) IHC for SDF-1 in control and treated xenografts at the end of treatments. On the right, quantification of SDF-1+ tumor cells counted in five random fields of n = 5 xenograft sections/group is shown. ∗∗∗p ≤ 0.0001. (C) FACS analysis for CD133+ subsets evaluated in the H460 cell line 72 h after treatments with different chemotherapeutic drugs compared to the untreated control. Bars are the mean ± SD of n = 3 independent experiments. ∗p ≤ 0.05. (D) Median percentage of CD133+CXCR4+ MICs evaluated by FACS in primary NSCLC cultures (n = 4) treated with cisplatin (10 μM) and combination with CXCR4 inhibitor for 72 h. ∗p ≤ 0.05. (E) IHC for F4/80 macrophage marker in control or treated xenografts. (F) Percentage of tumor-associated macrophages (TAMs) detected in xenografts at the end of treatment. Bars are the mean value ± SD. n = 3 mice/group, two independent experiments. ∗p ≤ 0.05. (G) Frequency of CXCR4+ cells within gated TAMs, detected in (F). (H) Gene expression analysis for murine Vegfr and Vegfd in n = 3 treated xenografts (the same evaluated in F and G). Untreated xenografts were used as a calibrator. (I) FACS analysis for CD133+ cells evaluated in H460 xenografts and corresponding CTC-derived xenografts (CDXs). Bars are the mean value ± SD of n = 4 independent analysis, in duplicate. ∗p ≤ 0.05. (J) Percentage of DTCs detected by FACS in lungs of mice bearing H460 xenografts and CDXs evaluated in (I). Bars are the mean percentage ± SD. N = 3 independent analysis, in technical duplicate. (K) Content of CD133+CXCR4+ MICs in PDX LT111 at the end of treatments. Bars are the mean value ± SD. n = 5 mice/group. ∗p ≤ 0.05. (L) Relative percentage of CXCR4+CCR2+ cells within gated tumor-associated macrophages (TAMs) detected in PDX LT111 at the end of treatments. Bars are the mean value ± SD. n = 5 mice/group. ∗p ≤ 0.05. (M) Gene expression analysis for ATF3 in n = 10 treated tumors (five H460 xenografts and 5 LT111 PDXs). Untreated tumors were used as a calibrator. ∗p ≤ 0.05.
Interestingly, in vitro treatment of H460 cells and others NSCLC cell lines (H1299/A549/LT73) with different chemotherapeutic agents (80% inhibitory concentration [IC80] dose) demonstrated the unique ability of cisplatin to induce MIC expansion along with an CXCL12 increase (Figure 5C; Figures S6C and S6D). Nevertheless, in cell lines treated with different chemotherapeutic agents we detected an increase in the expression of other inflammatory cytokines, some of which were able to expand in vitro the subset of CD133+CXCR4− cells in the H460 cell line without modifying MICs (Figure 5C; Figures S6D and S6E). Finally, cisplatin treatment of short-term cultures of primary NSCLC also confirmed MIC expansion that can be impaired by CXCR4 blockade (Figure 5D).
Increased SDF-1 levels in cisplatin-treated xenografts also promoted the recruitment and the peritumoral accumulation of a specific subset of CXCR4+ TAMs (F480highGR-1−CD206+), known to promote intravasation and angiogenesis via the VEGF/VEGF receptor (VEGFR) axis.17,19 This accumulation was prevented by peptide R (Figures 5E–5G). Additionally Vegf/Vegfr modulation was observed in treated xenografts, coherently with different content of CXCR4+ TAMs (Figure 5H).
Moreover, by performing in vivo tumorigenic assays, we demonstrated that only circulating tumor cells (CTCs) isolated from blood of cisplatin-treated mice were able to generate tumors when injected subcutaneously in new recipient SCID mice. This evidence functionally suggests that the increase in MICs and TAMs induced in xenografts by cisplatin treatment can promote escape of primary tumor cells with enhanced metastatic potential. Ensuing CTC-derived xenografts (CDXs) showed an increased content of CD133+ CSCs compared to original xenografts and demonstrated enhanced lung dissemination ability, likely reflecting the properties of circulating MICs originally expanded in primary tumors and primed by cisplatin treatment (Figures 5I and 5J).
To further validate these observations in a different pre-clinical model, we treated a patient-derived xenograft that was derived from a primary lung adenocarcinoma (PDX-LT111). We confirmed that cisplatin was able to increase the frequency of CD133+CXCR4+ MICs within the tumor and the number of CXCR4+CCR2+ TAMs (Figures 5K and 5L), along with the upregulation of tumor CXCL12 expression (Figure S6F), and peptide R partially prevented such effects (Figures 5K and 5L).
Mechanistically, we found that cisplatin treatment induced the upregulation in tumor cells of the stress-inducible gene ATF3, a transcription factor that can directly regulate SDF-1.40 Peptide R prevented an ATF3 increase, thus possibly explaining the efficacy of CXCR4 inhibitor in controlling a cisplatin-induced SDF-1 increase (Figure 5M).
Cisplatin treatment of lung cancer xenografts co-recruits MICs and MAMs at distant sites through CXCR4/SDF-1 activation, favoring metastasis formation
Cisplatin treatment of primary tumors caused a boost in the formation of lung micro-metastases in mice bearing subcutaneous H460 xenografts, coupled with a significant enrichment in MIC content (1.8-fold change) (Figures 6A–6C). We confirmed in cisplatin-treated mice an increased stromal SDF-1 expression in the lung parenchyma interstitium and a significant higher level of plasmatic CCL2 compared to controls (Figures 6D and 6E; Figure S7A). Analysis of metastatic lungs demonstrated that nestin+ stromal cells are the main source of SDF-1, which only marginally co-localizes with CKs+ tumor cells (Figure 6D).
Figure 6.

Cisplatin treatment fosters spontaneous metastasis formation co-recruiting MICs and IMs
(A) Percentage ± SD of H460 lung metastatic cells detected in mice-bearing subcutaneous xenografts treated with cisplatin or combination treatment. n = 20 mice/group, three independent experiments. ∗∗∗p ≤ 0.001. (B) H&E of lungs of mice-bearing H460 xenografts analyzed in (A). (C) CD133+CXCR4+ MIC content analysis by FACS in H460 lung metastasis (as detected in A). n = 20 mice/group, three independent experiments. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001. (D) Double immunofluorescence for SDF-1 and murine nestin stroma cells or human CK tumor cells performed on metastatic lung tissue, as in (B). Quantification of SDF-1+ cells was performed both in stroma and tumor compartment of lungs of treated mice, by counting positive cells in n = 5 random fields in n = 3 lung sections/group. (E) ELISA quantification of CCL2 in plasma of mice-bearing xenografts, at the end of treatments. Bars are the mean value ± SD of duplicate experiments, performed on plasma sample, pooled from n = 2 mice/group. (F) Percentage of MAMs detected in murine lungs analyzed in (C). Bars are the mean value ± SD. n = 20 mice/group, three independent experiments.∗p ≤ 0.05. (G) Frequency of CXCR4+ MAMs detected in (F). ∗p ≤ 0.05.
Consistent with an SDF-1 and CCL2 increase, we observed an augmented lung recruitment of CCR2+CXCR4+ monocytes/macrophages (Figures 6F and 6G), which can promote the extravasation and expansion of the MIC subset, as we demonstrated above. Combination with the CXCR4 inhibitor impaired an SDF-1 increase, preventing the recruitment of CXCR4+ MICs and CXCR4+ MAMs, overall resulting in diminished metastasis formation (Figures 6A–6G).
Similarly, in the lungs of mice-bearing PDX LT111 treated with cisplatin we found an increased number of lung disseminated cells accompanied by a higher frequency of MAMs (Figures S7B and S7C).
We also investigated the metastatic dissemination from primary treated tumors to the liver, another preferential site of lung cancer metastasis. We observed in both models (H460 and LT111), using real-time PCR and flow cytometry analyses,31 that cisplatin only slightly increased the number of liver disseminated tumor cells (DTCs) compared to untreated controls. This effect was not counteracted by the combination with peptide R (Figures S7D and S7E). Notably, Cxcl12 and Ccl2 were not significantly modulated in cisplatin-treated liver tissue and consequently no specific chemoattraction for CD133+CXCR4+ MICs nor for CCR2+CXCR4+ macrophages was observed (Figures S7F–S7H). However, cisplatin increased the total number of liver macrophages through a CXCR4/SDF-1-independent mechanism, to be further elucidated (Figure S7I).
SDF-1 levels are increased in platinum-treated clinical samples of NSCLC and correlate with poor clinical outcome
To evaluate the clinical relevance of our pre-clinical data, we assessed by immunohistochemical (IHC) staining the expression of SDF-1 in surgical specimens from chemo-naive NSCLC patients (n = 57) and NSCLC patients treated with platinum-based chemotherapy in the neoadjuvant setting (n = 57), matched for stage (IIIA), histological subtypes, sex, and age (Table S1). The frequency of SDF-1-positive tumors was significantly higher in the neoadjuvant-treated group than in untreated tumors (67% versus 26%; p < 0.0001).
To address a possible prognostic role for SDF-1, patients treated with neoadjuvant platinum-based chemotherapy (n = 39, Table S2) were categorized according to the SDF-1 IHC score (percentage of positive cells × staining intensity) (Figure 7A): patients with a tumor SDF-1 score > 6 exhibited a significant shorter disease-free survival (DFS) (p = 0.0056; hazard ratio = 3.1) and overall survival (OS) (p = 0.029; hazard ratio = 3.46) compared to patients with an SDF-1 score < 6 (Figure 7B).
Figure 7.

High SDF-1 levels correlate with poor prognosis in NSCLC patients treated with neoadjuvant cisplatin
(A) IHC for SDF-1 of platinum-based neo-adjuvant-treated NSCLC tumors, representative of different score calculated as: % positive cells × intensity. Areas within dashed lines are shown at higher magnification. (B) Kaplan-Meier curves for DFS and OS, respectively, of n = 39 and n = 36 NSCLC patients treated with platinum-based neo-adjuvant therapy, categorized according to SDF-1 tumor score >6. Reported p values were calculated by a log-rank (Mantel-Cox) test.
Discussion
Herein, we demonstrate the crucial role of CXCR4/SDF-1 axis activation in both stromal and tumor cells in mediating the paradoxical pro-metastatic activity of cisplatin and provide evidence for a novel combination therapy based on CXCR4 inhibition that effectively controls the development of metastases. Early responses to cisplatin-induced damages include BM expansion of CCR2+CXCR4+ IMs and their recruitment into the lungs (the most common metastatic site for NSCLC),41 driven by the increased stromal release of SDF-1 and CCL2. These effects can be effectively prevented by blocking CXCR4 using a novel peptide inhibitor (peptide R).26
The CCR2+ subset of IMs has previously been described as an important player in the early stages of metastasis formation14,38,42 and has a phenotype that overlaps with M-MDSCs in mice.37 This subset can in fact favor the extravasation of tumor cells at distant sites through the release of VEGF-A,38 which can support their growth.43 Notably, chemotherapy may exacerbate the tumor release of CCL2 and consequently recruitment of CCR2+ IMs to both primary and metastatic sites.44 Interestingly, extracellular vesicles released by chemotherapy-treated mouse breast cancer cells have been shown to promote CCL2 release from endothelial cells and subsequent recruitment of CCR2+ IMs at the lung pre-metastatic niche, overall fostering metastasis development.15
Our data corroborate the pro-metastatic effects of CCR2+ IMs and further show that compared to other chemotherapeutic agents used for the treatment of NSCLC, cisplatin has the unique effect of generating stromal reactions that induce chemoattraction to the pre-metastatic niche of the population of CCR2+ IMs co-expressing CXCR4. This observation was confirmed both in SCID and immunocompetent mice, proving the central role of innate immune responses in mediating the pro-metastatic effects of cisplatin, as previously suggested.45 Moreover, our data support the pro-tumorigenic/metastatic properties of CXCR4 myeloid cells, already verified in a transgenic mouse model with genetic deletion of CXCR4 in the myeloid compartment.46
Our data on tumor-free mice also indicate that the acute response to cisplatin may preferentially trigger the generation of an inflammatory state that promotes the expansion of IMs rather than M-MDSCs, which share a similar phenotype but possess different immunosuppressive properties.47 Interactions with metastatic cells may then eventually induce the differentiation of IMs into immunosuppressive MAMs, as already described.39
We show that both CCL2 and CXCR4 inhibition in combination with cisplatin can impair IM recruitment and prevent cisplatin-induced metastasis formation. Although CCL2 blockade impairs metastasis formation,13,14,38 discontinuation of CCL2 neutralization in vivo has been shown to cause massive release of BM monocytes, which paradoxically exacerbates metastasis development by promoting cancer cell mobilization and formation of blood vessels.13 Altogether, these observations point to inhibition of CXCR4 as a potentially safer and more effective strategy to use in combination with cisplatin to prevent pro-metastatic mobilization of CXCR4+ IMs.
We verified, both in vivo and in vitro, that cisplatin can induce endothelial leakiness favoring tumor cell extravasation, and data are in line with the reported altered vascular permeability and overexpression of VEGFR-1 caused by chemotherapy, associated with augmented lung metastases.16,44,45 Moreover, we showed that the increase of IMs recruited by cisplatin can additionally foster tumor cells transendothelial migration via VEGF release, in accordance with previously reported observations.14
To the best of our knowledge, this is the first time that IMs, recruited by cisplatin into the lungs, are reported to select/expand MICs by releasing high levels of SDF-1 overall, promoting metastasis development.
At the primary tumor site, cisplatin often effectively reduces tumor size but specifically selects for a chemoresistant MIC subset.31,32 The cisplatin-induced increase of tumor SDF-1 levels can contribute to MIC expansion through activation of the CXCR4 pathway and also can possibly convert non-MICs into MICs, as we recently demonstrated.12
Among different chemotherapeutic agents routinely used for NSCLC treatments, we show in the present study that only cisplatin robustly induces SDF-1 expression and consequently can efficiently expand CXCR4+ MICs. Nevertheless, other drugs can also expand the fraction of CD133+CXCR4− CSCs, in line with previous data,48 by inducing the release of different inflammatory and pro-tumorigenic cytokines, as described in several reports.7,10,16,49
The cisplatin-induced increase of SDF-1 also fosters the recruitment at the primary tumor site of CXCR4+ TAMs that have been shown to favor tumor cell intravasation, neo-angiogenesis, and lymphangiogenesis and to promote tumor relapse after chemotherapy.19,50, 51, 52 Notably, we provide a functional proof of concept that the expansion of MICs at the primary site, along with a CXCR4+ TAM increase, enhances the ability of tumor cells to escape from primary tumors, as assessed by the unique ability of CTCs isolated from blood of xenograft-bearing cisplatin-treated mice to efficiently generate secondary tumors when transplanted into new recipient mice.
We provide preliminary evidence suggesting that cisplatin-induced damage in normal tissue may trigger differential effects according to the metastatic site; indeed, analysis of liver explants showed only a marginal increase in tumor cell dissemination and no activation of the CXCR4/SDF-1 axis.
We also obtained initial evidence that the link between cisplatin activity and activation of the CXCR4/SDF-1 pathway could be mediated at least in part by ATF3 at the primary tumor site and by HMGB1 in the lungs. ATF3 is a stress-inducible transcription factor, known to be involved in mitogen-activated protein kinase (MAPK)-dependent induction of cisplatin toxicity in cancer cells,34 and which has been shown to be able to orchestrate a tumor-promoting program directly involving SDF-1 induction.40 Interestingly, upregulation of ATF3 in stromal cells is observed in several types of cancers40 and has been shown to be necessary for paclitaxel-induced metastasis in breast cancer.53 In the present study, we show that cisplatin treatment induces ATF3 upregulation at the primary tumor site predominantly in tumor cells, which also represent the main source of local SDF-1. At distant sites we observed instead upregulation of HMGB1 as a potential mediator of cisplatin-induced damage in stromal cells. The increase and release of HMGB1 from damaged cells is an important pro-inflammatory mechanism with complex effects in cancer.54,55 Additionally, its activity as a recruiter of inflammatory cells has been shown to be dependent on the regulation of SDF-1 levels56,57 and on the formation of a heterocomplex with SDF-1, resulting in stabilization of the protein.58 Chemotaxis stimulated by HMGB1 also induces and relies on an SDF-1 autocrine loop.59 The limitation of this part of the study is related to the correlative nature of the observations, and further evidence is needed to confirm mechanistic links between this preliminary evidence and cisplatin pro-tumorigenic activity. Our results indicate, however, that CXCR4 signaling inhibition with peptide R could influence both local and systemic pathways, sustaining the generation of pro-metastatic microenvironments.
Finally, we prove in clinical samples from NSCLC patients that SDF-1 is significantly increased in tumor cells after platinum-based neo-adjuvant chemotherapy and, remarkably, that a high SDF-1 expression in post-treatment tumors is correlated with shorter progression-free survival and OS.
Very few studies have investigated the prognostic value of SDF-1 in primary NSCLC, and among them no significant association between SDF-1 expression and DFS or OS was reported.60,61 Our data prove the prognostic relevance of the SDF-1 marker in the setting of neo-adjuvant-treated NSCLC patients, although a prospective longitudinal study on a larger cohort of patients is needed to confirm the clinical value of our preliminary observations.
Taken together, our findings prove that cisplatin treatment uniquely activates the CXCR4/SDF-1 axis in both stromal and tumor cells, resulting in multiple metastasis-promoting actions.
One limitation of our study is related to the main use of immunodeficient mice models, which were selected to evaluate effects on human cancer cells, in particular, on an MIC subset. The use of naive immunocompetent mice and a syngeneic lung cancer model confirmed the activation of the CXCR4/SDF-1 axis and the pro-metastatic effects of cisplatin that can be counteracted by CXCR4 inhibitor. These studies also confirmed a central role for CCR2+CXCR4+ IMs and showed modulation of immunosuppressive T cell subsets in the late phase of metastasis development. However, more studies are needed to fully elucidate the complex interplay between adaptive immunity and tumor cells in the context of chemotherapy treatment.
A novel use of CXCR4 blockade in combination with cisplatin may therefore control more effectively metastatic disease in NSCLC patients and could be also exploited in a chemotherapy/ICI doublets regimen, the future mainstay of therapy for locally advanced/metastatic NSCLC patients, without targetable mutations.3,41,62 Notably, some pre-clinical and clinical evidence already suggests that CXCR4 inhibition may increase the efficacy of immunotherapy in different solid tumor types by relieving immune suppression caused by Tregs and MDSCs that highly express CXCR4.28, 29, 30,63,64 In this scenario, the immunomodulatory effects of cisplatin could be exploited for innovative combination therapies based on CXCR4 inhibition to increase and extend the benefit of immunotherapy to a larger cohort of patients with lung cancer.
Materials and methods
Cell cultures and reagents
H460 (large cell carcinoma), A549 (adenocarcinoma), and H1299 (large cell carcinoma) NSCLC cell lines, authenticated by short tandem repeat (STR) profiling, and murine RAW 264.7, murine 3B11, and HUVECs (all purchased from ATCC) were cultured in RPMI 1640 + 10% fetal bovine serum (FBS) or in endothelial growth media (EGM-2, Lonza). The LT73 cell line was derived from a primary NSCLC adenocarcinoma and cultured in vitro in RPMI 1640 + 10% FBS.
CCR2+ cells were positively isolated from BM cells flushed from SCID mouse femurs using anti- CCR2−allophycocyanin (APC)+ anti-APC MicroBeads and an autoMACS Pro separator (Miltenyi Biotec). 1 × 105 sorted CCR2+ IMs or CCR2− myeloid cells were co-cultured at a 1:1 ratio with tumor cells or used to recover CM after 48 h.
Short-term NSCLC primary cultures were obtained from four early stage (I–IIb) NSCLC undergoing surgical resection using differential filtration of dissociated primary tumor tissues as described in Bertolini et al.31
For in vitro experiments, cells were treated with the following: peptide R (10 μM), recombinant human SDF-1α (25 ng/mL) (300-28A), cytoMCP-1 (CCL2) (25 ng/mL) (300-04), IL-8 (50 ng/mL) (200-08), GRO-α/MGSA (25 ng/mL) (300-11), and IL-6 (10 ng/mL) (200-06) (all from PeproTech); as well as neutralizing Ab anti-human/mouse SDF-1 (25 μg/mL) (MAB310) and mouse VEGF (150 ng/mL) (VEGF164) (both from R&D Systems).
Flow cytometry
Immune cell subsets (live cells/CD45+) were identified in BM and lungs as follows: low SSC/DX5+, NK cells; CD11b+/LY6G+, neutrophils; Ly6G−/Ly6C−/low/F480high/CD11b−/CD11c+, alveolar macrophages; Ly6G−/Ly6C−/low/F480high/CD11b+CD11c−, monocyte-derived macrophages; Ly6G−/Ly6C−/F480−/low/CD11b+/CD11c+, conventional dendritic cells; Ly6G−/F480−/low/CD11b+/CD11c−/Ly6Chigh, IMs or Ly6Cdim resident monocytes. Expression of CXCR4 and CCR2 was evaluated within monophage/macrophage subsets.
Lung-disseminated/metastatic tumor cells were identified in lung-dissociated tissues by fluorescence-activated cell sorting (FACS) as 7-aminoactinomycin D (7-AAD)−/mouse major histocompatibility complex (MHC) class I− cells or by real-time detection of human B2M gene expression in murine tissue.31 A list of all antibodies used is available in Supplemental materials and methods. Gallios (Beckman Coulter) or FACSCanto (BD Biosciences) flow cytometers were used for data acquisition, and FlowJo software v10 was used for data analysis.
Transendothelial migration assay
5 × 104 HUVECs were plated in EGM-2 medium into the upper chamber of FluoroBlok 24-well cell culture inserts (Corning Life Sciences) covered with 100 μL of Matrigel (Corning Life Sciences). In the lower chamber, 5 × 104 RAW 264.7 cells were plated in RPMI 1640 + 10% FBS or filled with CM from CCR2+ and CCR2− BM cells. The following day HUVECs and RAW cells were treated with cisplatin (5 μM) with and without peptide R (10 μM). 2 × 104 PKH-26 (Sigma)-stained H460 cells were plated onto the HUVEC layer, and after 48 h migrated H460 cells were fixed with 4% paraformaldehyde (PFA) and counted under a fluorescence microscope.
Animal studies
For lung conditioning experiments, naive female SCID mice and BALB/c mice (Charles River Laboratories) were treated with intraperitoneal (i.p.) cisplatin (Teva Pharma) (2.5 mg/kg), followed by i.p. administration of peptide R (2 mg/kg) once every 3 days, and next injected intravenously (i.v.) with 1 × 106 H460 cells or 5 × 105 A549 cells. A similar experiment was performed in C57BL/6 mice treated i.p. with cisplatin (5 mg/kg) with and without peptide R (2 mg/kg) and injected i.v. 72 h after treatment with 1 × 106 Lewis lung carcinoma cells. Mice were monitored for 4 weeks. Treatment schedules for other chemotherapeutic drugs are available in Supplemental materials and methods.
Anti-mouse CCL2 antibody (150 μg/mouse) (clone 2H5, Bio X Cell) was administered to SCID mice together with cisplatin and for the following 3 days, before H460 cell injection.
Mice with palpable tumors (tumor weight [TW] ≥ 50 mm3), generated by s.c. injection of 1 × 105 H460 cells with Matrigel (Corning Life Sciences) or by the implant of a small fragment (≃25 mm3) of LT111 (adenocarcinoma PDXs), were randomized into three groups: untreated control, treated with 5 mg/kg cisplatin (i.p.) every 7 days alone or with 2 mg/kg peptide R (i.p.) once every 5 days after cisplatin for 3 weeks. In vivo experiment protocols were revised by the Internal Ethics Committee for Animal Experimentation and approved by the Italian Ministry of Health.
SDF-1 IHC score
NSCLC specimens were obtained from consenting patients (clinicopathological characteristics are available in Tables S1 and S2) undergoing surgical tumor resection after platinum-based neo-adjuvant chemotherapy (study INT 201/18 approved by the Independent Ethics Committee). IHC score for tumor SDF-1 staining (clone 79018, R&D Systems; IHC staining details are available in Supplemental materials and methods) was obtained by multiplying the percentage of positive cells (p = 0, no positive cells; p = 1, 1%–25% positive cells; p = 2, 25% −50% positive cells; p = 3, 50%–75% positive cells; p = 4, >75% positive cells) by the staining intensity (I = 1–3). The pathologist (M. Milione) performed the histological blinded evaluation of SDF-1 staining.
Statistical analyses
Statistical analyses were performed using GraphPad Prism v5.0. Statistically significant differences were determined with Student’s t tests when comparing two groups or an ANOVA test for multiple comparisons. Sample size and number of replicates are indicated in the figure legends for each experiment. Data are presented as mean (±SD), unless otherwise indicated. SDF-1 expression association with categorical clinical-pathological characteristics of NSCLC patients was calculated with Pearson’s chi-square test. Survival data were analyzed using Kaplan-Meier log-rank tests. All tests were two-sided, and statistical significance was defined as a p value less than 0.05.
Acknowledgments
This work was supported by Roche Italia (Roche per la Ricerca 2017 to G.B.); the Fondazione Umberto Veronesi (fellowship 2017-18 to G.B.); the Italian Association for Cancer Research (AIRC) (IG21431 to L.R., IG18812 to G.S., IG22145, 5x1000 12162 to C.T.); the Italian Ministry of Health (RF-2016-02362946 to L.R., RF-2018-12366714 to G.B., M 2/9 and M 2/6 to S.S.); and by the EuroNanoMed III-METASTARG Project (JTC2018-045; ERP-2018-23671123 to L.R.).
Author contributions
G.B., L.R., G.S., S.S., C.T., C. Chiodoni, and N.Z. conceived the study. G.B., V.C., O.F., M.T., G.C., C. Camisaschi, F.F., G.P., G.T., F.G., M. Moro, C. Chiodoni, and C.D.A. performed the experiments and analyzed the data. M. Milione performed pathological evaluation of human specimens. C.T. performed pathological evaluation of murine specimens. G.L.R., A.D.T., and U.P. selected case series and collected clinical information. G.B., L.R., and S.S. wrote the manuscript. All authors revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.014.
Contributor Information
Giulia Bertolini, Email: giulia.bertolini@istitutotumori.mi.it.
Stefania Scala, Email: s.scala@istitutotumori.na.it.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch F.R., Scagliotti G.V., Mulshine J.L., Kwon R., Curran W.J., Jr., Wu Y.L., Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311. doi: 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 3.Chatwal M.S., Tanvetyanon T. Combination chemotherapy and immunotherapy in metastatic non-small cell lung cancer: A setback for personalized medicine? Transl. Lung Cancer Res. 2018;7(Suppl 3):S208–S210. doi: 10.21037/tlcr.2018.07.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kris M.G., Gaspar L.E., Chaft J.E., Kennedy E.B., Azzoli C.G., Ellis P.M., Lin S.H., Pass H.I., Seth R., Shepherd F.A. Adjuvant systemic therapy and adjuvant radiation therapy for stage I to IIIA completely resected non-small-cell lung cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline update. J. Clin. Oncol. 2017;35:2960–2974. doi: 10.1200/JCO.2017.72.4401. [DOI] [PubMed] [Google Scholar]
- 5.Francolini G., Ferrari K., Scotti V. Neoadjuvant approach for nonsmall cell lung cancer: Overview of the current issues. Curr. Opin. Oncol. 2017;29:123–128. doi: 10.1097/CCO.0000000000000354. [DOI] [PubMed] [Google Scholar]
- 6.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 7.Vyas D., Laput G., Vyas A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther. 2014;7:1015–1023. doi: 10.2147/OTT.S60114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El Sharouni S.Y., Kal H.B., Battermann J.J. Accelerated regrowth of non-small-cell lung tumours after induction chemotherapy. Br. J. Cancer. 2003;89:2184–2189. doi: 10.1038/sj.bjc.6601418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karagiannis G.S., Condeelis J.S., Oktay M.H. Chemotherapy-induced metastasis: Molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res. 2019;79:4567–4576. doi: 10.1158/0008-5472.CAN-19-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaked Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer. 2019;19:667–685. doi: 10.1038/s41568-019-0209-6. [DOI] [PubMed] [Google Scholar]
- 11.Karagiannis G.S., Condeelis J.S., Oktay M.H. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin. Exp. Metastasis. 2018;35:269–284. doi: 10.1007/s10585-017-9870-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Alterio C., Scala S., Sozzi G., Roz L., Bertolini G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020;60:351–361. doi: 10.1016/j.semcancer.2019.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Bonapace L., Coissieux M.M., Wyckoff J., Mertz K.D., Varga Z., Junt T., Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–133. doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 14.Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keklikoglou I., Cianciaruso C., Güç E., Squadrito M.L., Spring L.M., Tazzyman S., Lambein L., Poissonnier A., Ferraro G.B., Baer C. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019;21:190–202. doi: 10.1038/s41556-018-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu G., Chen Y., Qi F., Jia L., Lu X.A., He T., Fu Y., Li L., Luo Y. Specific chemotherapeutic agents induce metastatic behaviour through stromal- and tumour-derived cytokine and angiogenic factor signalling. J. Pathol. 2015;237:190–202. doi: 10.1002/path.4564. [DOI] [PubMed] [Google Scholar]
- 17.Venneri M.A., De Palma M., Ponzoni M., Pucci F., Scielzo C., Zonari E., Mazzieri R., Doglioni C., Naldini L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276–5285. doi: 10.1182/blood-2006-10-053504. [DOI] [PubMed] [Google Scholar]
- 18.Squadrito M.L., De Palma M. Macrophage regulation of tumor angiogenesis: implications for cancer therapy. Mol. Aspects Med. 2011;32:123–145. doi: 10.1016/j.mam.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Hughes R., Qian B.Z., Rowan C., Muthana M., Keklikoglou I., Olson O.C., Tazzyman S., Danson S., Addison C., Clemons M. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75:3479–3491. doi: 10.1158/0008-5472.CAN-14-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scala S. Molecular pathways: Targeting the CXCR4-CXCL12 axis—Untapped potential in the tumor microenvironment. Clin. Cancer Res. 2015;21:4278–4285. doi: 10.1158/1078-0432.CCR-14-0914. [DOI] [PubMed] [Google Scholar]
- 21.Kawaguchi N., Zhang T.T., Nakanishi T. Involvement of CXCR4 in normal and abnormal development. Cells. 2019;8:185. doi: 10.3390/cells8020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao H., Guo L., Zhao H., Zhao J., Weng H., Zhao B. CXCR4 over-expression and survival in cancer: A system review and meta-analysis. Oncotarget. 2015;6:5022–5040. doi: 10.18632/oncotarget.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cojoc M., Peitzsch C., Trautmann F., Polishchuk L., Telegeev G.D., Dubrovska A. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013;6:1347–1361. doi: 10.2147/OTT.S36109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drenckhan A., Kurschat N., Dohrmann T., Raabe N., Koenig A.M., Reichelt U., Kaifi J.T., Izbicki J.R., Gros S.J. Effective inhibition of metastases and primary tumor growth with CTCE-9908 in esophageal cancer. J. Surg. Res. 2013;182:250–256. doi: 10.1016/j.jss.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 25.Tahirovic Y.A., Pelly S., Jecs E., Miller E.J., Sharma S.K., Liotta D.C., Wilson L.J. Small molecule and peptide-based CXCR4 modulators as therapeutic agents. A patent review for the period from 2010 to 2018. Expert Opin. Ther. Pat. 2020;30:87–101. doi: 10.1080/13543776.2020.1707186. [DOI] [PubMed] [Google Scholar]
- 26.Portella L., Vitale R., De Luca S., D’Alterio C., Ieranò C., Napolitano M., Riccio A., Polimeno M.N., Monfregola L., Barbieri A. Preclinical development of a novel class of CXCR4 antagonist impairing solid tumors growth and metastases. PLoS ONE. 2013;8:e74548. doi: 10.1371/journal.pone.0074548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alterio C., Zannetti A., Trotta A.M., Ieranò C., Napolitano M., Rea G., Greco A., Maiolino P., Albanese S., Scognamiglio G. New CXCR4 antagonist peptide R (Pep R) improves standard therapy in colorectal cancer. Cancers (Basel) 2020;12:1952. doi: 10.3390/cancers12071952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Alterio C., Buoncervello M., Ieranò C., Napolitano M., Portella L., Rea G., Barbieri A., Luciano A., Scognamiglio G., Tatangelo F. Targeting CXCR4 potentiates anti-PD-1 efficacy modifying the tumor microenvironment and inhibiting neoplastic PD-1. J. Exp. Clin. Cancer Res. 2019;38:432. doi: 10.1186/s13046-019-1420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng Y., Li B., Liang Y., Reeves P.M., Qu X., Ran C., Liu Q., Callahan M.V., Sluder A.E., Gelfand J.A. Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J. 2019;33:6596–6608. doi: 10.1096/fj.201802067RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang K., Li J., Zhang J., Wang L., Zhang Q., Ge J., Guo Y., Wang B., Huang Y., Yang T. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int. Immunopharmacol. 2019;75:105818. doi: 10.1016/j.intimp.2019.105818. [DOI] [PubMed] [Google Scholar]
- 31.Bertolini G., D’Amico L., Moro M., Landoni E., Perego P., Miceli R., Gatti L., Andriani F., Wong D., Caserini R. Microenvironment-modulated metastatic CD133+/CXCR4+/EpCAM− lung cancer-initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res. 2015;75:3636–3649. doi: 10.1158/0008-5472.CAN-14-3781. [DOI] [PubMed] [Google Scholar]
- 32.Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertolini G., Gatti L., Roz L. The “stem” of chemoresistance. Cell Cycle. 2010;9:628–629. doi: 10.4161/cc.9.4.10821. [DOI] [PubMed] [Google Scholar]
- 34.St Germain C., Niknejad N., Ma L., Garbuio K., Hai T., Dimitroulakos J. Cisplatin induces cytotoxicity through the mitogen-activated protein kinase pathways and activating transcription factor 3. Neoplasia. 2010;12:527–538. doi: 10.1593/neo.92048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandke P., Vasquez K.M. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: Implications in DNA repair and immune responses. DNA Repair (Amst.) 2019;83:102701. doi: 10.1016/j.dnarep.2019.102701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krynetskaia N.F., Phadke M.S., Jadhav S.H., Krynetskiy E.Y. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response to chemotherapy-induced DNA damage. Mol. Cancer Ther. 2009;8:864–872. doi: 10.1158/1535-7163.MCT-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bronte V., Brandau S., Chen S.H., Colombo M.P., Frey A.B., Greten T.F., Mandruzzato S., Murray P.J., Ochoa A., Ostrand-Rosenberg S. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitamura T., Qian B.Z., Soong D., Cassetta L., Noy R., Sugano G., Kato Y., Li J., Pollard J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015;212:1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura T., Doughty-Shenton D., Cassetta L., Fragkogianni S., Brownlie D., Kato Y., Carragher N., Pollard J.W. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front. Immunol. 2018;8:2004. doi: 10.3389/fimmu.2017.02004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buganim Y., Madar S., Rais Y., Pomeraniec L., Harel E., Solomon H., Kalo E., Goldstein I., Brosh R., Haimov O. Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis. 2011;32:1749–1757. doi: 10.1093/carcin/bgr203. [DOI] [PubMed] [Google Scholar]
- 41.Postmus P.E., Kerr K.M., Oudkerk M., Senan S., Waller D.A., Vansteenkiste J., Escriu C., Peters S., ESMO Guidelines Committee Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017;28(suppl_4):iv1–iv21. doi: 10.1093/annonc/mdx222. [DOI] [PubMed] [Google Scholar]
- 42.Linde N., Casanova-Acebes M., Sosa M.S., Mortha A., Rahman A., Farias E., Harper K., Tardio E., Reyes Torres I., Jones J. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018;9:21. doi: 10.1038/s41467-017-02481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian B., Deng Y., Im J.H., Muschel R.J., Zou Y., Li J., Lang R.A., Pollard J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakasone E.S., Askautrud H.A., Kees T., Park J.H., Plaks V., Ewald A.J., Fein M., Rasch M.G., Tan Y.X., Qiu J. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daenen L.G., Roodhart J.M., van Amersfoort M., Dehnad M., Roessingh W., Ulfman L.H., Derksen P.W., Voest E.E. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011;71:6976–6985. doi: 10.1158/0008-5472.CAN-11-0627. [DOI] [PubMed] [Google Scholar]
- 46.Yang J., Kumar A., Vilgelm A.E., Chen S.C., Ayers G.D., Novitskiy S.V., Joyce S., Richmond A. Loss of CXCR4 in myeloid cells enhances antitumor immunity and reduces melanoma growth through NK cell and FASL mechanisms. Cancer Immunol. Res. 2018;6:1186–1198. doi: 10.1158/2326-6066.CIR-18-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cassetta L., Baekkevold E.S., Brandau S., Bujko A., Cassatella M.A., Dorhoi A., Krieg C., Lin A., Loré K., Marini O. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019;68:687–697. doi: 10.1007/s00262-019-02302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levina V., Marrangoni A.M., DeMarco R., Gorelik E., Lokshin A.E. Drug-selected human lung cancer stem cells: Cytokine network, tumorigenic and metastatic properties. PLoS ONE. 2008;3:e3077. doi: 10.1371/journal.pone.0003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia D., Li L., Andrew S., Allan D., Li X., Lee J., Ji G., Yao Z., Gadde S., Figeys D., Wang L. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017;8:e2932. doi: 10.1038/cddis.2017.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Palma M., Lewis C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Karagiannis G.S., Pastoriza J.M., Wang Y., Harney A.S., Entenberg D., Pignatelli J., Sharma V.P., Xue E.A., Cheng E., D’Alfonso T.M. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017;9:eaan0026. doi: 10.1126/scitranslmed.aan0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alishekevitz D., Gingis-Velitski S., Kaidar-Person O., Gutter-Kapon L., Scherer S.D., Raviv Z., Merquiol E., Ben-Nun Y., Miller V., Rachman-Tzemah C. Macrophage-Induced lymphangiogenesis and metastasis following paclitaxel chemotherapy is regulated by VEGFR3. Cell Rep. 2016;17:1344–1356. doi: 10.1016/j.celrep.2016.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang Y.S., Jalgaonkar S.P., Middleton J.D., Hai T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA. 2017;114:E7159–E7168. doi: 10.1073/pnas.1700455114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang R., Zhang Q., Zeh H.J., 3rd, Lotze M.T., Tang D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang D., Kang R., Zeh H.J., 3rd, Lotze M.T. High-mobility group box 1 and cancer. Biochim. Biophys. Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Penzo M., Molteni R., Suda T., Samaniego S., Raucci A., Habiel D.M., Miller F., Jiang H.P., Li J., Pardi R. Inhibitor of NF-κB kinases α and β are both essential for high mobility group box 1-mediated chemotaxis [corrected] J. Immunol. 2010;184:4497–4509. doi: 10.4049/jimmunol.0903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venereau E., Schiraldi M., Uguccioni M., Bianchi M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013;55:76–82. doi: 10.1016/j.molimm.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 58.Schiraldi M., Raucci A., Muñoz L.M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012;209:551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kew R.R., Penzo M., Habiel D.M., Marcu K.B. The IKKα-dependent NF-κB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J. Immunol. 2012;188:2380–2386. doi: 10.4049/jimmunol.1102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner P.L., Hyjek E., Vazquez M.F., Meherally D., Liu Y.F., Chadwick P.A., Rengifo T., Sica G.L., Port J.L., Lee P.C. CXCL12 and CXCR4 in adenocarcinoma of the lung: Association with metastasis and survival. J. Thorac. Cardiovasc. Surg. 2009;137:615–621. doi: 10.1016/j.jtcvs.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 61.Kadota K., Nitadori J.I., Ujiie H., Buitrago D.H., Woo K.M., Sima C.S., Travis W.D., Jones D.R., Adusumilli P.S. Prognostic impact of immune microenvironment in lung squamous cell carcinoma: tumor-infiltrating CD10+ neutrophil/CD20+ lymphocyte ratio as an independent prognostic factor. J. Thorac. Oncol. 2015;10:1301–1310. doi: 10.1097/JTO.0000000000000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsao A.S., Jolly S., Lee J.M. Updates in local-regionally advanced non-small cell lung cancer. Am. Soc. Clin. Oncol. Educ. Book. 2019;39:553–562. doi: 10.1200/EDBK_237839. [DOI] [PubMed] [Google Scholar]
- 63.Chen I.X., Chauhan V.P., Posada J., Ng M.R., Wu M.W., Adstamongkonkul P., Huang P., Lindeman N., Langer R., Jain R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA. 2019;16:4558–4566. doi: 10.1073/pnas.1815515116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bockorny B., Semenisty V., Macarulla T., Borazanci E., Wolpin B.M., Stemmer S.M., Golan T., Geva R., Borad M.J., Pedersen K.S. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020;26:878–885. doi: 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.