

Abstract
Xanthine oxidase (XO) is the enzyme responsible for the conversion of endogenous purines into uric acid. Therefore, this enzyme has been associated with pathological conditions caused by hyperuricemia, such as the disease commonly known as gout. Barbiturates and their congeners thiobarbiturates represent a class of heterocyclic drugs capable of influencing neurotransmission. However, in recent years a very large group of potential pharmaceutical and medicinal applications have been related to their structure. This great diversity of biological activities is directly linked to the enormous opportunities found for chemical change off the back of these findings. With this in mind, sixteen bis-thiobarbiturates were synthesized in moderate to excellent reactional yields, and their antioxidant, anti-proliferative, and XO inhibitory activity were evaluated. In general, all bis-thiobarbiturates present a good antioxidant performance and an excellent ability to inhibit XO at a concentration of 30 µM, eight of them are superior to those observed with the reference drug allopurinol (Allo), nevertheless they were not as effective as febuxostat. The most powerful bis-thiobarbiturate within this set showed in vitro IC50 of 1.79 μM, which was about ten-fold better than Allo inhibition, together with suitable low cytotoxicity. In silico molecular properties such as drug-likeness, pharmacokinetics, and toxicity of this promising barbiturate were also analyzed and herein discussed.
Keywords: bis-thiobarbiturates, xanthine oxidase, antioxidant, cytotoxicity, in silico evaluation
1. Introduction
Xanthine oxidase (XO) is a molybdoflavoprotein widely disseminated throughout the human body, and is present in the liver, intestine, lungs, kidneys, heart, brain, and plasma [1]. Physiologically, XO catalyzes the oxidative hydroxylation of hypoxanthine and xanthine into uric acid (UA) with the concomitant production of reactive oxygen species (ROS) [1]. Therefore, this enzyme is an important source of superoxide radicals and hydrogen peroxide, which contributes to oxidative stress and takes part in the aging process. In addition, these ROS are involved in several pathological processes such as atherosclerosis and cancer [2,3]. Moreover, the excessive activity of XO leads to the overproduction of UA, increasing its concentration in the bloodstream and can result in hyperuricemia [4]. Hyperuricemia is a predisposing factor of gout, whether by excessive production of UA or under-excretion by the kidneys and is also considered a lifestyle-related syndrome. In fact, it has been associated with a high intake of foods rich in nucleic acids, such as red meats and seafood, leading to excessive production of UA [5,6].
Gout is a form of inflammatory arthritis characterized by the chronic deposition of monosodium urate crystals on the joints [4,6]. Hyperuricemia is a critical factor, not only for the development of gout but also for chronic nephritis, cardiovascular diseases, hypertension, type II diabetes mellitus, and metabolic syndrome [6]. The standard therapy for gout relies on urate-lowering drugs, especially those that target XO [4,6], such as allopurinol (Allo), febuxostat, and topiroxostat [1,6,7,8].
The synthesis and biological evaluation of nitrogen-based heterocyclic compounds has received increasing attention over the years [9]. Barbituric and thiobarbituric acids are examples of these heterocycles. Firstly synthesized by Adolph von Baeyer in 1864, barbituric acid was the key molecule for developing new derivatives with central nervous system (CNS) depressing activity in the twentieth century [10]. Initially used as anticonvulsant, anxiolytic, sedative-hypnotic and anesthetics [10], barbituric and thiobarbituric acid derivatives have recently been associated with several potential industrial and pharmaceutical/biological applications [10,11] namely as antifungal [12], antibacterial [13,14,15], anticancer [11,13,16,17], antiviral [18], as well as urease [19], XO [13], and helicase inhibitors [20]. Additionally, it has been established that this pyrimidine class of compounds undergoes Knoevenagel condensations with aldehydes to give 5-ylidene derivatives [13,21]. Furthermore, the formation of Michael adducts often follows Knoevenagel condensations with the formation of novel bis-thiobarbiturates [21]. On the other hand, bis-thiobarbiturates have been scarcely explored except that they have been established as urease inhibitors [22] and were tested as antibacterials but without any relevant activity [15].
2. Materials and Methods
2.1. Chemicals and Instrumentation
All reagents and solvents used were analytically pure and were used without purification. Ethanol was purchased from Honeywell (Paris, France), acetic acid from José M. Vaz Pereira, S. A. (Sintra, Portugal) and petroleum ether from Chem-Lab (Zedelgem, Belgium). Barbituric and 2-thiobarbituric acids and 2,4-dinitrobenzaldehyde were obtained from Alfa Aesar (Kandel, Germany), while 2-nitrobenzaldehyde was from Maybridge (Loughborough, United Kingdom). N,N-Diethyl-2-thiobarbituric acid, 4-formylbenzonitrile, 4-methoxybenzaldehyde, 5-hydroxy-2-nitrobenzaldehyde, 6-nitrobenzo[d][1,3]diaxole-5-carbaldehyde and 3-pyridinecarboxaldehyde were acquire from Acros Organics (Geel, Belgium). Benzaldehyde, 4-methylbenzaldehyde, 4-nitrobenzaldehyde, N-(4-formylphenyl)acetamide, 4-bromobenzaldehyde, 3-hydroxybenzaldehyde, 4-(dimethylamino)-2-nitrobenzaldehyde and α-methyl-trans-cinnamaldehyde were obtained from Sigma-Aldrich (St. Louis, MO, USA).
All reactions were monitored by thin-layer chromatography on precoated silica-gel aluminum plates of 0.2 mm (Macherey-Nagel 60 G/UV254, Düren, Germany). After the elution, the plates’ observation was performed under ultraviolet (UV) light with a wavelength of 254 and/or 365 nm.
The melting points (mp) were determined in open capillary tubes using a Büchi B-540 mp apparatus and were not corrected.
Proton (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were performed on a Bruker Avance III 400 MHz spectrophotometer and were processed by the software MestReNova 14.2.0 lite (Mestrelab Research S.L., Santiago de Compostela, Spain). Deuterated chloroform (CDCl3, Acros Organics, Geel, Belgium) or hexadeuterodimethyl sulfoxide (DMSO-d6, Eurisotop, Gif-sur-Yvette, France) were used as a solvent and internal standard, δ = 7.26 and 77.16 ppm or 2.50 and 39.52 ppm in 1H and 13C NMR, respectively. The chemical shift (δ) values are given in parts per million (ppm) and coupling constants (J) in Hertz (Hz). The multiplicity of the signals is reported as singlet (s), doublet (d), doublet of doublets (dd), doublet of triplets (dt), doublet of doublet of triplets (ddt), triplet (t), triplet of doublets (td), triplet of triplets (tt), quartet (q), quartet of doublets (qd), or multiplet (m).
High-resolution mass spectrometry (HRMS) was performed for new compounds by electrospray ionization time-of-flight (ESI-TOF) at CACTI services from the University of Vigo (Spain).
2.2. Synthesis of Bis-thiobarbiturates 3–19
A mixture of barbituric acid 1c or thiobarbituric acids 1a-b (2.0 mmol) and a benzaldehyde 2a-m or aldehydes 2n-o (1.0 mmol) in ethanol (5 mL) was stirred for two to six hours, at room temperature (rt) or by reflux [22] or alternatively in acetic acid (5 mL) at 80 °C [23]. The obtained solid was filtered, washed with cold ethanol and petroleum ether 40–60 °C, dried, and recrystallized from ethanol to afford the following bis-barbiturate and bis-thiobarbiturates 3–19.
5,5′-(Phenylmethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (3)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and benzaldehyde (2a, 1.0 mmol, 106.1 mg, 101.6 µL) in ethanol at rt; yellow solid (434.9 mg, 89% yield); mp: 177–179 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.32 (t, J = 7.7 Hz, 2H, 2 × ArCH), 7.26 (t, J = 7.4 Hz, 1H, ArCH), 7.13 (d, J = 7.9 Hz, 2H, 2 × ArCH), 5.68 (s, 1H, 5-CCH), 4.76–4.50 (m, 8H, 4 × NCH2CH3), 1.38 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S4. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.71 (2 × 2-CS), 163.84 (2 × CO), 162.38 (2 × CO), 135.62 (ArC), 128.58 (2 × ArCH), 126.90 (ArCH), 126.43 (2 × ArCH), 97.55 (2 × 5-C), 45.26 (2 × NCH2CH3), 44.70 (2 × NCH2CH3), 35.03 (5-CCH), 12.20 (2 × NCH2CH3), 12.12 (2 × NCH2CH3); Figure S5.
5,5′-(p-Tolymethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (4)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-methylbenzaldehyde (2b, 1.0 mmol, 120.2 mg, 118.0 µL) in ethanol at rt; yellow solid (397.1 mg, 79% yield); mp: 163–164 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.12 (d, J = 8.1 Hz, 2H, 2 × ArCH), 7.00 (d, J = 8.3 Hz, 2H, 2 × ArCH), 5.63 (s, 1H, 5-CCH), 4.91–4.47 (m, 8H, 4 × NCH2CH3), 2.34 (s, 3H, ArCCH3), 1.38 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S6. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.66 (2 × 2-CS), 163.79 (2 × CO), 162.34 (2 × CO), 136.46 (ArC), 132.44 (ArC), 129.28 (2 × ArCH), 126.29 (2 × ArCH), 97.67 (2 × 5-C), 45.23 (2 × NCH2CH3), 44.65 (2 × NCH2CH3), 34.71 (5-CCH), 21.09 (ArCCH3), 12.19 (2 × NCH2CH3), 12.11 (2 × NCH2CH3); Figure S7.
4-(Bis(1,3-diethyl-6-hydroxy-4-oxo-2-thioxo-1,2,3,4-tetrtahydropyrimidin-5-yl)methyl)benzonitrile (5)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-formylbenzonitrile (2c, 1.0 mmol, 131.1 mg) in ethanol at rt; white solid (400.6 mg, 78% yield); mp: 199–200 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.62 (d, J = 8.5 Hz, 2H, 2 × ArCH), 7.27 (d, J = 8.5 Hz, 2H, 2 × ArCH), 5.66 (s, 1H, 5-CCH), 4.90–4.42 (m, 8H, 4 × NCH2CH3), 1.37 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.28 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S8. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.71 (2 × 2-CS), 163.93 (2 × CO), 162.35 (2 × CO), 141.76 (ArC), 132.40 (2 × ArCH), 127.42 (2 × ArCH), 118.74 (ArCCN), 110.92 (ArC), 96.73 (2 × 5-C), 45.35 (2 × NCH2CH3), 44.80 (2 × NCH2CH3), 35.43 (5-CCH), 12.14 (2 × NCH2CH3), 12.12 (2 × NCH2CH3); Figure S9.
5,5′-((4-Nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropirimidin-4(1H)-one) (6)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-nitrobenzaldehyde (2d, 1.0 mmol, 151.1 mg) in ethanol at rt; yellow solid (416.2 mg, 78% yield); mp: 202–204 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.18 (d, J = 8.9 Hz, 2H, 2 × ArCH), 7.33 (d, J = 8.9 Hz, 2H, 2 × ArCH), 5.69 (s, 1H, 5-CCH), 4.77–4.38 (m, 8H, 4 × NCH2CH3), 1.37 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S10. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.72 (2 × 2-CS), 163.93 (2 × CO), 162.39 (2 × CO), 146.92 (ArC), 143.88 (ArC), 127.56 (2 × ArCH), 123.82 (2 × ArCH), 96.82 (2 × 5-C), 45.38 (2 × NCH2CH3), 44.82 (2 × NCH2CH3), 35.43 (5-CCH), 12.14 (2 × NCH2CH3), 12.12 (2 × NCH2CH3); Figure S11.
5,5′-((4-Nitrophenyl)methylene)bis(6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (7)
From 2-thiobarbituric acid (1b, 2.0 mmol, 258.6 mg) 4-nitrobenzaldehyde (2d) (1.0 mmol, 152.3 mg) in ethanol at reflux; pale yellow solid (303.5 mg; 72% yield); mp: 225 °C dec.; 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.76 (s, 4H, 4 × NH), 8.07 (d, J = 8.7 Hz, 2H, 2 × ArCH), 7.27 (d, J = 8.5 Hz, 2H, 2 × ArCH), 6.04 (s, 1H, 5-CCH); Figure S12. 13C NMR (101 MHz, DMSO-d6) δ (ppm) 173.16 (2 × 2-CS), 163.04 (4 × CO), 151.99 (ArC), 145.34 (ArC), 127.88 (2 × ArCH), 123.16 (2 × ArCH), 95.28 (2 × 5-C), 31.29 (5-CCH); Figure S13.
5,5′-((4-Nitrophenyl)methylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) (8)
From barbituric acid (1c, 2.0 mmol, 231.8 mg) 4-nitrobenzaldehyde (2d) (1.0 mmol, 152.3 mg) in ethanol at reflux during six hours; white solid (319.2 mg, 82% yield); mp: 227 °C dec.; 1H NMR (400 MHz, DMSO-d6) δ (ppm) 10.74 (s, 4H, 4 × NH), 8.08 (d, J = 8.7 Hz, 2H, 2 × ArCH), 7.37 (d, J = 8.4 Hz, 2H, 2 × ArCH), 5.82 (s, 1H, 5-CCH); Figure S14.13C NMR spectra was not herein present due to a rapid product decomposition in DMSO-d6 solution.
N-(4-(Bis(1,3-diethyl-6-hydroxy-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)phenyl)acetamide (9)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and N-(4-formylphenyl)acetamide (2e, 1.0 mmol, 167.2 mg) in ethanol at rt; yellow solid (485.6 mg, 89% yield); mp: 178–179 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.47 (d, J = 8.3 Hz, 2H, 2 × ArCH), 7.17 (s, 1H, NHCOCH3), 7.06 (d, J = 8.1 Hz, 2H, 2 × ArCH), 5.62 (s, 1H, 5-CCH), 4.74–4.52 (m, 8H, 4 × NCH2CH3), 2.17 (s, 3H, NHCOCH3), 1.37 (t, J = 6.9 Hz, 6H, 2 × NCH2CH3), 1.28 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S15. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.47 (2 × 2-CS), 168.76 (NHCOCH3), 163.59 (2 × CO), 162.13 (2 × CO), 137.25 (ArC), 130.59 (ArC), 126.72 (2 × ArCH), 119.64 (2 × ArCH), 97.40 (2 × 5-C), 45.06 (2 × NCH2CH3), 44.48 (2 × NCH2CH3), 34.45 (5-CCH), 24.37 (NHCOCH3), 12.01 (2 × NCH2CH3), 11.95 (2 × NHCH2CH3); Figure S16. HMRS (ESI-TOF): m/z [M + H]+ calcd for C25H32N5O5S2: 546.1839; found: 546.1840.
5,5′-((4-Methoxyphenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (10)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-methoxybenzaldehyde (2f, 1.0 mmol, 136.2 mg) in acetic acid at 80 °C; yellow solid (352.7 mg, 68% yield); mp: 139–140 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.02 (dd, J = 8.8, 1.0 Hz, 2H, 2 × ArCH), 6.84 (d, J = 8.8 Hz, 2H, 2 × ArCH), 5.62 (s, 1H, 5-CCH), 4.75–4.45 (m, 8H, 4 × NCH2CH3), 3.80 (s, 3H, OCH3), 1.37 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S17. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.67 (2 × 2-CS), 163.78 (2 × CO), 162.34 (2 × CO), 158.44 (ArC), 127.54 (2 × ArCH), 127.34 (ArC), 113.95 (2 × ArCH), 97.76 (2 × 5-C), 55.39(OCH3), 45.26 (2 × NCH2CH3), 44.69 (2 × NCH2CH3), 34.36 (5-CCH), 12.20 (2 × NCH2CH3), 12.14 (2 × NCH2CH3); Figure S18.
5,5′-((4-Bromophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (11)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-bromobenzaldehyde (2g, 1.0 mmol, 185.0 mg) in ethanol at rt; white solid (402.9 mg, 64% yield); mp: 169–170 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.43 (dt, J = 8.6, 2.6, 1.9 Hz, 2H, 2 × ArCH), 7.01 (dt, J = 8.6, 2.7, 1.8 Hz, 2H, 2 × ArCH), 5.59 (s, 1H, 5-CCH), 4.81–4.45 (m, 8H, 4 × NCH2CH3), 1.37 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S19. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.71 (2 × 2-CS), 163.87 (2 × CO), 162.33 (2 × CO), 134.91 (ArC), 131.66 (2 × ArCH), 128.33 (2 × ArCH), 120.79 (ArC), 97.19 (2 × 5-C), 45.31 (2 × NCH2CH3), 44.75 (2 × NCH2CH3), 34.78 (5-CCH), 12.18 (2 × NCH2CH3), 12.13 (2 × NCH2CH3); Figure S20.
5,5′-((3-Hydroxyphenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (12)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 3-hydroxybenzaldehyde (2h, 1.0 mmol, 123.9 mg) in ethanol at rt; yellow solid (183.9 mg, 73% yield); mp: 191–193 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.12 (t, J = 7.9 Hz, 1H, ArCH), 6.68 (ddt, J = 8.0, 2.1, 1.0 Hz, 1H, ArCH), 6.60 (dt, J = 8.2, 1.9, 0.9 Hz, 1H, ArCH), 6.58 (dd, J = 2.1, 1.0 Hz, 1H, ArCH), 5.57 (s, 1H, 5-CCH), 4.75–4.44 (m, 8H, 4 × NCH2CH3), 1.34 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.26 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S21. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.58 (2 × 2-CS), 163.61 (2 × CO), 162.28 (2 × CO), 157.36 (ArC), 137.22 (ArC), 129.35 (ArCH), 117.49 (ArCH), 113.93 (ArCH), 113.77 (ArCH), 97.52 (2 × 5-C), 45.11 (2 × NCH2CH3), 44.60 (2 × NCH2CH3), 34.85 (5-CCH), 12.12 (2 × NCH2CH3), 12.04 (2 × NCH2CH3); Figure S22.
5,5′-((2-Nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (13)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 2-nitrobenzaldehyde (2i, 1.0 mmol, 151.1 mg) in ethanol at rt; rose solid (474.9 mg, 89% yield); mp: 172–173 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.56 (dd, J = 7.8, 1.4 Hz, 1H, ArCH), 7.52 (td, J = 7.7, 1.5 Hz, 1H, ArCH), 7.42 (tt, J = 7.6, 1.1 Hz, 1H, ArCH), 7.28 (dt, J = 7.9, 1.1 Hz, 1H, ArCH), 6.11 (s, 1H, 5-CCH), 4.63 (q, J = 7.0 Hz, 4H, 2 × NCH2CH3), 4.59–4.47 (m, 4H, 2 × NCH2CH3), 1.36 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.28 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S23. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.56 (2 × 2-CS), 163.77 (2 × CO), 162.15 (2 × CO), 150.18 (ArC), 131.44 (ArCH), 129.64 (ArCH), 129.42 (ArC), 128.18 (ArCH), 124.24 (ArCH), 96.73 (2 × 5-C), 45.22 (2 × NCH2CH3), 44.73 (2 × NCH2CH3), 32.75 (5-CCH), 12.04 (2 × NCH2CH3), 11.87 (2 × NCH2CH3); Figure S24.
5,5′-((4-(Dimethylamino)-2-nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (14)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 4-(dimethylamino)-2-nitrobenzaldehyde (2j, 1.0 mmol, 200.2 mg) in ethanol at rt; red solid (490.2 mg, 85% yield); mp: 143–145 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.10 (d, J = 8.3 Hz, 1H, ArCH), 7.06–6.96 (m, 2H, 2 × ArCH), 6.01 (s, 1H, 5-CCH), 4.63 (q, J = 7.1 Hz, 4H, 2 × NCH2CH3), 4.57–4.45 (m, 4H, 2 × NCH2CH3), 3.03 (s, 6H, N(CH3)2), 1.36 (t, J = 6.8 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.1 Hz, 6H, 2 × NCH2CH3); Figure S25. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.54 (2 × 2-CS), 163.71 (2 × CO), 162.12 (2 × CO), 150.87 (ArC), 147.87 (ArC), 130.62 (ArCH), 118.95 (ArC), 116.46 (ArCH), 109.51 (ArCH), 97.00 (2 × 5-C), 45.22 (2 × NCH2CH3), 44.72 (2 × NCH2CH3), 41.78 (N(CH3)2), 32.12 (5-CCH), 12.08 (2 × NCH2CH3), 11.94 (2 × NCH2CH3); Figure S26. HMRS (ESI-TOF): m/z [M + H]+ calcd for C25H33N6O6S2: 577.1898; found: 577.1891.
5,5′-((2,4-Dinitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (15)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 2,4-dinitrobenzaldehyde (2k, 1.0 mmol, 196.1 mg) in ethanol at rt; yellow solid (434.0 mg, 75% yield); mp: 169–170 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.41 (d, J = 2.3 Hz, 1H, ArCH), 8.37 (dd, J = 8.7, 2.4 Hz, 1H, ArCH), 7.52 (dd, J = 8.7, 1.3 Hz, 1H, ArCH), 6.11 (s, 1H, 5-CCH), 4.63 (q, J = 7.0 Hz, 4H, 2 × NCH2CH3), 4.59–4.46 (m, 4H, 2 × NCH2CH3), 1.37 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.29 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S27. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.57 (2 × 2-CS), 163.85 (2 × CO), 162.21 (2 × CO), 149.93 (ArC), 146.87 (ArC), 137.06 (ArC), 131.25 (ArCH), 125.67 (ArCH), 119.58 (ArCH), 95.99 (2 × 5-C), 45.34 (2 × NCH2CH3), 44.87 (2 × NCH2CH3), 33.34 (5-CCH), 12.01 (2 × NCH2CH3), 11.89 (2 × NCH2CH3); Figure S28.
5,5′-((5-Hydroxy-2-nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (16)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 5-hydroxy-2-nitrobenzaldehyde (2l, 1.0 mmol, 167.1 mg) in ethanol at rt; yellow solid (505.7 mg, 92% yield); mp: 167–169 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.53 (d, J = 8.6 Hz, 1H, ArCH), 6.74 (dd, J = 8.6, 2.5 Hz, 1H, ArCH), 6.70 (d, J = 2.2 Hz, 1H, ArCH), 6.10 (s, 1H, 5-CCH), 4.58 (q, J = 7.0 Hz, 4H, 2 × NCH2CH3), 4.49 (m, 4H, 2 × NCH2CH3), 1.31 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.22 (t, J = 6.9 Hz, 6H, 2 × NCH2CH3); Figure S29. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.35 (2 × 2-CS), 163.51 (2 × CO), 161.86 (2 × CO), 160.76 (ArC), 141.96 (ArC), 132.45 (ArC), 127.17 (ArCH), 116.99 (ArCH), 113.93 (ArCH), 97.01 (2 × 5-C), 45.04 (2 × NCH2CH3), 44.60 (2 × NCH2CH3), 33.01 (5-CCH), 11.96 (2 × NCH2CH3), 11.76 (2 × NCH2CH3); Figure S30. HMRS (ESI-TOF): m/z [M + H]+ calcd for C23H28N5O7S2: 550.1425; found: 550.1417.
5,5′-((6-Nitrobenzo[d][1,3]dioxol-5-yl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (17)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 6-nitrobenzo[d][1,3]diaxole-5-carbaldehyde (2m, 1.0 mmol, 199.1 mg) in ethanol at rt; yellow solid (429.4 mg, 77% yield); mp: 148–149 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.11 (s, 1H, ArCH), 6.67 (d, J = 1.1 Hz, 1H, ArCH), 6.11 (s, 2H, OCH2O), 6.10 (d, J = 1.1 Hz, 1H, 5-CCH), 4.68–4.46 (m, 8H, 4 × NCH2CH3), 1.36 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.28 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S31. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.50 (2 × 2-CS), 163.79 (2 × CO), 162.06 (2 × CO), 150.44 (ArC), 146.72 (ArC), 144.08 (ArC), 125.75 (ArC), 109.24 (OCH2O), 105.84 (ArCH), 103.09 (ArCH), 97.06 (2 × 5-C), 45.24 (2 × NCH2CH3), 44.73 (2 × NCH2CH3), 32.83 (5-CCH), 12.03 (2 × NCH2CH3), 11.88 (2 × NCH2CH3); Figure S32. HMRS (ESI-TOF): m/z [M + H]+ calcd for C24H28N5O8S2: 578.1374; found: 578.1381.
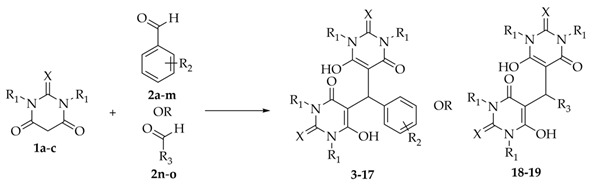
5,5′-(2-Methyl-3-phenylprop-2-ene-1,1-diyl)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (18)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and α-methyl-trans-cinnamaldehyde (2n, 1.0 mmol, 150.5 mg, 145.1 µL) in ethanol at rt; yellow solid (264.3 mg, 50% yield); mp: 145–146 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.34 (t, J = 7.8 Hz, 2H, 2 × ArCH), 7.25–7.19 (m, 3H, 3 × ArCH), 6.27 (s, 1H, 5-CCH), 5.01 (q, J = 2.7, 1.4 Hz, 1H, C=CH), 4.77–4.51 (m, 8H, 4 × NCH2CH3), 1.77 (t, J = 1.3 Hz, 3H, CCH3), 1.36 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3), 1.31 (t, J = 7.0 Hz, 6H, 2 × NCH2CH3); Figure S33. 13C NMR (101 MHz, CDCl3) δ (ppm) 174.62 (2 × 2-CS), 163.61 (2 × CO), 162.39 (2 × CO), 138.02 (ArC), 131.24 (C=CH), 129.04 (2 × ArCH), 128.26 (2 × ArCH), 126.72 (ArCH), 126.60 (C=CH), 97.53 (2 × 5-C), 45.26 (2 × NCH2CH3), 44.72 (2 × NCH2CH3), 38.27 (5-CCH), 17.19 (CCH3), 12.19 (2 × NCH2CH3), 12.15 (2 × NCH2CH3); Figure S34. HMRS (ESI-TOF): m/z [M + H]+ calcd for C26H33N4O4S2: 529.1938; found: 529.1939.
5,5′-(Pyridin-3-ylmethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (19)
From N,N-diethyl-2-thiobarbituric acid (1a, 2.0 mmol, 400.6 mg) and 3-pyridinecarboxaldehyde (2o, 1.0 mmol, 109.3 mg, 95.9 µL) in ethanol at rt; yellow solid (264.4 mg, 54% yield); mp: 253–254 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.69 (d, J = 5.6 Hz, 1H, ArCH), 8.58 (s, 1H, ArCH), 8.27 (d, J = 8.2 Hz, 1H, ArCH), 7.92 (dd, J = 8.2, 5.5 Hz, 1H, ArCH), 6.43 (s, 1H, 5-CCH), 4.44 (qd, J = 13.0, 6.4 Hz, 8H, 4 × NCH2CH3), 1.17 (t, J = 6.8 Hz, 12H, 4 × NCH2CH3); Figure S35. 13C NMR (101 MHz, DMSO-d6) δ (ppm) 174.65 (2 × 2-CS), 161.13 (4 × CO), 144.71 (ArCH), 143.15 (ArC), 140.14 (ArCH), 139.24 (ArCH), 126.70 (ArCH), 94.24 (2 × 5-C), 43.12 (4 × NCH2CH3), 32.24 (5-CCH), 12.30 (4 × NCH2CH3); Figure S36.
2.3. In Vitro Studies
XO inhibitory and antioxidant assays were performed in triplicate while antiproliferative assay was conducted in quadruplicate. For each assay, at least two independent experiments were performed. An initial screening at the concentration of 30 μM for all compounds under study was performed in each in vitro study. A second screening at 5 μM was performed for compounds that originated an inhibitory potential higher than 80% for the XO activity at 30 μM. Half-maximal inhibitory concentration (IC50) studies were performed for drugs used as a reference and for the most promising bis-thiobarbiturate in XO inhibitory, antioxidant, and cytotoxicity assays.
2.3.1. Solutions Preparation
For in vitro studies, all bis-thiobarbiturates, Allo, febuxostat, Trolox, and 5-fluorouracil (5-FU) were dissolved in DMSO at the concentration of 10 mM. Additionally, a 10 mM xanthine solution was prepared in a 25 mM sodium hydroxide solution. All solutions were kept at a temperature of 4 °C before each experiment. Allo, febuxostat, Trolox, and 5-FU were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.3.2. XO Inhibitory Assay
The XO inhibitory activity was evaluated by spectrophotometric quantification of uric acid formation [13]. The 50 mM dihydrogen phosphate buffer (pH 7.4) was used to dilute all solutions. For each assay performed, 50 μL of the test solution and 50 μL of the 0.1 U/mL XO (Sigma-Aldrich X4875, St. Louis, MO, USA) solution were added in each well of an Elisa microplate (96 wells) followed by 5 min of incubation at 37 °C. Final concentrations of 50, 25, 10, 5, 1, and 0.5 µM for Allo, 0.5, 0.075, 0.05, 0.025, 0.01, and 0.001 µM for febuxostat and 10, 5, 2.5, 1, 0.5, and 0.1 µM for bis-thiobarbiturate 11 were used for IC50 determinations. The reaction started with the addition of 150 μL of a xanthine solution (420 μM). The absorbance was recorded at a wavelength of 295 nm every minute for 10 min. To obtain only the absorbance associated with uric acid, a solution consisting of 50 μL of the test solution, 150 μL of the xanthine solution, and 50 μL of buffer were used as blank. Additionally, the dihydrogen phosphate buffer was used as a negative control and Allo and febuxostat as positive controls. For each compound, the percentage of enzyme inhibition was calculated according to the following formula:
| % of XO inhibition = [1 − (ABSsample − ABSblank of sample)/ABSnegative control] × 100 |
2.3.3. Antioxidant Assay
Antioxidant potential was spectrophotometrically evaluated by the 2,2-diphenyl-1-picrylhydrazyl (DPPH, Sigma-Aldrich, St. Louis, MO, USA) method [13]. All solutions and dilutions were prepared in 99.5% ethanol before each experiment. For each assay, 100 μL of the test solution and 100 μL of DPPH solution (0.2 mM) were added to each well of a 96 wells Elisa microplate. Final concentrations of 120, 60, 60, 15, 7.5, 3.75, and 1 µM for Trolox and bis-thiobarbiturate 11 were used for IC50 determinations. After 60 min of incubation in dark at rt, the capacity of each compound to reduce DPPH was followed by measuring the absorbance at 517 nm. Ethanol was used as a negative control and Trolox as the positive control. To discount the absorbance of each compound at 517 nm, a blank was performed with 100 μL of each test compound and 100 μL of ethanol. The antioxidant capacity of each sample was calculated according to the following formula:
| % DPPH scavenging = [1 − (ABSsample − ABSblank of sample)/ABSnegative control] × 100 |
2.3.4. Cytotoxicity Assay
The cytotoxic potential of the compounds being studied was evaluated by quantifying the extent of the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, VWR, Radnor, PA, USA) [24] on tumor cell lines of the colon (Caco-2) and breast (MCF-7) adenocarcinoma and non-tumor human dermal fibroblasts (NHDF). All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in 75 cm2 culture flasks in a humidified air incubator with 5% CO2 at 37 °C. NHDF cells have grown in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 mM L-glutamine, 1 mM sodium pyruvate, and 1% antibiotic/antimycotic (Ab: 10,000 units/mL penicillin G, 100 mg/mL streptomycin and 25 μg/mL amphotericin B). MCF-7 cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS, and 1% Ab. The Caco-2 cell line was cultured in a high glucose DMEM supplemented with 20% FBS and 1% of the antibiotic mixture (Sp: 10,000 units/mL penicillin G and 100 mg/mL of streptomycin). For the assay, cells were seeded in 96-well plates (2 × 104 cells/mL) in the culture medium. After 48 h of adherence, cells were treated with the test solutions and incubated for 72 h. Final concentrations of 200, 100, 50, 10, 1, and 0.1 µM for bis-thiobarbiturate 11 were used for IC50 determinations. Untreated cells were used as a negative control and 5-FU as the positive control. Following incubation, the medium was removed and replaced with a fresh incomplete culture medium (without FBS and Ab or Sp) and MTT solution [5 mg/mL in phosphate buffer saline (PBS)]. After further incubation for 4 h at 37 °C, the medium with MTT was removed, formazan crystals were dissolved in DMSO, and the absorbance was read at 570 nm. Results were expressed as the relative cell proliferation in comparison with the negative control cells.
2.3.5. Statistics
All in vitro results are expressed as mean values ± standard deviation (SD) of at least two independent determinations. The difference between groups was analyzed for each assay by Student’s t-test. The IC50 values were calculated by sigmoidal fitting analysis considering a 95% confidence interval.
2.4. In Silico Studies
Drug-likeness of bis-thiobarbiturate 11 was verified by the free web tool SwissADME [25]. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters were evaluated at SwissADME [25] and pkCSM [26] web tools.
3. Results and Discussion
Bis-diethylthiobarbiturates 3–6, 9, and 11–19 were synthesized by the method previously described [22]; although, and to the best of our knowledge, compounds 9, 14, 16–18 have never been described in the literature. These conditions are characterized by a short reaction time and simplicity of reaction conditions, affording the desired products with a high degree of purity even without crystallization. Furthermore, this method was carried out at rt, and the product was easily isolated by filtration, in moderate to excellent reactional yields, from 50 to 92% (Table 1). Bis-diethylthiobarbiturate 10, bis-thiobarbiturate 7, and bis-barbiturate 8 were synthesized by modified processes once the conditions used at rt were not successfully in affording the respective arylidene. Thus, bis-diethylthiobarbiturate 10 was synthesized in acetic acid at 80 °C [23], and bis-(thio)barbiturates 7–8 were synthesized in ethanol at reflux temperature. The formation of bis-(thio)barbiturates 3–19 was confirmed by NMR, since a singlet in the range of 5.58 to 6.43 ppm and a signal from 31.3 to 38.3 ppm in 1H and 13C NMR spectra, respectively, were obtained for the aryltrisubstituted methyne group. On the other hand, when arylidene derivatives were formed, a singlet at 8 ppm in 1H NMR and 153 ppm in 13C NMR were observed [13]. Despite the synthesis and isolation of bis-barbiturate 8, a DMSO-d6 solution rapid decomposition to the barbituric acid (1c) and respective arylidene was observed (Scheme S1), in accordance with the literature [27]. Therefore, bis-barbiturate 8 was not used in further biological studies.
Table 1.
Chemical synthesis, structure and reactional yields of bis-(thio)barbiturates 3–19.
| ||||||
|---|---|---|---|---|---|---|
| Bis-(thio) Barbiturate | Starting Material | X | R1 | R2 | R3 | Yield (%) |
| 3 | 1a + 2a | S | Et | H | - | 89 |
| 4 | 1a + 2b | S | Et | 4-CH3 | - | 79 |
| 5 | 1a + 2c | S | Et | 4-CN | - | 78 |
| 6 | 1a + 2d | S | Et | 4-NO2 | - | 78 |
| 7 | 1b + 2d | S | H | 4-NO2 | - | 72 |
| 8 | 1c + 2d | O | H | 4-NO2 | - | 82 |
| 9 | 1a + 2e | S | Et | 4-NHCOCH3 | - | 89 |
| 10 | 1a + 2f | S | Et | 4-OCH3 | - | 68 |
| 11 | 1a + 2g | S | Et | 4-Br | - | 71 |
| 12 | 1a + 2h | S | Et | 3-OH | - | 73 |
| 13 | 1a + 2i | S | Et | 2-NO2 | - | 78 |
| 14 | 1a + 2j | S | Et | 2-NO2, 4-N(CH3)2 | - | 85 |
| 15 | 1a + 2k | S | Et | 2,4-NO2 | - | 75 |
| 16 | 1a + 2l | S | Et | 2-NO2, 5-OH | - | 92 |
| 17 | 1a + 2m | S | Et | 2-NO2, 4,5-OCH2O | - | 77 |
| 18 | 1a + 2n | S | Et | - |
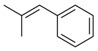
|
50 |
| 19 | 1a + 2o | S | Et | - |
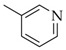
|
54 |
The XO inhibitory assay of the synthesized bis-thiobarbiturates was performed by a spectrophotometric method at 295 nm, and commercial febuxostat and Allo were used as positive controls. The study started with a screening at the concentration of 30 µM, and all sixteen bis-thiobarbiturates demonstrated potential as XO inhibitors (Figure 1 and Table S1), with inhibitions from 14.92 to 95.72% (88.51% for Allo and 95.03% for febuxostat). After this study, an additional screening at 5 µM was performed for the target compounds that presented an inhibition higher than 80% at 30 µM. Thus, the identification of the most promising bis-thiobarbiturates under study for further concentration-response studies was performed.
Figure 1.

In vitro XO inhibitory activity of bis-thiobarbiturates 3–7 and 9–19 and references febuxostat and Allo. Results are expressed as average values ± SD of two independent determinations, and each one was performed in triplicate. A p < 0.01 versus the negative control in the statistical significance analysis (Student’s t-test) was observed for all compounds.
In order to perform some structure-activity relationships inferences, firstly the influence of N substitution in the thiobarbituric acid moiety was analyzed. Results demonstrated that N substitution with ethyl group at thiobarbiturate moiety led to an increment in XO inhibitory activity from 20.86 to 92.25% (7 versus 6). Taking both results in mind, several bis-diethylthiobarbiturates with different substituent groups mainly at para position of phenyl moiety were analyzed in detail. In this context, electron-withdrawing groups appear to intensify the XO inhibitory potential in relation to electron-donating counterparts. In fact, the presence of nitrile, nitro, or bromo groups at para position increases the inhibitory activity from 80.15% (unsubstituted 3) to 86.12, 92.25, and 95.72% at 30 µM, and from 10.42% to 52.92, 56.82, and 80.92% at 5 µM, for 5, 6, and 11, respectively. On the other hand, the addition of a methyl, acetamide or methoxy group at the same position reduces the inhibitory activity to 36.47, 45.61, and 23.53 at 30 µM, for 4, 9, and 10 respectively. Besides these effects observed for different groups in the para position, a small decrease in the inhibitory activity was observed when the same group was in the ortho position. This effect is notoriously noticed for the bis-thiobarbiturates para-nitro and ortho-nitro substituted pair, where the values of XO inhibitions are 56.82 and 37.42% at 5 µM, for 6 and 13, respectively.
The analysis of the influence of the presence of di and tri substitutions in phenyl moiety on the percentage of XO inhibition seems not to be straightforward. Indeed, the additional presence of a dimethylamine group at the para position or two methylenedioxy groups at the meta and para positions of the ortho-nitro group substituted seems to increase the XO inhibitory activity at 5 µM (14 and 17 versus 13). However, the additional presence of a para-nitro or meta -hydroxyl decreases the XO inhibitory activity (15 and 16 versus 13). A deeply notorious reduction of XO inhibitory activity at 30 µM was observed with a double nitro substitution (14.92%; 15) concerning the related ortho- (89.73%; 13) or para- (92.25%; 6) mono-nitro congener. Additionally, this effect was also noted for the bis-thiobarbiturates 12 (72.25%; 3-hydroxyl) and 13 (89.73%; 2-nitro) when compared with their related bis-thiobarbiturates 16 (61.44%; 2-nitro, 5-hydroxyl).
Finally, the replacement of the phenyl (bis-thiobarbiturate 3) moiety for 2-methyl-3-phenylprop-2-enyl or pyridin-3-yl showed a promising increment for activity at 5 µM by about six-fold (3 versus 18 and 19).
In conclusion, bis-thiobarbiturates 5, 6, 11, 13, 14, 17, 18, and 19 evidence a percentage of XO inhibition higher than Allo at 5 µM; however, this is still lower than the second positive control, febuxostat. Generally, the most active compounds present electron-withdrawing or halogen substituents on the phenyl ring. Nevertheless, the effect of further electron-withdrawing and/or electron-donating group substitutions must be carefully balanced.
The halogenated bis-thiobarbiturate 11 demonstrated that it was the most promising within this set of tested compounds. Further concentration-response studies for 11 showed the inhibition of XO was activity-dependent on the concentration, presenting a calculated IC50 value of 1.79 μM (Table 2 and Figure S1), being approximately ten-fold more active than the positive control Allo (IC50 of 10.73 μM), one of the reference drugs used for gout treatment.
Table 2.
In vitro IC50 values (µM) for XO inhibition, DPPH radical scavenging activity and cytotoxicity on NHDF cell line of bis-thiobarbiturate 11 and respective references Allo and Trolox a.
| XO Inhibition | DPPH Scavenging | Cytotoxicity on NHDF | ||||
|---|---|---|---|---|---|---|
| IC50 | R2 | IC50 | R2 | IC50 | R2 | |
| Febuxostat | 0.03 ± 0.01 | 0.9732 | n.d. b | - | n.d. b | - |
| Allo | 10.73 ± 0.81 | 0.9959 | n.d. b | - | n.d. b | - |
| Trolox | n.d. b | - | 23.82 ± 2.13 | 0.9947 | n.d. b | - |
| 11 | 1.79 ± 0.07 | 0.9986 | 24.67 ± 0.88 | 0.9992 | 93.15 ± 5.54 | 0.8913 |
a IC50 value ± SD represents as mean at least two independent determinations. b n.d. is not determined.
Since both steps of XO purine catabolism led to ROS generation (Scheme S2), a dual effect as an XO inhibitor and antioxidant can be profitable for new antigout drugs, since ROS generated by XO can produce cytotoxic effects in many circumstances and thus can promote mutagenesis and tumor development [3]. Therefore, molecules capable of this dual XO inhibitory and radical scavenging activity could be even more advantageous for gout treatment.
The antioxidant potential of all synthesized bis-thiobarbiturates 3–7 and 9–19 was evaluated by the DPPH method at a concentration of 30 µM, and results were compared with a reference compound, Trolox. The analysis of results (Figure 2 and Table S1) showed the high antioxidant potential of tested 3–7 and 9–18. The only exception was 19 that shows a low DPPH scavenging activity. Withal, the weak antioxidant activity of Allo was also evidenced, as expected [14]. Although a modest influence of several substituents on DPPH scavenging activity at 30 µM was noticed, some interesting conclusions can be inferred. Aligned with XO inhibitory results, the most DPPH scavenging bis-thiobarbiturate is once again the halogenated derivative 11. In fact, the IC50 value determined for 11 is similar to that obtained for Trolox (24.67 and 23.82 µM, respectively) in DPPH radical scavenging activity concentration-response studies (Table 2 and Figure S2).
Figure 2.

In vitro DPPH radical scavenging activity of bis-thiobarbiturates 3–7 and 9–19, Allo and reference Trolox. Results are expressed as average values ± SD of two independent determinations, and each one was performed in triplicated. A p < 0.001 versus the negative control in the statistical significance analysis (Student’s t-test) was observed for all compounds except for Allo and 19.
Regarding the promising results of the bis-thiobarbiturate 11 on both antioxidant and XO inhibitory activity, the in vitro biosafety effectiveness was further analyzed by the MTT method in a non-tumoral cell line. A calculated IC50 of 93.15 µM (Table 2 and Figure S3) clearly demonstrated the low cytotoxicity of this compound on NHDF cells at concentrations where XO inhibitory activity was effective. Thus, despite being used in vitro data, it was possible to calculate the selectivity index for bis-thiobarbiturate 11, with calculated values of 52.04 and 3.77 for XO inhibition and DPPH scavenging activity, respectively.
Although there is no direct relationship between the use of XO inhibitors and a good prognosis in cancer treatment, the expression and activity of this enzyme have been negatively associated with a high degree of malignancy and a worse prognosis in some types of cancer, namely of the breast and gastrointestinal tract, in recent years [28,29]. Considering the low cytotoxicity on NHDF cells observed for bis-thiobarbiturate 11, the interest of barbiturate and thiobarbiturate derivatives as anticancer agents [11,13] and the recent work on XO inhibitors with anticancer activity [30], we decided to evaluate the cytotoxicity on NHDF as well as the antiproliferative effects on colorectal adenocarcinoma Caco-2 and breast cancer MCF-7 cell lines for all bis-thiobarbiturates under study. To get a strengthened term of comparison, the anticancer drug 5-FU was used as a positive control. From the results (Figure 3 and Table S1), the not-marked cytotoxicity of almost all tested compounds was noted. In fact, only bis-thiobarbiturates 10, 13, 16, and 17 demonstrated a relevant effect on NHDF cells, with cell viability below 80%. On the other hand, all of the tested bis-thiobarbiturates did not present antiproliferative effects on the breast cancer MCF-7 cell line. Nonetheless, on the colorectal Caco-2 cancer cell line, bis-thiobarbiturates 5, 9, 10, and 11 showed a moderate effect on their cell viability; however, it was weaker than the positive control 5-FU. In addition, compounds 5, 9, and 11 demonstrated some selectivity for cancer Caco-2 versus normal NHDF cells.
Figure 3.

In vitro effects of bis-thiobarbiturates 3–7 and 9–19 and 5-FU on cell viability of non-tumor cell lines of normal human dermal fibroblasts (NHDF), and human adenocarcinoma tumor cell lines of colorectal (Caco-2) and breast (MCF-7), at the single concentration of 30 µM. Results are expressed as average values ± standard deviation of at least two independent assays performed in quadruplicate. * p < 0.05 versus the negative control by Student’s t-test.
In silico approaches remain a critical tool for drug discovery since their use can be a determinant for a cost-effective identification of promising drug candidates and to reduce the use of animal models [31,32]. In this context, the most promising bis-thiobarbiturate herein tested, 11, was in silico assessed to verify its drug-likeness characteristics with the applicability of Lipinski’s rule of five [33] and Veber’s parameters [34] by the free web tool SwissADME [25]. These two rules are essential tools in drug discovery to predict the potential bioavailability of new drug candidates. Therefore, it was determined that compound 11 respect Lipinski’s rule parameters for hydrogen bond donors (nOHNH), hydrogen bond acceptors (nON) and octanol-water partition coefficient (logP) (Table 3). The exception to this rule was only a molecular weight larger than 500. However, this is not a preponderant factor, since it has been considered that one violation of this rule is acceptable [33]. The alternative analysis of the variant of Lipinski’s rule of five by Veber et al. even claims that a molecular weight cutoff at 500 by itself does not suggest compounds with low bioavailability. Veber’s parameters defend a simple analysis of the number of rotatable bonds (n-rot; 10 or fewer) and topological polar surface area (TPSA; equal to or less than 140 Å2 or 12 or fewer nON and nOHNH) [34]. Taking this consideration in mind leads us to conclude that bis-thiobarbiturate 11 has a high probability of presenting good bioavailability. Despite presenting TPSA superior to 140 Å2, bis-thiobarbiturate 11 does not exceed the allowed number of nON and nOHNH.
Table 3.
In silico molecular properties of bis-thiobarbiturate 11 using the SwissADME predictive database a.
| Descriptor | Value | |
|---|---|---|
| Molecular weight | 567.52 g/mol | |
| Log P | 4.01 | |
| nON | 6 | |
| nOHNH | 2 | |
| n-rot | 7 | |
| TPSA | 158.50 Å2 | |
| Drug-likeness | Lipinski | Yes; 1 violation: MW > 500 |
| Veber | Yes | |
| Medicinal Chemistry | PAINS | 0 alert |
a Octanol-water partition coefficient (logP); number of hydrogen bond acceptors (nON); number of hydrogen bond donors (nOHNH); number of rotatable bonds (n-rot); topological polar surface area (TPSA); Lipinski’s rule of five: molecular weight < 500 Da; logP < 5; n-OHNH < 5; n-OHNH < 10. A maximum of 1 violation is permitted [33]. Veber’s parameters: n-rot ≤ 10; TPSA ≤ 140 Å2 or total of nON and nOHNH ≤ 12 [34].
The identification of 11 as a potential promiscuous compound with reactivity on several biological targets [35] was also crucial for their potential interest in medicinal chemistry. The results obtained eliminate the potential of 11 to be a pan-assay interference compound (PAINS), since no significate alerts were observed. Although some barbituric and thiobarbituric acid derivatives are considered to be highly reactive with numerous biological targets [35], the hypothesis of the bis-thiobarbiturate 11 as PAINS is thus removed.
The ADMET parameters of the bis-thiobarbituric 11 were then evaluated on SwissADME [25] and pkCSM [26] web tools. As expected from the fulfillment of Lipinski’s and Veber’s rules, 11 presents potentially good intestinal absorption (71.66%) with moderate solubility in water (Table 4). In addition, compound 11 should not be a potential substrate for P-glycoprotein, an important parameter to consider in the study of drug-drug interactions [36]. Besides, interactions with organic cation transporter 2 (OCT2) [36] and cytochrome P450 (CYP) enzymes [37] are two other important checks to take in mind. In accordance and as expected, 11 reveals not to be a substrate for renal OCT2 CYP1A2, CYP2C19, and CYP2D6 inhibitors. On the other hand, 11 could likely be a potential inhibitor for CYP2C9 and CYP3A4.
Table 4.
In silico pharmacokinetic (absorption, distribution, metabolism, and excretion) and toxicity parameters of bis-thiobarbiturate 11 using pkCSM and SwissADME predictive databases a.
| Property | Model Name | Predicted Value |
|---|---|---|
| Absorption | Water solubility | −4.66 b (Moderately soluble) |
| Intestinal absorption (human) | 71.66% | |
| P-glycoprotein substrate | No | |
| Distribution | BBB permeant | No |
| LogBB | −1.38 | |
| LogPS | −2.42 | |
| Metabolism | CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No | |
| CYP2C9 inhibitor | Yes | |
| CYP2D6 inhibitor | No | |
| CYP3A4 inhibitor | Yes | |
| Excretion | Log total clearance | −0.36 c |
| Renal OCT2 substrate | No | |
| Toxicity | AMES toxicity | No |
| hERG I inhibitor | No | |
| hERG II inhibitor | No | |
| Hepatotoxicity | Yes | |
| Skin Sensitisation | No |
a Blood-brain barrier (BBB); logarithm of permeability in blood-brain barrier (logBB) < −1 are poorly distributed; blood-brain permeability-surface area product (logPS) > −2 are considered to penetrate the central nervous system and logPS < −3 are considered as unable to penetrate the central nervous system [26]; human ether-a-go-go-related gene (hERG). b log(mol/L). c log(mL/min/kg).
As barbiturate derivatives are potential CNS depressants [10], 11′s distribution profile is additionally analyzed. In this context, in silico SwissADME predictions showed no expectable ability for 11 to permeate the blood-brain barrier (BBB). This low potential capacity of 11 to reach the CNS is corroborated by pkCSM calculated values of log BBB and log CNS.
Besides 11′s low cytotoxicity on NHDF cells previously established, predictions by pkCSM indicated that their manipulation should not cause skin sensitization. This bis-thiobarbiturate also would not be genotoxic or cardiotoxic since it does not have the potential to originate human ether-a-go-go-related gene’s (hERG’s) inhibition. Nonetheless, 11 can be a potential hepatotoxic compound and future studies will be necessary to evaluate their in vitro and/or in vivo toxicity profile.
4. Conclusions
Bis-barbiturates and bis-thiobarbiturates were easily and straightforwardly synthesized in moderate to excellent reactional yields. These bis-thiobarbiturates stood out by their antioxidant performance and excellent ability to inhibit the XO at a concentration of 30 µM. The most powerful bis-diethylthiobarbiturate within this set showed an XO inhibition IC50 of 1.79 μM, which was about ten-fold better than in vitro Allo inhibition, together with high DPPH radical scavenging activity and suitable low cytotoxicity. The in silico molecular properties such as druglikeness, pharmacokinetics and toxicity were fulfilled for this promising barbiturate, clearly pointing to the potential use of this class of molecules for the treatment of hyperuricemia diseases, such as gout.
5. Patents
“Bis-pirimidinonas como inibidores da xantina oxidase para o tratamento de condições patológicas causadas por hiperuricemia” PT116062 (20 January 2020).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/biomedicines9101443/s1, Table S1: Full in vitro data, Scheme S1: Chemical mechanism for the formation of bis-thiobarbiturates; Scheme S2: Diagram of XO oxidative hydroxylation’s; Figures S1–S3: Curves for IC50 calculations; Figures S4–S36: 1H and 13C NMR spectra.
Author Contributions
Synthesis, J.L.S., D.L. and M.J.A.R.; XO inhibition studies and writing—original draft preparation, J.L.S. and D.L.; DPPH, Cytotoxic and in silico assays, J.L.S.; funding acquisition and writing—review and editing, R.E.F.B., S.S. and P.A.; supervision, S.S. and P.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by funds from the Health Sciences Research Center (CICS-UBI) (UID/Multi/00709/2019) through National Funds by Fundação para a Ciência e Tecnologia (FCT). FEDER funds also support this work through the POCI-COMPETE 2020-Operational Programme Competitiveness and Internationalization in Axis I–Strengthening research, technological development and innovation [Programa Operacional Competitividade e Internacionalização (COMPETE 2020), project No. 007491)]. It is also acknowledged funding from C4—Cloud Computing Competences Center project (CENTRO-01-0145-FEDER-000019). J. L. Serrano acknowledges a doctoral fellowship grant from FCT (SFRH/BD/148028/2019).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no knowledge of conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Šmelcerović A., Tomović K., Šmelcerović Ž., Petronijević Ž., Kocić G., Tomašič T., Jakopin Ž., Anderluh M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017;135:491–516. doi: 10.1016/j.ejmech.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 2.Cos P., Ying L., Calomme M., Hu J.P., Cimanga K., Van Poel B., Pieters L., Vlietinck A.J., Berghe D.V. Structure−activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998;61:71–76. doi: 10.1021/np970237h. [DOI] [PubMed] [Google Scholar]
- 3.Battelli M.G., Polito L., Bortolotti M., Bolognesi A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxid. Med. Cell. Longev. 2016;2016:3527579. doi: 10.1155/2016/3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalbeth N., Merriman T.R., Stamp L.K. Gout. Lancet. 2016;388:2039–2052. doi: 10.1016/S0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- 5.Martillo M.A., Nazzal L., Crittenden D.B. The crystallization of monosodium urate. Curr. Rheumat. Rep. 2013;16:400. doi: 10.1007/s11926-013-0400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benn C.L., Dua P., Gurrell R., Loudon P., Pike A., Storer R.I., Vangjeli C. Physiology of hyperuricemia and urate-lowering treatments. Front. Med. 2018;5:160. doi: 10.3389/fmed.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrano J.L., Figueiredo J., Almeida P., Silvestre S. From xanthine oxidase inhibition to in vivo hypouricemic effect: An integrated overview of in vitro and in vivo studies with focus on natural molecules and analogues. Evid-Based. Complement. Alternat. Med. 2020;2020:9531725. doi: 10.1155/2020/9531725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh J.V., Bedi P.M.S., Singh H., Sharma S. Xanthine oxidase inhibitors: Patent landscape and clinical development (2015–2020) Expert Opin. Ther. Pat. 2020;30:769–780. doi: 10.1080/13543776.2020.1811233. [DOI] [PubMed] [Google Scholar]
- 9.Kerru N., Gummidi L., Maddila S., Gangu K.K., Jonnalagadda S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules. 2020;25:1909. doi: 10.3390/molecules25081909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nusrat S., Uzma A., Gul Z., Shagufta P., Irum J., Aisha A. A comprehensive review: Bio-potential of barbituric acid and its analogues. Curr. Org. Chem. 2020;24:129–161. [Google Scholar]
- 11.Prasher P., Sharma M., Singh S.P., Rawat D.S. Barbiturate derivatives for managing multifaceted oncogenic pathways: A mini review. Drug Develop. Res. 2021;82:364–373. doi: 10.1002/ddr.21761. [DOI] [PubMed] [Google Scholar]
- 12.Shabeer M., Barbosa L.C.A., Karak M., Coelho A.C.S., Takahashi J.A. Thiobarbiturates as potential antifungal agents to control human infections caused by Candida and Cryptococcus species. Med. Chem. Res. 2018;27:1043–1049. doi: 10.1007/s00044-017-2126-0. [DOI] [Google Scholar]
- 13.Figueiredo J., Serrano J.L., Cavalheiro E., Keurulainen L., Yli-Kauhaluoma J., Moreira V.M., Ferreira S., Domingues F.C., Silvestre S., Almeida P. Trisubstituted barbiturates and thiobarbiturates: Synthesis and biological evaluation as xanthine oxidase inhibitors, antioxidants, antibacterial and anti-proliferative agents. Eur. J. Med. Chem. 2018;143:829–842. doi: 10.1016/j.ejmech.2017.11.070. [DOI] [PubMed] [Google Scholar]
- 14.Figueiredo J., Serrano J.L., Soares M., Ferreira S., Domingues F.C., Almeida P., Silvestre S. 5-Hydrazinylethylidenepyrimidines effective against multidrug-resistant Acinetobacter baumannii: Synthesis and in vitro biological evaluation of antibacterial, radical scavenging and cytotoxic activities. Eur. J. Pharm. Sci. 2019;137:104964. doi: 10.1016/j.ejps.2019.104964. [DOI] [PubMed] [Google Scholar]
- 15.Sharma A., Noki S., Zamisa S.J., Hazzah H.A., Almarhoon Z.M., El-Faham A., de la Torre B.G., Albericio F. Exploiting the thiobarbituric acid scaffold for antibacterial activity. ChemMedChem. 2018;13:1923–1930. doi: 10.1002/cmdc.201800414. [DOI] [PubMed] [Google Scholar]
- 16.Ortega J.A., Riccardi L., Minniti E., Borgogno M., Arencibia J.M., Greco M.L., Minarini A., Sissi C., De Vivo M. Pharmacophore hybridization to discover novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 2018;61:1375–1379. doi: 10.1021/acs.jmedchem.7b01388. [DOI] [PubMed] [Google Scholar]
- 17.Bhatt P., Kumar M., Jha A. Design, synthesis and anticancer evaluation of oxa/thiadiazolylhydrazones of barbituric and thiobarbituric acid: A collective in vitro and in silico approach. ChemistrySelect. 2018;3:7060–7065. doi: 10.1002/slct.201800832. [DOI] [Google Scholar]
- 18.Cagno V., Tintori C., Civra A., Cavalli R., Tiberi M., Botta L., Brai A., Poli G., Tapparel C., Lembo D., et al. Novel broad spectrum virucidal molecules against enveloped viruses. PLoS ONE. 2018;13:e0208333. doi: 10.1371/journal.pone.0208333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rauf A., Shahzad S., Bajda M., Yar M., Ahmed F., Hussain N., Akhtar M.N., Khan A., Jończyk J. Design and synthesis of new barbituric- and thiobarbituric acid derivatives as potent urease inhibitors: Structure activity relationship and molecular modeling studies. Bioorg. Med. Chem. 2015;23:6049–6058. doi: 10.1016/j.bmc.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 20.Marecki J.C., Aarattuthodiyil S., Byrd A.K., Penthala N.R., Crooks P.A., Raney K.D. N-Naphthoyl-substituted indole thio-barbituric acid analogs inhibit the helicase activity of the hepatitis C virus NS3. Bioorg. Med. Chem. Lett. 2019;29:430–434. doi: 10.1016/j.bmcl.2018.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson J., Coe J.B., Grassam L.H., Jeffery C.J., Coles J.S., Hursthouse B.M. Reactions of 1,3-diethyl-2-thiobarbituric acid with aldehydes: Formation of arylbis(1,3-diethyl-2-thiobarbitur-5-yl)methanes † and crystallographic evidence for ground state polarisation in 1,3-diethyl-5-[4-(dimethylamino)benzylidene]-2-thiobarbituric acid. J. Chem. Soc. Perkin Trans. I. 1999;1:2483–2488. doi: 10.1039/a904015c. [DOI] [Google Scholar]
- 22.Rahim F., Ali M., Ullah S., Rashid U., Ullah H., Taha M., Javed M.T., Rehman W., Khan A.A., Abid O.U.R., et al. Development of bis-thiobarbiturates as successful urease inhibitors and their molecular modeling studies. Chin. Chem. Lett. 2016;27:693–697. doi: 10.1016/j.cclet.2015.12.035. [DOI] [Google Scholar]
- 23.Sachar A., Gupta P., Gupta S., Sharma R.L. A novel approach towards the synthesis of tricyclic systems based on pyridine, pyran, thiopyran, azepine, oxepin, thiepin, and pyrimidine rings under different solvent conditions. Can. J. Chem. 2010;88:478–484. doi: 10.1139/V10-015. [DOI] [Google Scholar]
- 24.Matias M., Campos G., Santos A.O., Falcão A., Silvestre S., Alves G. Potential antitumoral 3,4-dihydropyrimidin-2-(1H)-ones: Synthesis, in vitro biological evaluation and QSAR studies. RSC Adv. 2016;6:84943–84958. doi: 10.1039/C6RA14596E. [DOI] [Google Scholar]
- 25.Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jursic B.S., Neumann D.M. Preparation of 5,5′-pyrilidene and 5,5′-quinolidene bis-barbituric acid derivatives. J. Het. Chem. 2003;40:465–474. doi: 10.1002/jhet.5570400310. [DOI] [Google Scholar]
- 28.Battelli M.G., Bortolotti M., Polito L., Bolognesi A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019;21:101070. doi: 10.1016/j.redox.2018.101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Battelli M.G., Polito L., Bortolotti M., Bolognesi A. Xanthine oxidoreductase in cancer: More than a differentiation marker. Cancer Med. 2016;5:546–557. doi: 10.1002/cam4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi G., Sharma M., Kalra S., Gavande N.S., Singh S., Kumar R. Design, synthesis, biological evaluation of 3,5-diaryl-4,5-dihydro-1H-pyrazole carbaldehydes as non-purine xanthine oxidase inhibitors: Tracing the anticancer mechanism via xanthine oxidase inhibition. Bioorg. Chem. 2021;107:104620. doi: 10.1016/j.bioorg.2020.104620. [DOI] [PubMed] [Google Scholar]
- 31.Brogi S., Ramalho T.C., Kuca K., Medina-Franco J.L., Valko M. Editorial: In silico methods for drug design and discovery. Front. Chem. 2020;8:612. doi: 10.3389/fchem.2020.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rifaioglu A.S., Atas H., Martin M.J., Cetin-Atalay R., Atalay V., Doğan T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019;20:1878–1912. doi: 10.1093/bib/bby061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 34.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 35.Baell J.B., Holloway G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 36.Gessner A., König J., Fromm M.F. Clinical aspects of transporter-mediated drug–drug interactions. Clin. Pharmacol. Ther. 2019;105:1386–1394. doi: 10.1002/cpt.1360. [DOI] [PubMed] [Google Scholar]
- 37.Manikandan P., Nagini S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets. 2018;19:38–54. doi: 10.2174/1389450118666170125144557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.