

Abstract
The evolutionary history of the Malus genus has not been well studied. In the current study, we presented genetic evidence on the origin of the Malus genus based on genome sequencing of 297 Malus accessions, revealing the genetic relationship between wild species and cultivated apples. Our results demonstrated that North American and East Asian wild species are closer to the outgroup (pear) than Central Asian species, and hybrid species including natural (separated before the Pleistocene, about 2.5 Mya) and artificial hybrids (including ornamental trees and rootstocks) are between East and Central Asian wild species. Introgressions from M. sylvestris in cultivated apples appeared to be more extensive than those from M. sieversii, whose genetic background flowed westward across Eurasia and eastward to wild species including M. prunifolia, M. × asiatica, M. × micromalus, and M. × robust. Our results suggested that the loss of ancestral gene flow from M. sieversii in cultivated apples accompanied the movement of European traders around the world since the Age of Discovery. Natural SNP variations showed that cultivated apples had higher nucleotide diversity than wild species and more unique SNPs than other apple groups. An apple ERECTA‐like gene that underwent selection during domestication on 15th chromosome was identified as a likely major determinant of fruit length and diameter, and an NB‐ARC domain‐containing gene was found to strongly affect anthocyanin accumulation using a genome‐wide association approach. Our results provide new insights into the origin and domestication of apples and will be useful in new breeding programmes and efforts to increase fruit crop productivity.
Keywords: Malus, apple, sequencing, evolution, domestication
Introduction
The genus Malus belongs to the family Rosaceae and comprises ~30 species of deciduous trees and shrubs native to the temperate regions of the northern hemisphere. Given the diversity of the Malus genus in the wild and cultivation (Robinson et al., 2001; Rohrer et al., 1994), it is unsurprising that its evolutionary origins are controversial. The current mainstream view is that the genus originated in the so‐called Chuan‐Dian Palaeoland, corresponding to Southern China, Northern Vietnam, and Northern Laos. This is partly because this region is the geographic centre for over two‐thirds of extant wild Malus species, including the oldest species such as M. yunnanensis, M. sikkimensis, M. kansuensis, M. prattii, M. sieboldii , and M. hupehensis (Jiang, 1986). It is widely believed that these progenitor species spread across Eurasia in the aftermath of the Ice Ages as the glaciers melted and temperatures increased (Langenfeld, 1991). However, some scientists have suggested that the genus Malus had multiple genetic centres (Ferree and Warrington, 2003; Li, 1989). The wild species M. sieversiiis distributed within the Tian Shan Mountains; its distribution has remained largely unchanged from antiquity to the present day (Li, 1989). Europe, North America, and the Far East have also been considered as genetic centres for Malus (Ferree and Warrington, 2003; Li, 1989) due to their distinct native species. These studies have sought to clarify the origin of the genus Malus by considering its morphology, geography, and cytology. While their results have largely been inconclusive, these investigations have yielded preliminary insights into the relationships between wild species from different regions. Studies on allelic variants can provide genetic evidence regarding a species’ origins and facilitate understanding of its evolutionary history. This approach is particularly powerful when paired with genomic approaches using second‐generation sequencing technologies that enable high‐throughput genotyping and large‐scale surveys of genetic variation (Weigel and Mott, 2009).
The cultivated apple (Malus × domestica Borkh) has greatly influenced human history. Its fruit is widely consumed and is regarded as a symbol of wisdom and love (Juniper and Mabberley, 2006). Previous studies on the biogeography, isozyme polymorphisms, cytological markers, and simple sequence repeat markers of cultivated apples (Cornille et al., 2014; Gross et al., 2014) have suggested that cultivated apples were first domesticated in Central Asia from M. sieversii and were brought to Europe about 3000 years ago via migration and trade (Cornille et al., 2014; Harris et al., 2002; Juniper and Mabberley, 2006; Velasco et al., 2010). Analyses of microsatellite markers showed that the European species M. sylvestris and M. orientalis contributed to the genetic make‐up of domesticated apples (Cornille et al., 2012; Harrison and Harrison, 2011). A re‐sequencing of 117 diverse Malus accessions (Duan et al., 2017) indicated that cultivated apples were domesticated from M. sieversii and received a strong introgression from M. sylvestris. Genomic analysis of 49 cultivars revealed that M. sieversii and M. sylvestris contributed significantly to these cultivars (Sun et al., 2020). In addition, a recent study indicated that dessert and cider apples may have independent domestication events (Liao et al., 2021). However, the genetic flow and dispersal routes of these two wild apple species in worldwide cultivars remain unknown.
Apple domestication involved many changes in fruit morphology and biochemistry. The most pronounced morphological difference between cultivated and wild species is that the former produce much larger fruit, probably because of selection for consumption. In addition, most modern cultivars have skin that is deep red in colour, as opposed to the green or yellow skin of many wild species. Colour is an important variable in modern breeding programmes because it strongly affects consumer appeal (King and Cliff, 2002). The main contributors to red skin, anthocyanins, have strong antioxidant activity (Eberhardt et al., 2000; Wolfe et al., 2003) and are popularly believed to benefit human health (Butelli et al., 2008; Toufektsian et al., 2008). In addition to fruit size and colour, selected attributes include fruit firmness, flavour, acid, and sugar content. The major organic acid in cultivated apples is malic acid (Beruter, 2004; Ma et al., 2015), while their main soluble sugars are fructose and sucrose (Ma et al., 2015).
Genetic loci controlling domestication traits can be identified in populations through genome‐wide association studies (GWAS). GWAS involve saturating the genome with molecular markers, typically SNPs. They were employed to study traits in agronomic crops (Chen et al., 2014; Cui et al., 2016; Tieman et al., 2017) and have recently been applied to fruit trees (Cao et al., 2016; Mariette et al., 2016). Previous GWAS on apple trees has used SNP arrays or re‐sequencing (Bianco et al., 2016; Duan et al., 2017; Farneti et al., 2017). Other studies have made use of genotyping‐by‐sequencing using restriction endonucleases (Amyotte et al., 2017; McClure et al., 2018; Migicovsky et al., 2016; Migicovsky et al., 2017). These investigations provided valuable information on genes that may affect fruit quality traits. However, genes associated with other valuable agronomic traits such as sugar and organic acid content were not identified, and there is still a need for experimental verification of the relationship between the identified genes and fruit quality traits.
Here, we analyse whole‐genome genetic variation from 297 Malus accessions using high‐throughput re‐sequencing technology and use these data to evaluate the geographic origins of the Malus genus and domesticated apples. With a total of ~4.4 million high‐quality SNPs, our results uncover the relationships among wild species of the genus Malus through population structure, and introgressions from the wild progenitors, M. sieversii and M. sylvestris, to domesticated apples from different continents. In addition, we apply these data in a GWAS focusing on fruit size and fruit flesh colour. Our findings provide genetic evidence regarding the origins of apple trees and will facilitate trait improvement through breeding in this important crop.
Results
Genetic diversity and structure
To study Malus evolution and domestication, we sequenced 297 Malus accessions, including 20 species from East Asia (38 accessions), four species from Central Asia (43 accessions), four species from North America (four accessions), two species from Europe (two accessions), 12 hybrid species (57 accessions), dwarfing rootstock cultivars (20 accessions), and commercial cultivated apples (128 accessions) (Table S1a). Sequencing was performed using the Illumina HiSeq 4000 system to a mean depth of ~11×, generating >two‐Tb high‐quality data. The genome coverage was over 70% across all chromosomes for most accessions, several accession of artificial hybrid and East Asia showed comparatively low coverage (>40%) (Figure S1). A total of ~4.4 million high‐quality SNPs (Table S2) were identified after mapping against an apple reference genome (Daccord et al., 2017).
We used this whole‐genome SNP data to assess phylogenetic relationships among the accessions (Figure 1a and Figure S2). A few accessions were grouped into other clusters of different genetic backgrounds, suggesting that some accessions may have been misclassified or that there may be previously unrecognized genetic differences between some accessions. Three wild species from North America (M. angustifolia, M. ioensis and M. fusca) and one wild European species (M. florentina) were genetically proximal to East Asian species and closer to the outgroup (pear) than East Asian species. The data set included several accessions representing hybrid species including both natural and artificial hybrids. The included species M. robusta, M. asiatica, M. micromalus, and M. prunifolia, which separated before the Pleistocene, about 2.5 Mya (Kumar et al., 2017), have been identified as natural hybrids. M. × domestica subsp. Chinensis was hybridized mainly from M. prunifolia and M. × asiatica in ancient China over 2000 years ago (Luo, 2014). These accessions are also considered natural hybrids and clustered with M. prunifolia and M. × asiatica. Artificial hybrids include ornamental trees (such as M. hybrid cv. Prairie Fire and M. hybrid cv. Robinson) and dwarfing rootstocks (such as M. × domestica cv. M26 and M. × domestica cv. Budagovsky57‐233). Ornamental trees have been grown in orchards and gardens in North America since the beginning of the 19th century, when North American local species were hybridized with wild East Asian species by American botanists to obtain ornamental trees with flowers having a more desirable appearance (Jefferson et al., 1970). Dwarfing rootstocks were bred from wild apples for dwarfing or resistance to environmental stresses. These hybrid species can be traced to wild parent species and clustered with wild species from East Asia, North America, and Central Asia. M. sieversii from Central Asia clustered close to M. × domestica, implying that, as previously thought, cultivated apples originated from M. sieversii. Twelve red‐fleshed cultivated accessions were clustered with M. Niedzwetzkyana, in keeping with the genetic evidence that a presumed natural form of M. sieversii native to central Asia (‘Niedzwetzkyana’) is a major contributor to most red‐fleshed apples (van Nocker et al., 2011). Based on the evolutionary tree derived from our genomic data and the available information on each accession’s region of origin (Table S1a) (Li, 2001), we divided the accessions into four groups for subsequent analysis: East Asia, Natural Hybrids, Central Asia, and Domestica (Table S1a). Artificial hybrid species were ignored because they provide no useful information in the context of evolutionary analysis.
Figure 1.
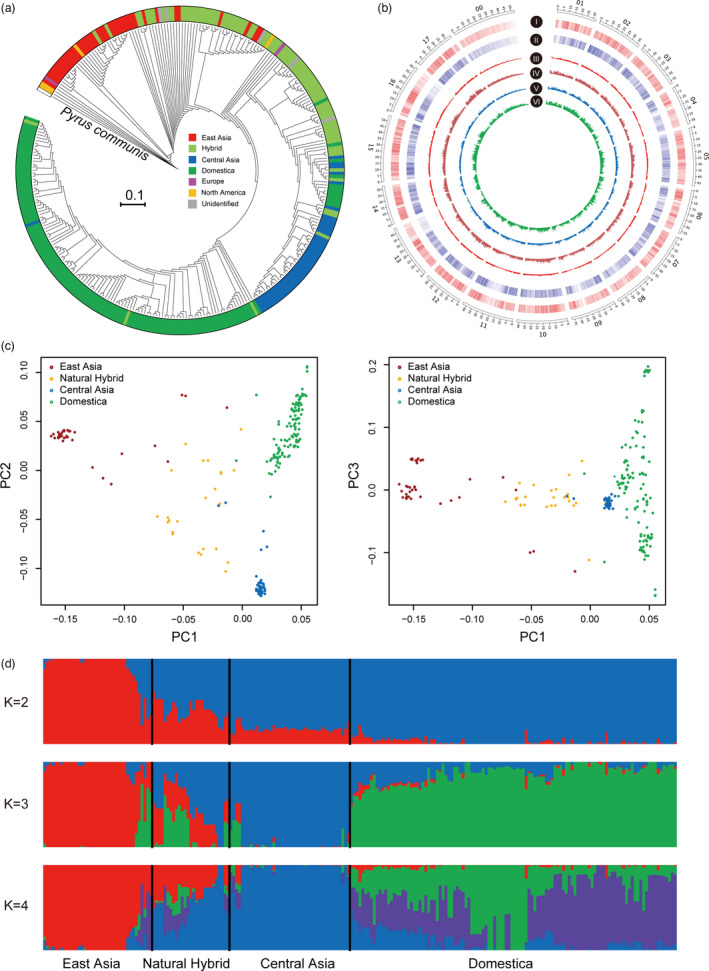
Overview of population genetic analysis of Malus accessions. (a) Phylogenetic tree (neighbour‐joining) of all accessions constructed using whole‐genome SNPs. Pyrus communis (cultivated pear) is included as an outgroup. Four main groups are: East Asia (red), Hybrids (light green), Central Asia (blue), and Domestica (dark green). Other samples: Europe (purple), North America (yellow), and Unidentified (grey). (b) A Circos image representing the diversity of different components: heat map view of SNPs (I) and InDels (II). Total genetic diversity of the East Asia (III), Natural Hybrids (IV), Central Asia (V), and Domestica (VI) groups. The outer rings indicate individual chromosomes. (c) PCA plots of the first three components for four groups: East Asia (brown), Natural Hybrids (yellow), Central Asia (blue), and Domestica (green). (d) Population structure of four groups inferred using ADMIXTURE for an assumed number of groups (K) from 2 to 4. Each colour represents one ancestral population. Each group is indicated by a vertical bar.
A survey of identified SNPs and InDels in the apple genome revealed that all its chromosomes have similar levels of SNP diversity and similar numbers of InDels (Figure 1b). An evaluation of global nucleotide diversity (π) across all Malus accessions revealed diverse genomic regions that were correlated to SNP density. Broadly speaking, we found that cultivated apples had higher nucleotide diversity than wild species (Figure 1b and Table S3) and more unique SNPs (12 721) than other apple groups (Figure S3). Principal component analysis (PCA) demonstrated that the East Asia, Central Asia, and Domestica groups clustered together, while the Natural Hybrids group was dispersed among the other groups (Figure 1c). Population structure analysis suggested that the similarity of the genetic backgrounds of the Domestica and Central Asia groups was higher than that for any other pair of groups (K = 2, Figure 1d). Together with the phylogenetic analysis (Figure 1a,c), these results suggest that domesticated apples originated from M. sieversii in Central Asia (Juniper and Mabberley, 2006; Velasco et al., 2010). The Domestica and Natural Hybrids groups exhibited clear patterns of relatively high genetic heterogeneity (K = 3, Figure 1d and Figure S4). Cultivated apples developed genetic polymorphism as a result of extensive human hybridization from wild apples, which was then maintained through asexual propagation. However, at higher K values (K ≥ 4) some natural hybrids (including M. × robusta, M. × asiatica, M. × micromalus, and M. prunifolia) exhibited a similar genetic background and level of genetic diversity to the Domestica group (Figure S4), implying that there was considerable gene flow between these natural hybrids and the Domestica group during apple domestication.
To further analyse the contributions of different Malus species and cultivars and the relationships between them, various Malus species were divided into 22 different groups according to the varieties and regions (Table S1b). We inferred patterns of population splits and mixtures from our genomic data and previously reported re‐sequencing data (Duan et al., 2017) for the major Malus accession groups using TreeMix (Pickrell and Pritchard, 2012). Using pear as the root of the likelihood tree, some native species (M. kansuensis, M. Fusca, M.toringgoides, M. florentina, M. angustifolia, M. ioensis and M. coronaria) demonstrated low levels of genetic drift relative to M. sieversii and M. × domestica (Figure 2a). The M. × domestica cultivars were close to M. sylvestris and M. sieversii in the tree (Figure 2a), indicating that the latter two species are closely related to domesticated apples. Interestingly, M. fusca from North America clustered with East Asian species rather than other North American species (Figure 2a). It was observed that an admixture event assigning 0.347357% of the ancestry of M. sieversii to M. × micromalus by TreeMix analysis (Figure 2a). M. × micromalus traced about 0.447285% of their ancestry to a population prior to the divergence of M. × asiatica. In addition, the analysis revealed frequent gene flow between wild species (M. prunifolia, M. × micromalus, M. × asiatica, and M. sieversii). Model‐based analyses of population admixture (K = 3, Figure 2b ) revealed that the M. × domestica had a more similar genetic background to M. sylvestris than to any other wild species. Population admixture analysis also suggested that M. sieversii had a similar genetic background to some of the M. × domestica and wild species (M. × asiatica, M. prunifolia, M. × micromalus, and M. baccata). This genetic background existed in M. × domestica from Europe and America but was rare in M. × domestica from Australia and New Zealand and absent in M. × domestica from Japan (K ≤ 6; Figure 2b). Higher K values (K ≥ 7; Figure 2b) reduced the similarity of the genetic background of M. × domestica to those of M. sylvestris and M. sieversii; this similarity was detectable in M. domestica from Europe and America but not in M. domestica from Australia, New Zealand, and Japan.
Figure 2.
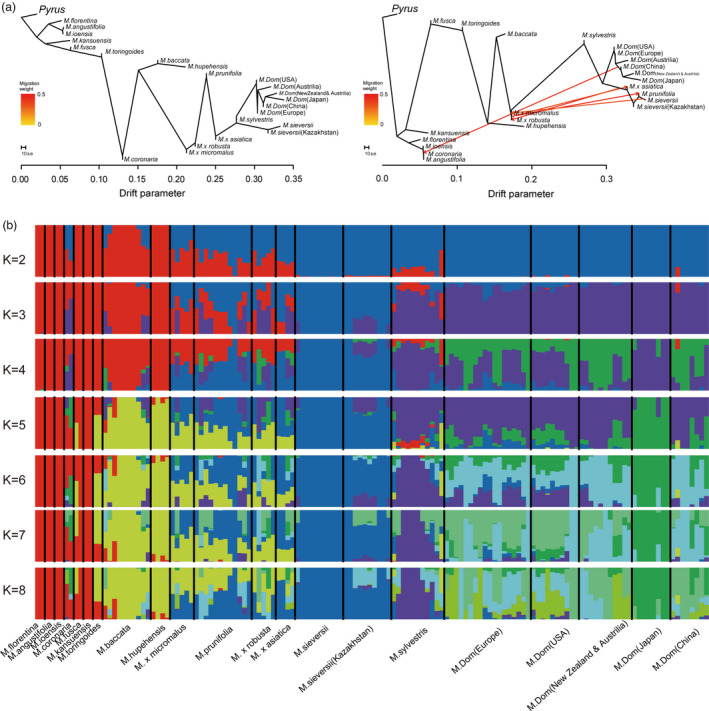
Introgression and structure of Malus accessions. (a) Maximum‐likelihood (ML) tree of Malus species with inferred migration edges. Arrows on the graph represent admixture events between different Malus species. (b) Population structure of Malus species inferred using ADMIXTURE for an assumed number of groups (K) from 2 to 8.
Selection signals of domestication
To identify signals of artificial selection during apple domestication (identified by comparing the Central Asia and Domestica groups) and improvement (identified by comparing the East Asia and Domestica groups), a 100‐kb sliding window with 10‐kb step approach was applied to quantify F ST and θπ, and the cross top 10% of two values was selected as selective signals. A total of 28 domestication‐selective sweeps and 51 improvement‐selective sweeps were detected (Tables S4 and S5). All chromosomes underwent selection, especially Chromosome 17 (Figure S6). Genes located in the selective sweep regions included those encoding disease resistance proteins, protein kinases, and transcription factors. GO enrichment (FDR ≤ 0.01) demonstrated that genes in domestication‐selected sweep regions were enriched mainly for the terms ‘cellulose biosynthetic and metabolic process’ and ‘glucosyltransferase activities’, both of which are potentially relevant to fruit development. Enriched genes in improvement‐selective sweep regions were mainly related to molecular functions, including ‘transmembrane’, ‘signaling receptor activities’, ‘tetrapyrrole’, ‘iron ion binding’, and ‘response to biotic stimulus’ (Table S6).
GWAS for agronomic traits
In order to identify the genes that associate with important agronomic traits of apple, we measured fruit weight, length, diameter, firmness, and flesh colour, as well as the soluble solid content (SSC), and contents of fructose, sucrose, and malic acid (Table S7), and performed a GWAS analysis. Fifteen to twenty mature fruits from more than two trees in 2015 and 2016 were collected based on Starch‐Iodine Index (Blanpied and Silsby, 1992). Only accessions showing consistent data for each trait in the two years were used for GWAS analysis. An obvious trait that is likely to have been selected during domestication and improvement is (increased) fruit size. Wild Malus species and cultivated apples show striking differences in fruit size. For example, the fruit of East Asian species tends to be <20 mm in length and diameter, whereas the fruit of M. × domestica is typically >50 mm on both measures (Figure 3e and Table S7). A significant GWAS signal (the threshold for GWAS was –log10 P = 5) related to fruit length and diameter was located in a 400‐kb region of Chromosome 15 (Figure 3a). This region showed extensive polymorphisms between wild species and cultivated apples and overlapped with the improvement‐selective sweeps (Figure 3b and Table S8). Within this region, strong GWAS signals were apparent for several SNPs within a gene designated MD15G1049300, which encodes a leucine‐rich receptor‐like protein kinase family protein homologous to Arabidopsis ERECTA (Figure 3c). This finding suggests that there was strong artificial selection acting on this gene during apple domestication. This was further supported by sequence analysis of ERECTA from F1 hybrids of M. prunifolia (small fruit) and M. × domestica (large fruit), which segregated for fruit size (fruit diameter and length). Two SNPs (Chr15:3358841 and Chr15:3359594) located in the intron of ERECTA strongly associated with fruit size showed the same genotype in both F1 hybrids and sequenced accessions (Figure 3d,e). To further verify whether ERECTA is a key gene controlling fruit size, we overexpressed apple ERECTA gene in tomato (Solanum lycopersicum cv. ‘Ailsa Craig’). The results showed that overexpression of MdERECTA could significantly increase the size and weight of tomato fruits (Figure 3f–h). In addition to ERECTA, 23 genes associated with fruit size and fruit weight were identified in a selective sweep region from 2.68 to 3.93 Mb in Chromosome 15 (Table S8). These results strongly suggest that these selected regions contributed to an increase in fruit size and weight during domestication. Other significant GWAS signals overlapping with selective sweep regions for improvement were also predicted to affect the traits of fruit length, diameter, weight, firmness, SSC, and contents of fructose and malic acid (Tables S6 and S9).
Figure 3.
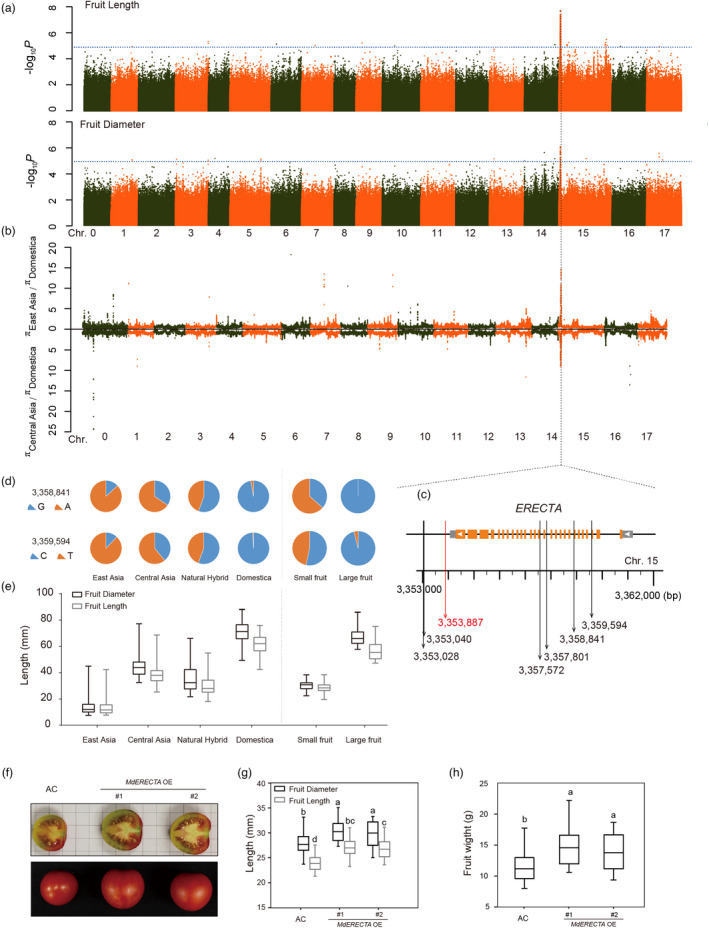
ERECTA was selected in artificial selection during apple domestication and affected fruit size. (a) GWAS analysis for apple fruit length and diameter. The dotted blue line represents the Bonferroni‐corrected significant threshold for GWAS (–log10 P = 5). (b) The region showed extensive polymorphisms between wild species and cultivated apples. (c) Location of ERECTA associated with fruit size GWAS and selection signals. The red arrow indicates the most significant SNP associated with fruit length and diameter. Black arrows indicate SNPs associated with fruit length. (d) Genotype of two SNPs associated with fruit length in ERECTA in both sequenced apple accessions and F1 hybrids. (e) Fruit size of sequenced apple accessions and F1 hybrids. (f) Phenotypes of transgenic tomato overexpressing MdERECTA (OE). (g) and (h) fruit size (g) and weight (h) of transgenic tomato overexpressing MdERECTA. Statistical analysis was performed using one‐way analysis of variance (ANOVA) followed by Duncan's multiple range test.
The GWAS analysis also identified several loci associated with fruit flesh colour located on Chromosome 9. A significant SNP signal was found in the promoter region of the MD09G1278600 gene, which encodes an R2R3 MYB transcription factor designated MdMYB10 (Figure 4a and Table S10). MdMYB10 was previously detected in a QTL region in the red‐fleshed apple genome (Chagne et al., 2007). Ectopic activation of MdMYB10 in apple induces anthocyanin accumulation in the fruit flesh and foliage (Allan et al., 2008; Espley et al., 2007), together with the transportation of anthocyanins into vacuoles (Hu et al., 2016). A rearrangement in the promoter region of MdMYB10 enhances the gene’s activation, leading to the phenotype of red foliage and red fruit flesh (Espley et al., 2009). MYB10 also strongly affects fruit colour in peach (Rahim et al., 2014), pear (Kusano et al., 2015), and strawberry (Medina‐Puche et al., 2014). A gene on Chromosome 1 encoding a UDP‐glycosyltransferase superfamily protein (UGT) was also detected in the GWAS analysis. This gene regulates the biosynthesis of flavonoids, thereby influencing pigmentation (Bowles et al., 2006; Caputi et al., 2012). However, the most significant SNP was located in a gene (MD09G1272500) encoding an NB‐ARC domain‐containing disease resistance protein (we named this gene NB‐ARC) about 20‐kb upstream of the MdMYB10 gene (Figure 4a). This gene was expressed significantly more strongly in red‐fleshed apples than typical white‐fleshed apples (Figure 4b). To investigate the function of this NB‐ARC gene in fruit coloration, we introduced a genomic copy of the NB‐ARC sequence into M. × domestica cv. Granny Smith by transient expression analysis. This resulted in the accumulation of red coloration in the fruit flesh and a change in petiole colour from green to red (Figure 4c,d). Moreover, high‐performance liquid chromatography (HPLC) analysis confirmed that the red components were anthocyanins (Figure 4e). In addition, we obtained transgenic plants overexpressing MdNB‐ARC in GL‐3. Under the treatment of 6% sucrose, anthocyanin accumulation was induced (Liu et al., 2017), We found that MdNB‐ARC OE transgenic apple plants exhibited greater anthocyanin accumulation than the wild‐type (GL‐3) plants when 6% sucrose was added to the medium (Figure 4f,g). These results indicated that increased expression of NB‐ARC led to anthocyanin accumulation.
Figure 4.
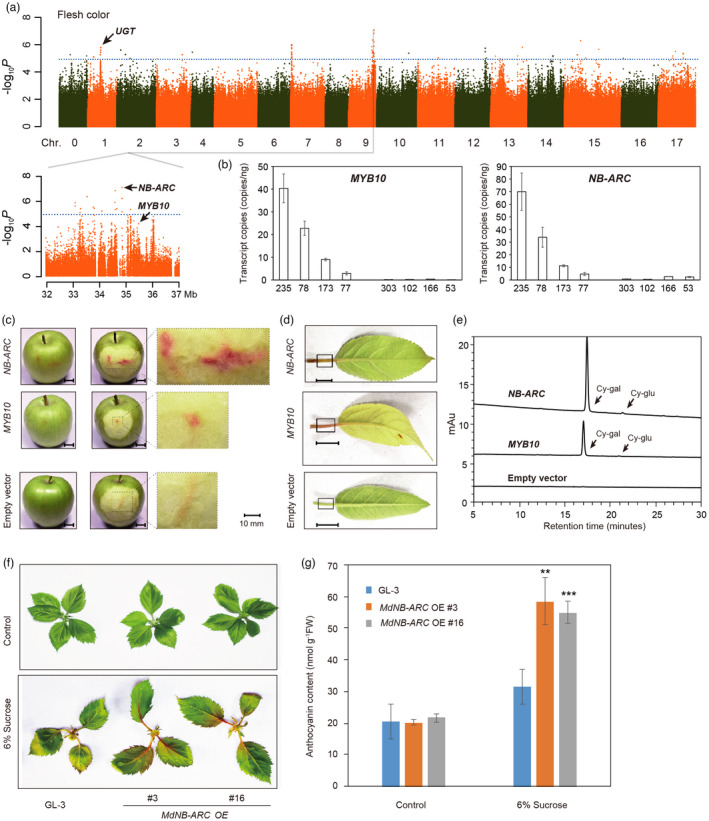
Identification and functional analyses of genes identified through GWAS analysis of flesh colour. (a) Manhattan plot showing the results of GWAS analysis of flesh colour and genomic locations of significant SNPs located around representative genes for flesh colour. The dotted blue line represents the Bonferroni‐corrected significant threshold for GWAS (–log10 P = 5). (b) Absolute quantification of MdMYB10 and MdNB‐ARC (MD09G1272500) expression in red‐ and white‐ fleshed accessions. 235, 78, 77, 173 are red‐fleshed apple accessions; 303, 102, 166, 53 are white‐fleshed accessions. (c) and (d) Phenotypes of apple fruit flesh (c) and foliage (d) transiently expressing 35S:MdMYB10 or 35S:MdNB‐ARC. Bars = 10 mm. (e) Flesh anthocyanin accumulation determined by HPLC analysis. Fruits were transiently transformed with 35S:MdMYB10 or 35S:MdNB‐ARC. Peaks were identified from HPLC traces at 520 nm. (f) Phenotypes of MdNB‐ARC overexpression (OE) plants and GL‐3 (the wild type) under sucrose treatment. (g) Anthocyanin content of the plants shown in (f). Statistical analysis was performed using one‐way analysis of variance (ANOVA) followed by Student's t‐test. **P < 0.01, ***P < 0.001.
The flavour of apples is strongly dependent on their malic acid content (Etienne et al., 2013). An aluminium‐activated malate transporter family protein (MD14G1135700) and a NAD‐dependent malic enzyme (MD07G1129000) related to malic acid metabolism were found in the GWAS results (Figure S7). Both genes have been reported to play key roles in malic acid biosynthesis (Bai et al., 2015; Etienne et al., 2013). The most significant SNP was located on Chromosome 10 and was associated with an integrin‐linked protein kinase family gene (ILK). ILKs transduce multiple ion‐mediated signalling pathways (Brauer et al., 2016; Popescu et al., 2017), including the potassium pathway, which influences plant photosynthesis and other metabolic processes affecting malic acid synthesis (Wang and Wu, 2017).
Strong GWAS signals for sucrose content were seen on multiple chromosomes (Figure S8). The most significant signal was associated with an F‐box family protein (MD09G1064500) located on Chromosome 9. GC‐MS analyses revealed two isomers of fructose in apples (fructose1 and fructose2) that had very similar GWAS patterns (Figure S9). MD04G1247700, which encodes an AP2‐like ethylene‐responsive transcription factor, was associated with the accumulation of both fructose1 and fructose2. The association of this gene with fructose (Figure S9), and with plant growth and fruit ripening (Gu et al., 2017; Phukan et al., 2017), may explain why ripe fruits accumulate more sucrose than immature ones and are thus sweeter (Ackermann et al., 1992). MD17G1149000, a gene encoding a synaptobrevin family protein, overlapped domestication‐selective sweep signals located on Chromosome 17, suggesting that increased fructose production was selected during apple domestication (Table S7). SSC is another quality trait that influences apple flavour. The most significant SNPs for SSC were within a gene (MD17G1286100) of unknown function on Chromosome 17 and a gene (MD05G1300000) encoding a cyclic nucleotide‐gated channel 1 protein on Chromosome 5 (Figure S10). Three significant SNPs were related to a gene encoding a sodium/calcium exchanger family protein (MD15G1340300) on Chromosome 15 and two genes encoding a Got1/Sft2‐like vesicle transport protein (MD01G1219600) (Figure S10). The two most significant SNPs for fruit firmness were located between the genes MD07G1071800 and MD07G1071900, which encode a Demeter‐like protein and a nucleotide diphospho sugar transferases superfamily protein, respectively. This suggests that fruit firmness may be linked to glycosylation (Figure S11). Taken together, these results indicate that genes associated with the agronomic traits of apples may participate in a complex regulatory network affecting sugar metabolism, biosynthesis, and other metabolic pathways.
Discussion
The comprehensive evaluation and utilization of large‐scale genomic data in this work has provided new insights into the origin of the Malus genus and the domestication history of cultivated apples. Our results indicate that the Central Asian species are genetically distinct from the East Asian species. Remarkably, M. angustifolia and M. ioensis from North America and M. florentina from Europe were more closely related to pears than to East Asian species. Unlike mammals, there is little fossil evidence available to support the hypothesis about the evolution of Malus, and it is difficult to define common ancestry (Li, 2001). In our study, wild Malus species from different continents were found to have very different genetic backgrounds, even those in Eurasia. We therefore conclude that wild Malus species from different continents may have origins that are independent of their current geographical distribution.
As a self‐incompatible perennial plant that has undergone frequent intraspecific and interspecific hybridization and adaptation to vegetative propagation, Malus has acquired an indistinct genetic background. Unlike many crops (such as wheat and rice), apples have not experienced domestication bottlenecks. The wild ancestors of wheat and rice have higher SNP diversity than cultivated species (Cavanagh et al., 2013; Pont et al., 2019; Wang et al., 2018; Wang et al., 2017a). This is because cultivated species have experienced domestication bottleneck in the process of artificial selection and have lost much ancestral SNP diversity. On the contrary, due to self‐incompatibility, apple trees need to be crossed to set offspring, resulting in higher SNP diversity. Wild apples, for instance, M. sieversii, maintain a natural community until today in Tianshan Mountain, resulting in lower SNP diversity by crossing within the population. Rice has experienced the doctrine of single origin or multiple origins (Huang et al., 2012; Wang et al., 2014). In recent years, it has been gradually confirmed that indica and japonica originated separately (van Andel et al., 2016; Zhao et al., 2018). Cultivated apples have also experienced the debate about whether they originated from European M. sylvestris or Central Asian M. sieversii. Present study suggests that cultivated apples were domesticated from M. sieversii in Central Asia and brought to Europe, where they obtained introgressions from M. sylvestris (Cornille et al., 2013; Cornille et al., 2014; Cornille et al., 2012; Juniper et al., 1998; Velasco et al., 2010). Our data indicate that the genetic background of M. sieversii has similarities with those of wild species including M. prunifolia, M. × asiatica, M. × micromalus and M. × robust, and with domesticated apples (Figure 2). This genetic background was present to a significant degree in European and American cultivars, to a lesser degree in cultivars from Oceania and completely absent in Japanese cultivars (Figure 2). We thus infer that the genetic background of M. sieversii flowed eastward to wild species including M. prunifolia, M. × asiatica, M. × micromalus and M. × robust, and westward across Eurasia to cultivated apples (Figure 2). Moreover, the genetic background of cultivated apples is more similar to that of M. sylvestris than M. sieversii, indicating that M. sylvestris contributed more to cultivated apples than M. sieversii (Figure 2). Although confidence in this conclusion is limited by the sizes of the available samples, the genetic backgrounds of M. sieversii and M. sylvestris, which are ancestors of cultivated apples, were present to varying degrees in cultivars from different continents. Interestingly, although Japan is very close to Eurasia in geographical terms, we found no evidence of gene flow between M. sieversii and cultivated apples from Japan (Figure 2). Our results further clarify how wild apples affect cultivated apples through extensive sequencing and how did the European cultivar apples spread to the world with the genetic background of the ancestors.
Interestingly, the genetic background of wild ancestor apples shows a trend of gradual loss along with the trajectory of human activities. Maritime trade between Europe, America, and Oceania has been ongoing since the so‐called Age of Discovery, which began in the 15th century (Diamond, 1997), whereas Europe’s maritime exchanges with China and Japan took place after the 18th century (Diamond, 1997). Before the rise of maritime trade, China mainly communicated with Europe via the Silk Road, which became an important trading route 2000 years ago. It is thus possible that the gene pool of M. sieversii first spread along the Silk Road (Juniper and Mabberley, 2006) and then spread around the world with the advent of maritime trade. We therefore infer that after western traders began trading with Asia by the sea in the 18th century, nearly 300 varieties were introduced into Japan by 1910 (Yi, 1989), prompting extensive hybrid breeding. The genetic background of cultivated apples in Japan was thus established much later than that of cultivars in Europe, America, and Oceania, resulting in the complete loss of the M. sieversii background. A roadmap illustrating the spread of ancestral species’ genetic backgrounds in modern apple cultivars is presented in Figure 5. The spread of food crops is considered to be an important symbol of the transition from nomadic to agricultural civilization. Dogs were domesticated by humans for hunting 16 000 years ago, and chickens and pigs were domesticated by farmers 7000 years ago (Kiple, 2007). Europe became the centre of civilization after the completion of the industrial revolution, which enabled marine trade and the spread of cultivated apples. This might be the reason why the gene flow from M. sieversii has declined gradually, while that from M. sylvestris has retained its dominant position during apple domestication. Our analysis indicates that cultivated apples traced the paths of human civilization from the 15th century to 19th century (Figure 5). In addition, humans have domesticated the genus Malus as ornamental plants, independently of its use as a source of food.
Figure 5.
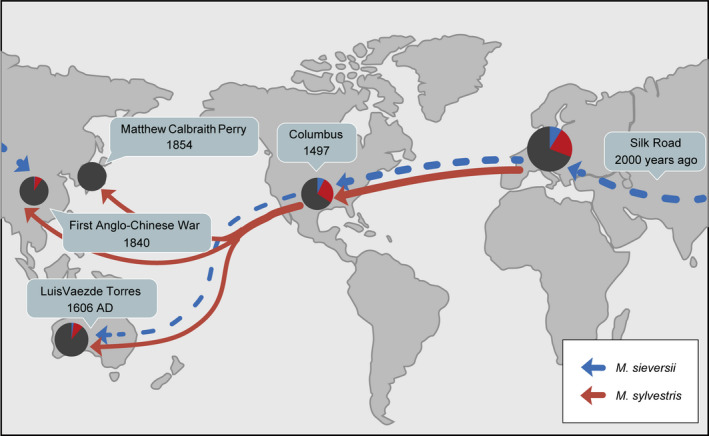
A roadmap illustrating distribution of genetic backgrounds of ancestral species in modern apple cultivars. The proportional pie charts showed the genetic backgrounds of M. sieversii (blue), M. sylvestris (red), and others (black) were defined by the ADMIXTURE analysis (K = 6). The areas of pie charts were proportional to the number of accessions. Arrows indicated potential spreading direction.
By combining GWAS with a selective sweep analysis, a signal related to fruit size was detected at the ERECTA gene locus on Chromosome 15 (Figure 3), indicating that ERECTA has important effects on fruit size and was subjected to artificial selection during apple domestication. In order to verify the GWAS signal of fruit size, we detected the genotypes of ERECTA from F1 hybrids of ‘Fuji’ x M. prunifolia, which showed a segregation of large and small fruits. Our results showed that ERECTA does have a preference genotype (Figure 3). ERECTA and its counterparts from other plants are critical modulators of growth and development (Cai et al., 2017; Shpak, 2013) that regulate cell elongation (Bundy et al., 2012; Uchida et al., 2012) and have particularly strong effects on plant architecture and flower and fruit morphology (Torii et al., 1996). In rice, increased expression of ERECTA is associated with increased grain weight (Shen et al., 2015). Manipulation of this gene may thus be a powerful tool for increasing crop productivity.
We also identified a previously unknown NB‐ARC domain‐containing gene in anthocyanin accumulation (Figure 4). It has been reported that pathogen infection often causes the fruits to ripen and turn red. The ripening process of grapes has been accelerated upon Botrytis cinerea infection (Blanco‐Ulate et al., 2015). Virus‐infected grapes exhibit uneven ripening and red spots (Blanco‐Ulate et al., 2017). Alternaria alternata infection can cause ‘red ring’, a typical phenotype of pathogenic infection on the jujube fruit (Yuan et al., 2019). Previous studies have shown that the NB‐ARC domain is important for NLRs (nucleotide‐binding, leucine‐rich repeat proteins) function as immune receptors of plants to confer resistance against pathogens through regulating activation of NLRs self‐association (Li et al., 2019). The genes containing the NB‐ARC domain play a molecular switch in plant resistance to pathogen infection. This domain acts as an ATPase module that can hydrolyse ATP, thus triggering defence signalling and unravelling many new proteins of the plant immune system (Tameling et al., 2006). NLRs can interact with WRKY proteins such as WRKY1/2, WRKY45, WRKY46, and WRKY72 to affect the expression of defence genes (Hu et al., 2017; Inoue et al., 2013; Shen et al., 2007). CaRGA (a CC‐NB‐ARC‐LRR protein) interacts with WRKY64 that stimulates EDS1 transcription in chickpea in response to Fusarium wilt infection (Chakraborty et al., 2018). Interestingly, WRKY family proteins play a role not only in plant disease resistance but also in anthocyanin accumulation. HpWRKY44 transcriptionally activates HpCytP450‐like1 in red pitaya fruit (Cheng et al., 2017). SlWRKYs is involved in colour change during tomato fruit ripening (Wang et al., 2017b). Apple MdWRKY40 promotes anthocyanin biosynthesis by interacting with MdMYB1 (An et al., 2019). In our study, overexpression of MdNB‐ARC involved in plant defence signalling pathway leading to red apple fruit maybe through the WRYK family. However, this needs to be studied further. Taken together, our results will facilitate the breeding of new apple cultivars and future studies on various aspects of apple biology.
Methods
Sample collection and sequencing
Young leaves and fruits of 297 Malus accessions were collected from plants maintained at the Horticulture Experimental Station of Northwest A&F University (34°20′N, 108°24′E), Yangling, China, except that M. baccata cv. ‘Dong Bei Shan Ding Zi’ was provided by China National Fruit Germplasm Repository, Huludao, China. The origin of all the germplasms is shown in Table S1a. About five young leaves were collected in June 2015 and preserved in liquid nitrogen. Fifteen to twenty fruits at mature stage were collected from at least two trees of each accession. Fruit maturity was determined using the Starch‐Iodine Index (Blanpied and Silsby, 1992). The fruits were collected in two years (2015 and 2016), from June to November each year. Fruits of F1 hybrids of M. prunifolia and M. × domestica were collected at an experimental field located 7.9 km east of the Horticulture Experimental Station. Genomic DNA was extracted using the CTAB method (Murray and Thompson, 1980). At least 5 µg of genomic DNA from each sample was used to construct a sequencing library using NEBNext Ultra DNA Library Prep Kit (NEB Inc., America) in accordance with the manufacturer’s instructions. Libraries with an insert size of approximately 500 bp were sequenced on an Illumina HiSeq 4000 sequencer by NowBio Company (Kunming) using Illumina TruSeq reagents and the paired‐end protocol.
Mapping
Paired‐end reads containing adapter sequences or low‐quality reads (reads with >30% bases having Phred quality ≤ 25) were removed using NGSQCToolkit_v2.3.3 (Patel and Jain, 2012). In addition, 5 bp were trimmed off the 5’ end of each read and 15 bp were trimmed from the 3’ end (parameters: ‐l 5 ‐r 15). All reads from each line were mapped to a Malus × domestica reference genome (GDDH13 (Daccord et al., 2017)) using BWA (version: 0.7.10‐r789) (Li and Durbin, 2009), allowing for mismatches of 4 bp in a single read (parameters: ‐t 10 –A 1 –B 4). The mapping results were then grouped by chromosome and sorted according to mapping coordinates. Only mapped paired‐end reads were used for variant detection. Duplicated reads were filtered with the Picard package (picard.sourceforge.net, version: 2.1.1).
SNP variant calling
Variants were detected using HaplotypeCaller from GATK (version 3.3‐0‐g37228af) (Mckenna et al., 2010) with parameters ‘‐T HaplotypeCaller ‐stand_call_conf 30.0 ‐stand_emit_conf 10.0’. The process was as follows: (i) after BWA alignment, the reads around InDels were realigned. Realignment was performed with GATK in two steps. The first step used the RealignerTargetCreator package to identify regions where realignment was needed, and the second step used IndelRealigner to realign the regions found in the first step, which produced a realigned BAM file for each accession. (ii) Variations were called at a population level with HaplotypeCaller of GATK. The HaplotypeCaller parameter ‐stand_call_conf was set to 30 and ‐stand_emit_conf was set to 10. (iii) After variation calling, the SNPs and InDels were extracted from the HaplotypeCaller results, respectively. The SNPs were filtered using the GATK criteria with parameters ‘QD < 20.0 || ReadPosRankSum < −8.0 || FS > 10.0 || QUAL < 6446.82’. SNPs with allele frequencies less than 5% and the proportion of missing data greater than 30% were ignored. SNPs were annotated against the apple genome (Daccord et al., 2017) using the package ANNOVAR (version: 2015‐12‐14) (Wang et al., 2010b). SNPs were categorized into exonic regions, splicing sites, 5’UTRs or 3’UTRs, intronic regions, upstream and downstream regions (within a 1‐kb region upstream or downstream from the transcription start site), or intergenic regions. SNPs in coding exons were further categorized as synonymous, nonsynonymous, stop‐gain or stop‐loss.
Population analysis
SNPs within single‐copy orthologous gene regions between pear (Chagné et al., 2014) and apple were used to construct the phylogenetic tree. First, OrthoMCL (version 1.4) (Li et al., 2003) was used to determine single‐copy orthologous genes between pear and apple genomes. Then, multiple single‐copy genes were aligned using Clustal W (version 2.1) (Larkin et al., 2007). Finally, SNPs within these regions of the pear genome were extracted from the corresponding positions and concatenated into a supergene to provide outgroup information. A neighbour‐joining tree was constructed using PHYLIP 3.696 (http://evolution.genetics.washington.edu/phylip.html) on the basis of a distance matrix. The bootstrap values on the tree are based on 1,000 replicates. The nucleotide diversity (π) was calculated using VCFtools (v0.1.13) (Danecek et al., 2011) with 100‐kb sliding windows and a 10‐kb step size. Population structure was investigated using ADMIXTURE (version: 1.3) (Alexander et al., 2009), which is a model‐based clustering method for inferring population structure assuming different numbers of clusters (K). The statistic ‘delta K’, that is, the change in likelihood upon varying the number of assumed groups, was calculated and used to assess the most likely number of populations. Principal component analysis (PCA) was performed using GCTA (version: 1.25.3) software (Yang et al., 2011), and the first three eigenvectors were plotted.
Conjoint analysis with Duan’s samples
To characterize the gene flow between wild Malus species and cultivated apples, SNP variant calling was applied to the sequencing data for the apple accessions in Duan’s analysis and our data using the previously discussed GATK method (Table S1b). In this case, the filtering criteria for GATT were set to ‘QD < 20.0 || ReadPosRankSum < −8.0 || FS > 10.0 || QUAL < 5187.41’. After filtering SNPs with allele frequencies less than 5% and the proportion of missing data greater than 30%, a total of 134,781 SNPs were identified. These SNPs were used to determine population structure using ADMIXTURE (Alexander et al., 2009) and to infer patterns of population splits and mixtures among wild Malus species and M. × domestica from different continents using TreeMix (Pickrell and Pritchard, 2012).
Selected sweeps
To minimize the influence of genetic drift, we combined the three cultivated groups into a single cultivated gene pool for the analysis. Genetic differentiation (FST ) and polymorphism level (θπ, a variable reflecting pairwise nucleotide variation as a measure of variability)‐based cross approaches were used to investigate selection signals across the whole genome. F ST and θπ were quantified with a 100‐kb sliding window with 10‐kb step approach using VCFtools software (v0.1.13) (Danecek et al., 2011), and the cross top 10% of two values were selected as selective signals. GO enrichment was performed using Ontologizer (http://ontologizer.de) with default parameters.
Evaluation of agronomic traits
Levels of malic acid, sucrose and fructose were measured using the protocol of Lisec and Wang (Lisec et al., 2006; Wang et al., 2010a). Briefly, freeze‐dried fruit powder (about 0.1 g) was extracted in 1.4 ml 75% methanol with ribitol as an internal standard. After adding chloroform to fractionated non‐polar metabolites, 5 ul of supernatant was transferred into a 1.5‐ml tube for drying under vacuum. After derivatization, metabolites were analysed by GC‐MS using a Trace GC ULTRA/ISQ MS detector (Thermo Scientific™,Waltham, MA, USA). Fruit weight, SSC, and firmness were measured sequentially using a balance (TP‐A500, HUAZHI®,Beijing,China), a refractometer (PAL‐5, ATAGO®,Tokyo,Japan) and a Fruit Hardness Tester (FHM‐5, Takemura Techno Works Co., Ltd., Tokyo, Japan), respectively. Fruit length and diameter were measured using a vernier calliper (MNT‐150, Shanghai, China).
Genome‐wide association study
GWAS was carried out using Genomic Association and Prediction Integrated Tool (GAPIT, version: 2016.03.01) (Lipka et al., 2012) with the previously determined population parameters and the first three principal components (PCs) and Kinship (K) matrixes as covariates. –log10 P > 5 was used to identify significant associations.
Transient expression in apple fruits, leaves and petioles
The coding sequence of MdMYB10 or MD09G1272500(MdNB‐ARC) was amplified from cDNA of ‘Golden Delicious’ and cloned into the pEarlygate203 vector under the control of 35S promoter, resulting in plasmids 35S:MdMYB10 and 35S:MD09G1272500. Then, 35S:MdMYB10, 35S:MD09G1272500 or the empty vector was transformed into Agrobacterium strain C58C1 and coinfiltrated with 35S:p19 (p19 is an RNA‐silencing repressor protein from Tomato bushy stunt virus) in Agrobacterium strain C58C1 into 130 DAF (days after flowering) apple fruit (M. × domestica cv. Granny Smith). The injected apple fruits were placed in PVC bags and kept in the darkness for 12 h at room temperature and then transferred to a light growth chamber without the bags at 17 °C for 4–7 days (Li et al., 2012) before observation. The apple leaves and petioles were transiently transformed with the same infiltration method.
Absolute quantitative PCR
Total RNA was extracted from apple fruits or leaves using Wolact®Plant RNA Isolation Kit (Hong Kong, China), following the manufacturer’s protocol. Total RNA was subjected to reverse transcription using the RevertAid RT Reverse Transcription Kit (Thermo Scientific) following the manufacturer’s protocol. Then, the cDNA was used for PCR or absolute quantitative PCR. The absolute quantification assay was performed as described (Shirima et al., 2017) with minor modifications. Briefly, specific primers were used to amplify the genes to create a standard curve after gel purification. Copy numbers (copies/ng) were then calculated based on the standard curve by SYBR Green Real‐Time PCR.
Generation of transgenic plants
To generate transgenic tomato plants, coding region of MdERECTA from Malus domestica cv. ‘Golden Delicious’ was cloned into pGWB414 vector by Gateway® recombinant cloning technology. The resulting plasmid was transferred into Agrobacterium tumefaciens GV3101. The genetic transformation of tomato plants was performed as described previously (Jones et al., 2002). Homozygous T2 plants were grown in a growth chamber for 30–40 days and then transplanted to a greenhouse. Fruits from 10 plants of each genotype were used to analysed the fruit length, diameter and fruit weight.
The coding region of MdNB‐ARC was cloned into the vector of pRI101 and the resulting plasmid was transformed into Agrobacterium tumefaciens GV3101. The stably transgenic apple plants were obtained as described previously (Chen et al., 2020). To analyse the anthocyanin accumulation of MdNB‐ARC transgenic plants, the shoot segments of tissue‐cultured plants were transferred to Murashige and Skoog (MS) media containing 3% (normal concentration) and 6% (high concentration) sucrose with a long day photoperiod (light:dark, 14 h:10 h) for 30 days.
Measurement of anthocyanin
Measurement of anthocyanin from transiently transformed apple fruits, leaves and petioles were carried out as described (Wang et al., 2010a). Briefly, fruit samples were ground into powder under liquid nitrogen using the extraction buffer (methanol: formic acid, 70%:2%) and then centrifuged at 12 500 g for 20 min. The upper aqueous phase was then filtered through 0.45‐µm syringe filters prior to high‐performance liquid chromatography (HPLC) analysis using a HP1200 Liquid Chromatography with a diode array detector (Agilent technologies, Santa Clara, CA, USA).
The fresh leaf samples from transgenic plants and wild‐type GL‐3 plants were ground into fine powder which was then resuspended in methanol‐HCl (1% v/v) buffer and transferred to 4°C for overnight. After centrifugation, supernatant was determined at 530, 620 and 650 nm. The concentration of anthocyanin was calculated according to the formula:
Funding
This work was supported by the Key S&T Special Projects of Shaanxi Province, China (2020zdzx03‐01‐02), and the earmarked fund for the China Agriculture Research System (CARS‐27).
Authors’ contributions
Q.G. and F.M. designed the project; P.C., D.Z., P.X., J.Z., L.J. X.L., X.S., D. G., L.W., C.N., C.B., M.Y., H.L., C.L., Y.Y. and Y.Z. collected samples and performed phenotyping; P.C., D.M., E.K. and Z.L. analysed data; P.C., D.Z and W.S. performed experiments; P.C., Z.L., D.Z., W.S., and Q.G. wrote the manuscript; all authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Supporting information
Figure S1. Heat map of the coverage of each accession across each chromosome of the apple genome. Artificial Hybrid, Central Asia, East Asia, and Other refer to the groups in Table S1a.
Figure S2. Unrooted phylogenetic tree of all accessions inferred from whole‐genome re‐sequencing, with Pyrus as an outgroup.
Figure S3. Venn diagram of SNPs among four groups.
Figure S4. Population structure of four groups inferred using ADMIXTURE. (a) The standard estimate of the cross‐validation (CV) error (b) ADMIXTURE analyses for an assumed number of subpopulations (K) from 5 to 9. Each colour represents one ancestral population. Each group is indicated by a vertical bar.
Figure S5. The standard estimate of the cross‐validation (CV) error of population structure of Malus species (Fig. 2B).
Figure S6. Genome‐wide distribution of F ST.
Figure S7. Manhattan plot showing the GWAS results for malic acid content. Gene abbreviations: ALMT, Aluminium‐activated malate transporter family protein; ILK, Integrin‐linked protein kinase family; NME, NAD‐dependent malic enzyme 2. The dotted grey line represents the significant threshold for GWAS (–log10 P = 5).
Figure S8. Manhattan plot showing the GWAS results of content for sucrose content. Gene abbreviations: FBX, F‐box family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S9. Manhattan plot showing the GWAS results for fructose content. Gene abbreviations: AP2L, AP2‐like ethylene‐responsive transcription factor; SFP, Synaptobrevin family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S10. Manhattan plot showing the GWAS results for soluble solid content. Gene abbreviations: CNGC1, cyclic nucleotide‐gated channel 1; Eu1035, Other Eukaryotes‐1035; GVTP, Got1/Sft2‐like vescicle transport protein family; SEFP, sodium/calcium exchanger family protein / calcium‐binding EF hand family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S11. Manhattan plot showing the GWAS results for fruit firmness. Gene abbreviations: DML1, demeter‐like 1; NTSP, Nucleotide‐diphospho‐sugar transferases superfamily protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S12. Manhattan plot showing the GWAS results for fruit weight. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Table S1. Details of all accessions and group information. (a) Details of 297 sequencing accessions for this study. (b) Details of accessions for Malus species analyses.
Table S2. Whole‐genome distribution of SNPs and InDels.
Table S3. Summary SNP statistics in different apple groups and species.
Table S4. Regions significantly associated with domestication‐selective sweeps and potential genes.
Table S5. Regions significantly associated with improvement‐selective sweeps and potential genes.
Table S6. Enrichment of genes from selective sweeps.
Table S7. Agronomic traits and phenotyping data used for GWAS.
Table S8. GWAS‐significant genes overlapping with improvement‐selective sweep signals.
Table S9. GWAS‐significant genes overlapping with domestication‐selective sweep signals.
Table S10. Information of the significant SNPs and associated genes from the GWAS.
Table S10. Information of the significant SNPs and associated genes from the GWAS.
Acknowledgements
We thank Dr. Kun Wang (China National Fruit Germplasm Repository, Huludao) for providing apple material (M. baccata cv. ‘Dong Bei Shan Ding Zi’) and Rui Zhai for helping with transient expression assays in apple fruit. We are very grateful to Dr. Pengmin Li for anthocyanin measurement.
Chen, P. , Li, Z. , Zhang, D. , Shen, W. , Xie, Y. , Zhang, J. , Jiang, L. , Li, X. , Shen, X. , Geng, D. , Wang, L. , Niu, C. , Bao, C. , Yan, M. , Li, H. , Li, C. , Yan, Y. , Zou, Y. , Micheletti, D. , Koot, E. , Ma, F. and Guan, Q. (2021) Insights into the effect of human civilization on Malus evolution and domestication. Plant Biotechnol. J. 10.1111/pbi.13648
Contributor Information
Fengwang Ma, Email: fwm64@nwafu.edu.cn.
Qingmei Guan, Email: qguan@nwafu.edu.cn.
Data Availability
The WGRS data set generated and analysed in the current study is available from NCBI under BioProject accession PRJNA728537.
References
- Ackermann, J. , Fischer, M. and Amado, R. (1992) Changes in sugars, acids, and amino acids during ripening and storage of apples (cv. Glockenapfel). J. Agric. Food Chem. 40, 1131–1134. [Google Scholar]
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan, A.C. , Hellens, R.P. and Laing, W.A. (2008) MYB transcription factors that colour our fruit. Trends Plant Sci. 13, 99–102. [DOI] [PubMed] [Google Scholar]
- Amyotte, B. , Bowen, A.J. , Banks, T. , Rajcan, I. and Somers, D.J. (2017) Mapping the sensory perception of apple using descriptive sensory evaluation in a genome wide association study. PLoS One, 12, e0171710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, J.P. , Zhang, X.W. , You, C.X. , Bi, S.Q. , Wang, X.F. and Hao, Y.J. (2019) MdWRKY40 promotes wounding‐induced anthocyanin biosynthesis in association with MdMYB1 and undergoes MdBT2‐mediated degradation. New Phytol. 224, 380–395. [DOI] [PubMed] [Google Scholar]
- van Andel, T.R. , Meyer, R.S. , Aflitos, S.A. , Carney, J.A. , Veltman, M.A. , Copetti, D. , Flowers, J.M. et al. (2016) Tracing ancestor rice of Suriname Maroons back to its African origin. Nat. Plants, 2, 16149. [DOI] [PubMed] [Google Scholar]
- Bai, Y. , Dougherty, L. , Cheng, L. , Zhong, G.Y. and Xu, K. (2015) Uncovering co‐expression gene network modules regulating fruit acidity in diverse apples. BMC Genom. 16, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beruter, J. (2004) Carbohydrate metabolism in two apple genotypes that differ in malate accumulation. J. Plant Physiol. 161, 1011–1029. [DOI] [PubMed] [Google Scholar]
- Bianco, L. , Cestaro, A. , Linsmith, G. , Muranty, H. , Denance, C. , Theron, A. , Poncet, C. et al. (2016) Development and validation of the Axiom®Apple480K SNP genotyping array. Plant J. 86, 62–74. [DOI] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Amrine, K.C. , Collins, T.S. , Rivero, R.M. , Vicente, A.R. , Morales‐Cruz, A. , Doyle, C.L. et al. (2015) Developmental and metabolic plasticity of white‐skinned grape berries in response to Botrytis cinerea during noble rot. Plant Physiol. 169, 2422–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Hopfer, H. , Figueroa‐Balderas, R. , Ye, Z. , Rivero, R.M. , Albacete, A. , Perez‐Alfocea, F. et al. (2017) Red blotch disease alters grape berry development and metabolism by interfering with the transcriptional and hormonal regulation of ripening. J. Exp. Bot. 68, 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied, G.D. and Silsby, K.J. (1992) Predicting harvest date windows for apples. In Cornell University Cooperative Extension Bulletin 221. New York: CCE Publications. https://ecommons.cornell.edu/handle/1813/3299 [Google Scholar]
- Bowles, D. , Lim, E.K. , Poppenberger, B. and Vaistij, F.E. (2006) Glycosyltransferases of lipophilic small molecules. Annu. Rev. Plant Biol. 57, 567–597. [DOI] [PubMed] [Google Scholar]
- Brauer, E.K. , Ahsan, N. , Dale, R. , Kato, N. , Coluccio, A.E. , Pineros, M.A. , Kochian, L.V. et al. (2016) The Raf‐like kinase ILK1 and the high affinity K+ transporter HAK5 are required for innate immunity and abiotic stress response. Plant Physiol. 171, 1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundy, M.G.R. , Thompson, O.A. , Sieger, M.T. and Shpak, E.D. (2012) Patterns of cell division, cell differentiation and cell elongation in epidermis and cortex of Arabidopsis pedicels in the wild type and in erecta. PLoS One, 7, e46262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli, E. , Titta, L. , Giorgio, M. , Mock, H.‐P. , Matros, A. , Peterek, S. , Schijlen, E.G.W.M. et al. (2008) Enrichment of tomato fruit with health‐promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 26, 1301–1308. [DOI] [PubMed] [Google Scholar]
- Cai, H. , Zhao, L. , Wang, L. , Zhang, M. , Su, Z. , Cheng, Y. , Zhao, H. et al. (2017) ERECTA signaling controls Arabidopsis inflorescence architecture through chromatin‐mediated activation of PRE1 expression. New Phytol. 214, 1579–1596. [DOI] [PubMed] [Google Scholar]
- Cao, K. , Zhou, Z. , Wang, Q. , Guo, J. , Zhao, P. , Zhu, G. , Fang, W. et al. (2016) Genome‐wide association study of 12 agronomic traits in peach. Nat Commun. 7, 13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi, L. , Malnoy, M. , Goremykin, V. , Nikiforova, S. and Martens, S. (2012) A genome‐wide phylogenetic reconstruction of family 1 UDP‐glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 69, 1030–1042. [DOI] [PubMed] [Google Scholar]
- Cavanagh, C.R. , Chao, S. , Wang, S. , Huang, B.E. , Stephen, S. , Kiani, S. , Forrest, K. et al. (2013) Genome‐wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl Acad. Sci. USA, 110, 8057–8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagne, D. , Carlisle, C.M. , Blond, C. , Volz, R.K. , Whitworth, C.J. , Oraguzie, N.C. , Crowhurst, R.N. et al. (2007) Mapping a candidate gene (MdMYB10) for red flesh and foliage colour in apple. BMC Genom. 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagné, D. , Crowhurst, R.N. , Pindo, M. , Thrimawithana, A. , Deng, C. , Ireland, H. , Fiers, M. et al. (2014) The draft genome sequence of European pear (Pyrus communis L. 'Bartlett'). PLoS One, 9, e92644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, J. , Priya, P. , Dastidar, S.G. and Das, S. (2018) Physical interaction between nuclear accumulated CC‐NB‐ARC‐LRR protein and WRKY64 promotes EDS1 dependent Fusarium wilt resistance in chickpea. Plant Sci. 276, 111–133. [DOI] [PubMed] [Google Scholar]
- Chen, P. , Yan, M. , Li, L. , He, J. , Zhou, S. , Li, Z. , Niu, C. et al. (2020) The apple DNA‐binding one zinc‐finger protein MdDof54 promotes drought resistance. Hortic Res. 7, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Gao, Y. , Xie, W. , Gong, L. , Lu, K. , Wang, W. , Li, Y. et al. (2014) Genome‐wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 46, 714–721. [DOI] [PubMed] [Google Scholar]
- Cheng, M.N. , Huang, Z.J. , Hua, Q.Z. , Shan, W. , Kuang, J.F. , Lu, W.J. , Qin, Y.H. et al. (2017) The WRKY transcription factor HpWRKY44 regulates CytP450‐like1 expression in red pitaya fruit (Hylocereus polyrhizus). Hortic. Res. 4, 17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornille, A. , Giraud, T. , Bellard, C. , Tellier, A. , Le Cam, B. , Smulders, M.J. , Kleinschmit, J. et al. (2013) Postglacial recolonization history of the European crabapple (Malus sylvestris Mill.), a wild contributor to the domesticated apple. Mol. Ecol. 22, 2249–2263. [DOI] [PubMed] [Google Scholar]
- Cornille, A. , Giraud, T. , Smulders, M.J. , Roldan‐Ruiz, I. and Gladieux, P. (2014) The domestication and evolutionary ecology of apples. Trends Genet. 30, 57–65. [DOI] [PubMed] [Google Scholar]
- Cornille, A. , Gladieux, P. , Smulders, M.J. , Roldan‐Ruiz, I. , Laurens, F. , Le Cam, B. , Nersesyan, A. et al. (2012) New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genet., 8, e1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Z. , Luo, J. , Qi, C. , Ruan, Y. , Li, J. , Zhang, A. , Yang, X. et al. (2016) Genome‐wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genom. 17, 946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daccord, N. , Celton, J.M. , Linsmith, G. , Becker, C. , Choisne, N. , Schijlen, E. , van de Geest, H. et al. (2017) High‐quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , Depristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, J. (1997) Guns, germs, and steel: the fates of human societies. New York, NY: W. W. Norton. [Google Scholar]
- Duan, N. , Bai, Y. , Sun, H. , Wang, N. , Ma, Y. , Li, M. , Wang, X. et al. (2017) Genome re‐sequencing reveals the history of apple and supports a two‐stage model for fruit enlargement. Nat. Commun. 8, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt, M.V. , Lee, C.Y. and Liu, R.H. (2000) Nutrition: antioxidant activity of fresh apples. Nature, 405, 903–904. [DOI] [PubMed] [Google Scholar]
- Espley, R.V. , Brendolise, C. , Chagne, D. , Kutty‐Amma, S. , Green, S. , Volz, R. , Putterill, J. et al. (2009) Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell, 21, 168–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espley, R.V. , Hellens, R.P. , Putterill, J. , Stevenson, D.E. , Kutty‐Amma, S. and Allan, A.C. (2007) Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 49, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne, A. , Génard, M. , Lobit, P. , Mbeguié‐A‐Mbéguié, D. and Bugaud, C. (2013) What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 64, 1451–1469. [DOI] [PubMed] [Google Scholar]
- Farneti, B. , Di Guardo, M. , Khomenko, I. , Cappellin, L. , Biasioli, F. , Velasco, R. and Costa, F. (2017) Genome‐wide association study unravels the genetic control of the apple volatilome and its interplay with fruit texture. J Exp Bot. 68, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferree, D.C. and Warrington, I.J. (2003) Apples: Botany, Production and Uses. Wallingford, UK: CABI Publishing. [Google Scholar]
- Gross, B.L. , Henk, A.D. , Richards, C.M. , Fazio, G. and Volk, G.M. (2014) Genetic diversity in Malus x domestica (Rosaceae) through time in response to domestication. Am. J. Bot. 101, 1770–1779. [DOI] [PubMed] [Google Scholar]
- Gu, C. , Guo, Z.‐H. , Hao, P.‐P. , Wang, G.‐M. , Jin, Z.‐M. and Zhang, S.‐L. (2017) Multiple regulatory roles of AP2/ERF transcription factor in angiosperm. Bot. Stud. 58, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, S.A. , Robinson, J.P. and Juniper, B.E. (2002) Genetic clues to the origin of the apple. Trends Genet. 18, 426–430. [DOI] [PubMed] [Google Scholar]
- Harrison, N. and Harrison, R.J. (2011) On the evolutionary history of the domesticated apple. Nat. Genet. 43, 1043–1044. author reply 1044–1045. [DOI] [PubMed] [Google Scholar]
- Hu, D.G. , Sun, C.H. , Ma, Q.J. , You, C.X. , Cheng, L. and Hao, Y.J. (2016) MdMYB1 regulates anthocyanin and malate accumulation by directly facilitating their transport into vacuoles in apples. Plant Physiol. 170, 1315–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L. , Wu, Y. , Wu, D. , Rao, W. , Guo, J. , Ma, Y. , Wang, Z. et al. (2017) The coiled‐coil and nucleotide binding domains of BROWN PLANTHOPPER RESISTANCE14 function in signaling and resistance against planthopper in rice. Plant Cell, 29, 3157–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Kurata, N. , Wei, X. , Wang, Z.X. , Wang, A. , Zhao, Q. , Zhao, Y. et al. (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature, 490, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, H. , Hayashi, N. , Matsushita, A. , Xinqiong, L. , Nakayama, A. , Sugano, S. , Jiang, C.J. et al. (2013) Blast resistance of CC‐NB‐LRR protein Pb1 is mediated by WRKY45 through protein‐protein interaction. Proc. Natl Acad. Sci. USA, 110, 9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson, R.M. , Division, C.R. and Sevice, A.R. (1970) History, Progeny, and Locations of Crabapples of Documented Authentic Origin. Washington, DC: U.S. Department of Agriculture. [Google Scholar]
- Jiang, N. (1986) A preliminary research on the original centre of genus Malus Miller . J. Southwest Agric. Univ. 1, 94–97. [Google Scholar]
- Jones, B. , Frasse, P. , Olmos, E. , Zegzouti, H. , Li, Z.G. , Latché, A. , Pech, J.C. et al. (2002) Down‐regulation of DR12, an auxin‐response‐factor homolog, in the tomato results in a pleiotropic phenotype including dark green and blotchy ripening fruit. Plant J. 32, 603–613. [DOI] [PubMed] [Google Scholar]
- Juniper, B.E. and Mabberley, D.J. (2006) The story of the apple. Portland, OR: Timber Press. [Google Scholar]
- Juniper, B.E. , Watkins, R. and Harris, S.A. (1998) The origin of the apple. Eucarpia Symp. Fruit Breed Genet, 484, 27–34. [Google Scholar]
- King, M.C. and Cliff, M.A. (2002) Development of a model for prediction of consumer liking from visual attributes of new and established apple cultivars. J. Amer. Pomol. Soc. 56, 223–229. [Google Scholar]
- Kiple, K.F. (2007) A Movable Feast: Ten Millennia of Food Globalization. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Kumar, S. , Stecher, G. , Suleski, M. and Hedges, S.B. (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- Kusano, M. , Yang, Z. , Okazaki, Y. , Nakabayashi, R. , Fukushima, A. and Saito, K. (2015) Using metabolomic approaches to explore chemical diversity in rice. Mol. Plant 8, 58–67. [DOI] [PubMed] [Google Scholar]
- Langenfeld, T.V. (1991) Apple‐tree. Morphology, Evolution, Phylogeny, Geography, Taxonomy. Riga: Zinatne. [Google Scholar]
- Larkin, M.A. , Blackshields, G. , Brown, N.P. , Chenna, R. , McGettigan, P.A. , McWilliam, H. , Valentin, F. et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Huang, H. , Zhu, M. , Huang, S. , Zhang, W. , Dinesh‐Kumar, S.P. and Tao, X. (2019) A plant immune receptor adopts a two‐step recognition mechanism to enhance viral effector perception. Mol. Plant, 12, 248–262. [DOI] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. (1989) An investigation of the genetic centre of M. Pumila and Malus in the world. Acta Hortic Sinica, 16, 101–108. [Google Scholar]
- Li, Y. (2001) Researches of Germplasm Resources of Malus Mill. Beijing, China: China Agriculture Press. [Google Scholar]
- Li, Y.Y. , Mao, K. , Zhao, C. , Zhao, X.Y. , Zhang, H.L. , Shu, H.R. and Hao, Y.J. (2012) MdCOP1 ubiquitin E3 ligases interact with MdMYB1 to regulate light‐induced anthocyanin biosynthesis and red fruit coloration in apple. Plant Physiol. 160, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, L. , Zhang, W. , Zhang, B. , Fang, T. , Wang, X.‐F. , Cai, Y. , Ogutu, C. et al. (2021) Unraveling a genetic roadmap for improved taste in the domesticated apple. Mol Plant. S1674‐2052(21)00179‐9. https://www.sciencedirect.com/science/article/pii/S1674205221001799 [DOI] [PubMed] [Google Scholar]
- Lipka, A.E. , Tian, F. , Wang, Q. , Peiffer, J. , Li, M. , Bradbury, P.J. , Gore, M.A. et al. (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics, 28, 2397–2399. [DOI] [PubMed] [Google Scholar]
- Lisec, J. , Schauer, N. , Kopka, J. , Willmitzer, L. and Fernie, A.R. (2006) Gas chromatography mass spectrometry‐based metabolite profiling in plants. Nat Protoc. 1, 387–396. [DOI] [PubMed] [Google Scholar]
- Liu, X.J. , An, X.H. , Liu, X. , Hu, D.G. , Wang, X.F. , You, C.X. and Hao, Y.J. (2017) MdSnRK1.1 interacts with MdJAZ18 to regulate sucrose‐induced anthocyanin and proanthocyanidin accumulation in apple. J. Exp. Bot. 68, 2977–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, G. (2014) The cultivation history of apple in China. J Beijing For. Univ. 13, 15–25. [Google Scholar]
- Ma, B. , Chen, J. , Zheng, H. , Fang, T. , Ogutu, C. , Li, S. , Han, Y. et al. (2015) Comparative assessment of sugar and malic acid composition in cultivated and wild apples. Food Chem. 172, 86–91. [DOI] [PubMed] [Google Scholar]
- Mariette, S. , Wong Jun Tai, F. , Roch, G. , Barre, A. , Chague, A. , Decroocq, S. , Groppi, A. et al. (2016) Genome‐wide association links candidate genes to resistance to Plum Pox Virus in apricot (Prunus armeniaca). New Phytol. 209, 773–784. [DOI] [PubMed] [Google Scholar]
- McClure, K.A. , Gardner, K.M. , Douglas, G.M. , Song, J. , Forney, C.F. , DeLong, J. , Fan, L. et al. (2018) A genome‐wide association study of apple quality and scab resistance. Plant Genome, 11, 170075. [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Puche, L. , Cumplido‐Laso, G. , Amil‐Ruiz, F. , Hoffmann, T. , Ring, L. , Rodríguez‐Franco, A. , Caballero, J.L. et al. (2014) MYB10 plays a major role in the regulation of flavonoid/phenylpropanoid metabolism during ripening of Fragaria × ananassa fruits. J. Exp. Bot. 65, 401–417. [DOI] [PubMed] [Google Scholar]
- Migicovsky, Z. , Gardner, K.M. , Money, D. , Sawler, J. , Bloom, J.S. , Moffett, P. , Chao, C.T. et al. (2016) Genome to phenome mapping in apple using historical data. Plant Genome, 9, plantgenome2015.11.0113. [DOI] [PubMed] [Google Scholar]
- Migicovsky, Z. , Li, M. , Chitwood, D.H. and Myles, S. (2017) Morphometrics reveals complex and heritable apple leaf shapes. Front. Plant Sci. 8, 2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M.G. and Thompson, W.F. (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nocker, S. , Berry, G. , Najdowski, J. , Michelutti, R. , Luffman, M. , Forsline, P. , Alsmairat, N. et al. (2011) Genetic diversity of red‐fleshed apples (Malus). Euphytica, 185, 281–293. [Google Scholar]
- Patel, R.K. and Jain, M. (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One, 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phukan, U.J. , Jeena, G.S. , Tripathi, V. and Shukla, R.K. (2017) Regulation of Apetala2/ethylene response factors in plants. Front. Plant Sci. 8, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell, J.K. and Pritchard, J.K. (2012) Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genet. 8, e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont, C. , Leroy, T. , Seidel, M. , Tondelli, A. , Duchemin, W. , Armisen, D. , Lang, D. et al. (2019) Tracing the ancestry of modern bread wheats. Nat. Genet. 51, 905–911. [DOI] [PubMed] [Google Scholar]
- Popescu, S.C. , Brauer, E.K. , Dimlioglu, G. and Popescu, G.V. (2017) Insights into the structure, function, and ion‐mediated signaling pathways transduced by plant integrin‐linked kinases. Front. Plant Sci., 8, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim, M.A. , Busatto, N. and Trainotti, L. (2014) Regulation of anthocyanin biosynthesis in peach fruits. Planta, 240, 913–929. [DOI] [PubMed] [Google Scholar]
- Robinson, J.P. , Harris, S.A. and Juniper, B.E. (2001) Taxonomy of the genus Malus Mill. (Rosaceae) with emphasis on the cultivated apple. Malus domestica Borkh. Plant Syst. Evol. 226, 35–58. [Google Scholar]
- Rohrer, J.R. , Robertson, K.R. and Phipps, J.B. (1994) Floral morphology of Maloideae (Rosaceae) and its systematic relevance. Am. J. Bot. 81, 574–581. [Google Scholar]
- Shen, H. , Zhong, X. , Zhao, F. , Wang, Y. , Yan, B. , Li, Q. , Chen, G. et al. (2015) Overexpression of receptor‐like kinase ERECTA improves thermotolerance in rice and tomato. Nat Biotechnol. 33, 996–1003. [DOI] [PubMed] [Google Scholar]
- Shen, Q.‐H. , Saijo, Y. , Mauch, S. , Biskup, C. , Bieri, S. , Keller, B. , Seki, H. et al. (2007) Nuclear activity of MLA immune receptors links isolate‐specific and basal disease‐resistance responses. Science 315, 1098. [DOI] [PubMed] [Google Scholar]
- Shirima, R.R. , Maeda, D.G. , Kanju, E. , Ceasar, G. , Tibazarwa, F.I. and Legg, J.P. (2017) Absolute quantification of cassava brown streak virus mRNA by real‐time qPCR. J. Virol. Methods 245, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpak, E.D. (2013) Diverse roles of ERECTA family genes in plant development. J. Integr. Plant Biol. 55, 1238–1250. [DOI] [PubMed] [Google Scholar]
- Sun, X. , Jiao, C. , Schwaninger, H. , Chao, C.T. , Ma, Y. , Duan, N. , Khan, A. et al. (2020) Phased diploid genome assemblies and pan‐genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameling, W.I. , Vossen, J.H. , Albrecht, M. , Lengauer, T. , Berden, J.A. , Haring, M.A. , Cornelissen, B.J. and et al. (2006) Mutations in the NB‐ARC domain of I‐2 that impair ATP hydrolysis cause autoactivation. Plant Physiol. 140, 1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieman, D. , Zhu, G. , Resende, M.F. Jr , Lin, T. , Nguyen, C. , Bies, D. , Rambla, J.L. et al. (2017) A chemical genetic roadmap to improved tomato flavor. Science, 355, 391–394. [DOI] [PubMed] [Google Scholar]
- Torii, K.U. , Mitsukawa, N. , Oosumi, T. , Matsuura, Y. , Yokoyama, R. , Whittier, R.F. and Komeda, Y. (1996) The Arabidopsis ERECTA gene encodes a putative receptor protein kinase with extracellular leucine‐rich repeats. Plant Cell, 8, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toufektsian, M.‐C. , de Lorgeril, M. , Nagy, N. , Salen, P. , Donati, M.B. , Giordano, L. , Mock, H.‐P. et al. (2008) Chronic dietary intake of plant‐derived anthocyanins protects the rat heart against ischemia‐reperfusion injury. J. Nutr. 138, 747–752. [DOI] [PubMed] [Google Scholar]
- Uchida, N. , Lee, J.S. , Horst, R.J. , Lai, H.‐H. , Kajita, R. , Kakimoto, T. , Tasaka, M. et al. (2012) Regulation of inflorescence architecture by intertissue layer ligand–receptor communication between endodermis and phloem. Proc. Natl Acd. Sci. USA, 109, 6337–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco, R. , Zharkikh, A. , Affourtit, J. , Dhingra, A. , Cestaro, A. , Kalyanaraman, A. , Fontana, P. et al. (2010) The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833–839. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Ma, F. and Cheng, L. (2010a) Metabolism of organic acids, nitrogen and amino acids in chlorotic leaves of 'Honeycrisp' apple (Malus domestica Borkh) with excessive accumulation of carbohydrates. Planta, 232, 511–522. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Vieira, F.G. , Crawford, J.E. , Chu, C. and Nielsen, R. (2017a) Asian wild rice is a hybrid swarm with extensive gene flow and feralization from domesticated rice. Genome Res. 27, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010b) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Zhang, X.L. , Wang, L. , Tian, Y. , Jia, N. , Chen, S. , Shi, N.B. et al. (2017b) Regulation of ethylene‐responsive SlWRKYs involved in color change during tomato fruit ripening. Sci Rep. 7, 16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Yu, Y. , Haberer, G. , Marri, P.R. , Fan, C. , Goicoechea, J.L. , Zuccolo, A. et al. (2014) The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 46, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Mauleon, R. , Hu, Z. , Chebotarov, D. , Tai, S. , Wu, Z. , Li, M. et al. (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature, 557, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. and Wu, W.H. (2017) Regulation of potassium transport and signaling in plants. Curr. Opin. Plant Biol. 39, 123–128. [DOI] [PubMed] [Google Scholar]
- Weigel, D. and Mott, R. (2009) The 1001 genomes project for Arabidopsis thaliana . Genome Biol. 10, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. , Wu, X. and Liu, R.H. (2003) Antioxidant activity of apple peels. J. Agric. Food Chem. 51, 609–614. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome‐wide complex trait analysis. Am. J. Hum. Genet. 88, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, Z. (1989) The historical changes, composition, and trends of Japanese cultivated apples. Deciduous Fruits, s1, 7–10. [Google Scholar]
- Yuan, S. , Yan, J. , Wang, M. , Ding, X. , Zhang, Y. , Li, W. , Cao, J. et al. (2019) Transcriptomic and metabolic profiling reveals 'Green Ring' and 'Red Ring' on jujube fruit upon postharvest Alternaria alternata infection. Plant Cell Physiol. 60, 844–861. [DOI] [PubMed] [Google Scholar]
- Zhao, Q. , Feng, Q. , Lu, H. , Li, Y. , Wang, A. , Tian, Q. , Zhan, Q. et al. (2018) Pan‐genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Heat map of the coverage of each accession across each chromosome of the apple genome. Artificial Hybrid, Central Asia, East Asia, and Other refer to the groups in Table S1a.
Figure S2. Unrooted phylogenetic tree of all accessions inferred from whole‐genome re‐sequencing, with Pyrus as an outgroup.
Figure S3. Venn diagram of SNPs among four groups.
Figure S4. Population structure of four groups inferred using ADMIXTURE. (a) The standard estimate of the cross‐validation (CV) error (b) ADMIXTURE analyses for an assumed number of subpopulations (K) from 5 to 9. Each colour represents one ancestral population. Each group is indicated by a vertical bar.
Figure S5. The standard estimate of the cross‐validation (CV) error of population structure of Malus species (Fig. 2B).
Figure S6. Genome‐wide distribution of F ST.
Figure S7. Manhattan plot showing the GWAS results for malic acid content. Gene abbreviations: ALMT, Aluminium‐activated malate transporter family protein; ILK, Integrin‐linked protein kinase family; NME, NAD‐dependent malic enzyme 2. The dotted grey line represents the significant threshold for GWAS (–log10 P = 5).
Figure S8. Manhattan plot showing the GWAS results of content for sucrose content. Gene abbreviations: FBX, F‐box family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S9. Manhattan plot showing the GWAS results for fructose content. Gene abbreviations: AP2L, AP2‐like ethylene‐responsive transcription factor; SFP, Synaptobrevin family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S10. Manhattan plot showing the GWAS results for soluble solid content. Gene abbreviations: CNGC1, cyclic nucleotide‐gated channel 1; Eu1035, Other Eukaryotes‐1035; GVTP, Got1/Sft2‐like vescicle transport protein family; SEFP, sodium/calcium exchanger family protein / calcium‐binding EF hand family protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S11. Manhattan plot showing the GWAS results for fruit firmness. Gene abbreviations: DML1, demeter‐like 1; NTSP, Nucleotide‐diphospho‐sugar transferases superfamily protein. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Figure S12. Manhattan plot showing the GWAS results for fruit weight. The dotted gray line represents the significant threshold for GWAS (–log10 P = 5).
Table S1. Details of all accessions and group information. (a) Details of 297 sequencing accessions for this study. (b) Details of accessions for Malus species analyses.
Table S2. Whole‐genome distribution of SNPs and InDels.
Table S3. Summary SNP statistics in different apple groups and species.
Table S4. Regions significantly associated with domestication‐selective sweeps and potential genes.
Table S5. Regions significantly associated with improvement‐selective sweeps and potential genes.
Table S6. Enrichment of genes from selective sweeps.
Table S7. Agronomic traits and phenotyping data used for GWAS.
Table S8. GWAS‐significant genes overlapping with improvement‐selective sweep signals.
Table S9. GWAS‐significant genes overlapping with domestication‐selective sweep signals.
Table S10. Information of the significant SNPs and associated genes from the GWAS.
Table S10. Information of the significant SNPs and associated genes from the GWAS.
Data Availability Statement
The WGRS data set generated and analysed in the current study is available from NCBI under BioProject accession PRJNA728537.