

Abstract
The gamma secretase catalytic subunit presenilin 1 (PS1) is expressed in the endoplasmic reticulum (ER) of neurons, where it regulates Ca2+ signaling. PS1 is also expressed in heart, but its role in regulation of cardiac Ca2+ transport remains unknown. Since the type 2 sarco/endoplasmic reticulum Ca2+ ATPase (SERCA2a) plays a central role in cardiac Ca2+ homeostasis, we studied whether PS1 regulates the cardiac SERCA2a function. The experiments were conducted in an inducible human SERCA2a stable T-Rex-293 cell line transfected with fluorescently labeled PS1 and the ER Ca2+ sensor R-CEPIA1er. Confocal imaging showed that that PS1 is localized predominantly in the ER membrane. Fluorescent resonance energy transfer (FRET) experiments in HEK293 cells transfected with fluorescently labeled SERCA2a and PS1 revealed that the two proteins directly interact with a 1:1 stoichiometry. The functional significance of this interaction was investigated in a heterologous cellular environment using a novel approach to directly measure ER Ca2+ dynamics. Measurements of SERCA2a-mediated Ca2+ transport showed that PS1 enhanced Ca2+ uptake at low ER Ca2+ loads (<0.15 mM) and reduced uptake at high loads (>0.35 mM). The results of this study revealed that PS1 could act as an important regulator of the cardiac Ca2+ pump function with a complex stimulatory/inhibitory profile.
Keywords: Ca2+ pump, Presenelin 1, Ca2+ transport, FRET, endoplasmic reticulum and HEK293 cells
Graphical Abstract
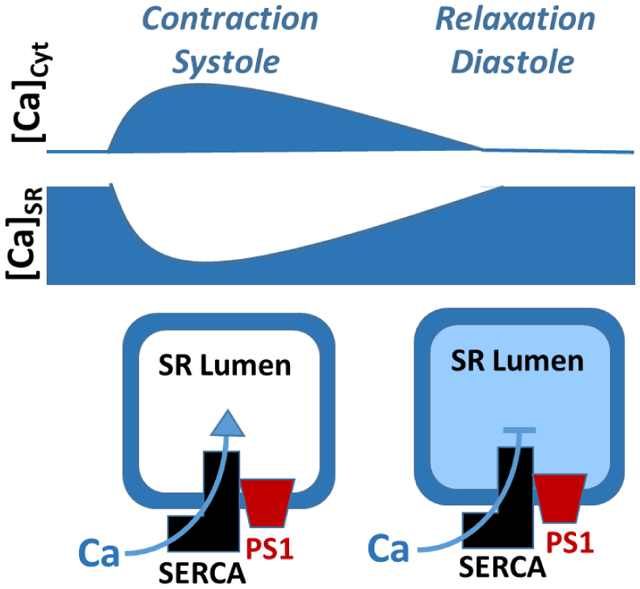
Mechanism of SERCA2a regulation by PS1
PS1 enhances SERCA2a Ca2+ transport low SR Ca2+ loads, whereas at high ER Ca2+ loads PS1 inhibits it. These two opposite PS1 effects on SERCA2a function play an important role in stability of the cardiac Ca2+ cycling. When [Ca2+]SR is depleted after the systolic SR Ca2+ release, PS1 enhances SERCA2a Ca2+ transport, causing faster recovery of SR Ca2+ load. During the diastolic relaxing phase, PS1 limits SERCA2a activity, preventing SR Ca2+ overload.
1. INTRODUCTION
In the heart, the type 2 sarco/endoplasmic reticulum (SR) Ca2+-ATPase (SERCA2a) plays a central role in regulation of Ca2+ homeostasis by transporting cytosolic Ca2+ into the sarcoplasmic reticulum (SR) after the systolic Ca2+ transient. As a result, the rate of SERCA2a Ca2+ transport determines the heart’s relaxation rate during diastole when the heart refills with blood. SERCA2a activity also sets SR Ca2+ load, which determines the magnitude of SR Ca2+ release and the strength of cardiac contraction during systole [1]. Due to its critical role in cardiac Ca2+ homeostasis and heart function, SERCA2a activity is tightly regulated. The main regulation of SERCA2a occurs via its interaction with the transmembrane peptide phospholamban (PLB). PLB reduces the pump apparent affinity for cytosolic Ca2+, decreasing SERCA2a Ca2+ transport [2]. This inhibition can be relieved after PLB phosphorylation by protein kinase A during adrenergic receptor activation [2]. In addition to PLB, additional regulatory partners have been shown to modulate SERCA2a function [3]. Recent discoveries of novel SERCA binding partners [4] have added to the growing appreciation of the complexity of SERCA2a regulation during the cardiac contraction-relaxation cycle.
Presenilin 1 (PS1) is a transmembrane protein that resides in the endoplasmic reticulum (ER) membrane of different cell types. The primary role of PS1 is to act as catalytic subunit of the gamma secretase complex and to cleave amyloid precursor protein (APP) [5]. Dysfunctional PS1 in neurons can affect APP processing and can lead to intracellular accumulation of amyloid β peptide plaques, which is recognized as the leading cause of neuron degradation during Alzheimer’s disease (AD) [6]. It has been shown that several PS1 mutations associated with AD can also cause intracellular Ca2+ dysregulation, including increased ER Ca2+ uptake [7]. Since PS1 is expressed in many different organs of human body including heart [8], we hypothesized that PS1 might also participate in regulation of cardiac Ca2+ homeostasis. Therefore, the aim of this study was to assess whether PS1 affects SERCA2a-mediated Ca2+ transport.
We used fluorescence resonance energy transfer (FRET) analysis in HEK293 cells to assess the interaction between SERCA2a and PS1. Using this assay together with our newly developed approach to measure the endoplasmic reticulum (ER) Ca2+ dynamics [9], we studied the effect of PS1 overexpression on ER Ca2+ transport in a heterologous cellular environment. The results of this study revealed new insight into mechanisms of SERCA2a regulation, suggesting that PS1 can act as an important regulator of the pump function and SR Ca2+ handling in cardiomyocytes.
2. MATERIAL and METHODS
2.1. Vectors and stable line cells production.
pCMV R-CEPIA1er was a gift from Dr. Masamitsu Iino (Addgene, USA) [10]. The vector encoding the human type 2 ryanodine receptor (RyR2) cDNA fused to GFP was kindly provided by Dr. Christopher George (University of Cardiff, UK). The vector encoding human SERCA2a cDNA was kindly provided by Dr. David Thomas (University of Minnesota, USA). The SERCA2a cDNA was cloned into the mCerluean-M1 (mCer) modified plasmid (Addgene, USA) as previously described [11]. The plasmid containing the PS1 coding sequence was a gift from Dr. Beth Stuzmann (Rosalind Franklin University, USA). The PS1 sequence was amplified through PCR with specific primers and cloned into pCMV-mCer. The final product resulted in the PS1 gene with mCer tag fused to the N-terminal domain of the PS1 gene. All the sequences were verified by single pass primer extension analysis (ACGT Inc., USA). Stable inducible Flp-In T-Rex-293 cell line expressing SERCA2a was generated using the Flp-In T-Rex Core Kit (Invitrogen, USA) as described before [11].
2.2. Western Blotting.
SERCA2a stable line cells were transfected with mCer-PS1 two days before experiments. Cells were collected and resuspended in lysis buffer as previously described [12]. After quantification of total protein concentration, equal amounts of total cell lysates were run on a 4–20% SDS-Page gel and blotted using the Turbo transfer system. PS1 was probed with a mouse monoclonal anti-PS1 antibody specific to the C-terminal fragment (Millipore, USA) at a dilution of 1:1000. This antibody recognizes both the full length PS1 and the endocleaved C-terminal fragment. Proteins were visualized using a goat anti-mouse secondary antibody conjugated to HRP at a dilution of 1:5000 and a chemiluminescence detection system (Biorad, USA).
2.3. Experimental protocol to measure FRET.
Progressive acceptor photobleaching was performed as described previously [13]. Briefly, we collected images of mCer and YFP fluorescence at intervals to establish a baseline and then initiated progressive acceptor photobleaching, acquiring successive images of mCer and YFP in between 10 sec of exposure to illumination through a 504/12 nm bandpass filter for selective photobleaching of YFP. The images were analyzed in Metamorph software (Molecular Devices, Sunnyvale, CA) and FRET was calculated from the pre- and post-bleach donor fluorescence intensity using the equation FRET=1 - (FDA/FD), where FDA = the intensity of the donor before bleaching and FD = the intensity of the donor after bleaching. To distinguish between 1:1 and higher order stoichiometry, the fluorescence of the donor was plotted against the fluorescence of the acceptor at the same time point during progressive bleaching.
2.4. Experimental protocol to measure [Ca2+]ER.
Similar experimental protocol as described in [9] was used to study SERCA2a function in living cells. Flp-In T-Rex-293 stably expressing SERCA2a were co-transfected with plasmids containing cDNA of PS1, RyR2 and R-CEPIA1er. PS1 expression and the luminal ER [Ca2+] ([Ca]ER) were measured with a confocal microscope (Radiance 2000 MP, Bio-Rad, UK) equipped with a ×40 oil-immersion objective lens. To verify PS1 expression, mCer was excited with the 458 nm line of the argon laser and signal collected at >485 nm. Saponin (0.005%) was used to permeabilize the surface membrane. Experiments were conducted after washing out of saponin with an experimental solution (in mM): K-aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.22; MgCl2 0.75; HEPES 10; dextran (MW: 40,000) 2% and pH 7.2. Free [Ca2+] and [Mg2+] of this solution were 200 nM and 1 mM, respectively. [Ca2+]ER was recorded as changes in fluorescence of the genetically encoded ER-targeted Ca2+ sensor R-CEPIA1er. R-CEPIA1er was excited with a 514 nm line of the argon laser and signal was collected at >560 nm. The R-CEPIA1er signal (F) was converted to [Ca2+]ER by the following formula: [Ca2+]SE = Kd × [(F - Fmin)/(Fmax - F)]. Fmin and Fmax were recorded in 0 and 5 mM Ca2+ and 5 μM ionomycin, respectively. The Ca2+ dissociation constant (Kd) is 564 μM [14]. SERCA2a-mediated Ca2+ uptake was calculated as the first derivative of [Ca2+]ER refilling (d[Ca2+]ER/dt) during RyR2 inhibition with ruthenium red (15 μM) and tetracaine (1 mM). ER Ca2+ leak was analyzed as the first derivative of [Ca2+]ER decline (d[Ca2+]ER/dt) after inhibition of SERCA with thapsigargin 10 μM. To quantify the rate of SERCA Ca2+ transport, the first derivative of ER Ca2+ uptake (d[Ca2+]ER/dt) was plotted against the corresponding [Ca2+]ER. All images were analyzed with ImageJ software (NIH, USA). All reagents were purchased from Sigma-Aldrich, USA.
2.5. Statistics.
Data are presented as mean +/− standard error of the mean (SEM) of n measurements. Two-sample comparisons were performed using Student’s t-test. Differences were considered statistically significant at P < 0.05. Statistical analysis and graphical representation of averaged data was carried out on OriginPro7.5 software (OriginLab, USA).
3. RESULTS
The following experiments were conducted in an inducible human SERCA2a stable cell line (T-Rex-293 cells) transfected with mCer-PS1. PS1 typically undergoes endoproteolytic cleavage that results in the formation of N-terminal (28 kDa) and C-terminal (18 kDa) fragments, associating with each other as a heterodimeric complex [15] [16]. Western blot analysis showed that transfection of cells with mCer-PS1 DNA significantly increased expression of both the full length (70 kDa) and the cleaved (18 kDa) PS1 species, while endogenous full length PS1 was not detected (Fig 1A). Since we used an antibody specific to the C-terminal region of PS1, the level of N-terminal fragment of PS1 could not be measured in these experiments. Using confocal microscopy, we found that mCer-PS1 expression in T-Rex-293 cells had a similar pattern as the ER-targeted Ca2+ sensor R-CEPIA1er (Fig 1B), indicating that PS1 is localized predominantly in the ER membrane.
Figure 1. Expression of PS1 in human SERCA2a stable cell line.

A, Western blot analysis of endogenous PS1 and mCer-PS1 expression levels in non-transfected and mCer-PS1 transfected cells. B, Confocal images of T-Rex-293 cells transfected with mCer-PS1 together with the ER-targeted Ca2+ indicator R-CEPIA1er. mCer-PS1 expression in these cells showed a similar pattern as R-CEPIA1er, suggesting mCer-PS1 is localized in the ER membrane.
Since both PS1 and SERCA2a are transmembrane proteins localizing in the ER membrane, we studied whether these two proteins directly interact with each other. Intermolecular FRET was measured between mCer-PS1 and YFP-SERCA2a in HEK293 cells. Photobleaching of the FRET acceptor (YFP-SERCA2a) eliminated energy transfer, dequenching the FRET donor (mCer-PS1) and increasing its brightness (Fig 2A). Progressive photobleaching of the acceptor (Fig 2B, arrow) resulted in a corresponding increase in the donor fluorescence. To determine whether PS1 formed regulatory heterodimers or higher-order complexes with SERCA2a, we plotted the donor fluorescence as a function of the acceptor fluorescence level. We observed a linear relationship between donor and acceptor fluorescence (Fig 2C), suggesting that PS1 binds SERCA2a with 1:1 stoichiometry.
Figure 2. Quantification of the PS1-SERCA2a interaction using FRET analysis.
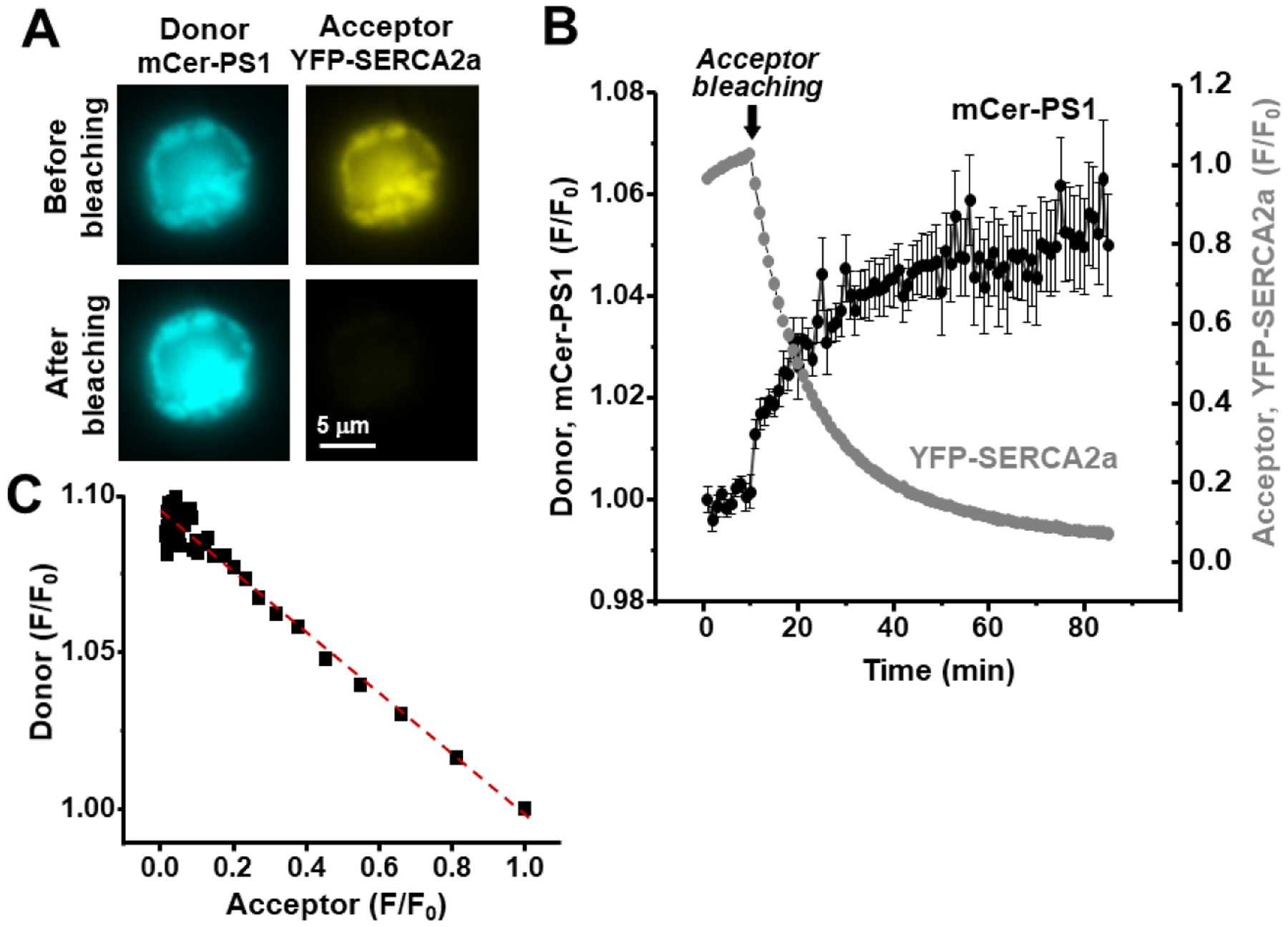
A, Images of HEK293 cells transfected with mCer-PS1 and YFP-SERCA2a. B, Progressive FRET acceptor (YFP-SERCA2a) photobleaching (starting at arrow; grey symbols) increased FRET donor (mCer-PS1) fluorescence (black symbols). C, The relationship between donor and acceptor fluorescence during the acceptor photobleaching protocol (B). The linear relationship (red dashed line) suggested that PS1 binds SERCA2a with a 1:1 stoichiometry.
Next, we used a newly developed approach to investigate the functional significance of PS1 and SERCA2a interaction. Changes in [Ca2+]ER were recorded with the ER Ca2+ sensor R-CEPIA1er in SERCA2a stable cell line as described before [9]. To manipulate ER Ca2+ load, the cells were also transfected with the Ca2+ release channel RyR2. The plasma membrane was permeabilized to control the cytosolic environment, including [Ca2+] and [ATP]. The SERCA2a function was quantified from the rate of [Ca2+]ER reuptake after full ER Ca2+ depletion caused by RyR2 activation with caffeine (Fig 3A; Caf) followed by RyR2 inhibition with ruthenium red (RR) and tetracaine. At the end of the experiment, the R-CEPIA1er fluorescence signal was calibrated with ionomycin to convert the fluorescent signal to [Ca2+]ER. To quantify the rate of SERCA Ca2+ transport, the first derivative of ER Ca2+ uptake (d[Ca2+]ER/dt) was plotted against the corresponding [Ca2+]ER. Since cells in the PS1 group were transfected with mCer-PS1, ER Ca2+ uptake rate was only analyzed in cells with the mCer signal. We found that that at low ER Ca2+ loads ([Ca2+]ER≤0.15 mM), PS1 overexpression enhances ER Ca2+ uptake by 39% (n=20 cells) compared to cells expressing SERCA2a alone (Ctrl, n=36 cells; Fig 3B). However, at high ER Ca2+ loads ([Ca2+]ER≥0.35 mM), PS1 inhibited ER Ca2+ uptake by 49% (n=20 cells; Fig 3B). As a result, PS1 overexpression reduced the maximal ER Ca2+ load by ~24% (Fig 3C). Since cytosolic [Ca2+] was held at a constant level throughout the experiment, we concluded that PS1 regulates SERCA2a function via the [Ca2+]ER-dependent mechanism.
Figure 3. Effect of PS1 overexpression on SERCA2a mediated ER Ca2+ uptake and ER Ca2+ load in human SERCA2a stable cell line.

A, Experimental protocol to measure ER Ca2+ uptake as a function of [Ca2+]ER. Changes in [Ca2+]ER during RyR2 activation with caffeine (Caf) followed by RyR2 inhibition with ruthenium red (RR) and tetracaine. The R-CEPIA1er fluorescence was calibrated with ionomycin to convert the recorded signal to [Ca2+]ER (see Material and Method). Experiments were conducted in cells expressing SERCA2a (black trace, Ctrl), and in cells expressing SERCA2a and PS1 (grey trace, PS1). B, Averaged data of the ER Ca2+ uptake rate at 0.15 mM [Ca2+]ER (left) and at 0.35 mM [Ca2+]ER (right). C, The maximum ER Ca2+ load in cells expressing SERCA2a alone (Ctrl; n=36 cells) and cells expressing SERCA2a and PS1 (PS1; n=20 cells).
It has been suggested that PS1 can aggregate forming ER Ca2+ leak channel [17]. Our ER Ca2+ uptake analysis requires similar RyR-independent Ca2+ leak rate between experimental groups. Increased ER Ca2+ leak in cells overexpressing PS1 could affect ER Ca2+ uptake measurements by offsetting the rate of [Ca2+]ER recovery after full ER Ca2+ depletion (Fig 3A). Therefore, we studied whether PS1 overexpression affects ER Ca2+ leak in SERCA2a stable cell line transfected with mCer-PS1 and R-CEPIA1er. To exclude ER Ca2+ leak via RyR2, cells were not transfected with RyR2. We measured the rate of [Ca2+]ER decline after SERCA2a inhibition with thapsigargin (10 μM) (Fig 4A). ER Ca2+ leak was quantified as the first derivative of the [Ca2+]ER decline. No significant difference was found in the Ca2+ leak rate in cells expressing PS1 compared to cells expressing SERCA2a alone (Fig 4B). These results illustrate that RyR-independent Ca2+ leak was not affected by the level of PS1 overexpression achieved in our expressing cell system.
Figure 4. Effect of PS1 overexpression on ER Ca2+ leak.

A, Experimental protocol to measure ER Ca2+ leak as a function of [Ca2+]ER. Changes of [Ca2+]SR in control conditions and after SERCA inhibition with thapsigargin (10 μM). The R-CEPIA1er fluorescence was calibrated with ionomycin to convert the recorded signal to [Ca2+]ER (see Material and Method). B, Average ER Ca2+ leak in cells expressing SERCA2a alone (Ctrl; n=8 cells) and cells expressing SERCA2a and PS1 (PS1; n=14 cells).
4. DISCUSSION
Previous studies have shown that several PS1 mutations associated with AD can cause intracellular Ca2+ dysregulation in neurons [18] [19]. Defective PS1 gamma secretase activity can affect Ca2+ homeostasis indirectly via an accumulation of amyloid β peptide plaques [20]. Other studies showed that the AD-related PS1 mutations can directly affect neuronal Ca2+ signaling by exacerbating ER Ca2+ leak [21] [22] [17] [23] or altering SERCA function [7]. While PS1 is also expressed in the heart, to our knowledge, its effect on the cardiac Ca2+ pump function has never been explored.
In this study, using a newly developed assay that directly measure ER Ca2+ uptake, we identified PS1 as a novel regulator of the cardiac SERCA2a function. We found that overexpression of PS1 in the human SERCA2a stable cell line modulates ER Ca2+ uptake by [Ca]ER-dependent mechanism. PS1 enhances the Ca2+ uptake rate at low ER Ca2+ loads, whereas at high ER Ca2+ loads PS1 inhibits the SERCA2a function (Fig 3). We hypothesized these two opposite effects of PS1 on SERCA2a Ca2+ transport might play an important role in the cardiac Ca2+ cycling. When [Ca2+]SR is depleted after the systolic SR Ca2+ release, PS1 would enhance SERCA2a Ca2+ reuptake, causing faster recovery of SR Ca2+ load. During the diastolic relaxing phase, however, PS1 would limit SERCA2a activity, preventing SR Ca2+ overload. The extreme SR Ca2+ load commonly caused by adrenergic receptor stimulation can trigger spontaneous SR Ca2+ release and cardiac arrhythmias [24] [25]. Thus, SERCA2a regulation by PS1 might represent an important molecular mechanism that insures a stability of the cardiac Ca2+ cycling.
Using FRET analysis, we found that SERCA2a regulation by PS1 involves a direct interaction of PS1 with the Ca2+ pump (Fig 2). To our knowledge, this is the first study that directly demonstrates the physical interaction between SERCA2a and PS1, as all the previous structural studies relied on indirect assays that cannot distinguish between direct and indirect protein interactions [7,8]. Furthermore, the linear donor-acceptor relationship during acceptor photobleaching indicates a 1:1 stoichiometry, suggesting that PS1 binds to SERCA2a as a monomer. However, we cannot rule out the possibility that PS1 oligomers bind to SERCA2a in a configuration that sets all but one acceptor beyond reach of FRET. The principal mechanism of SERCA2a regulation occurs through an interaction with PLB [4]. Since all experiments were conducted in the absence of PLB, we cannot conclude whether PS1 competes with PLB in SERCA2a regulation. However, the biphasic effect of PS1 on ER Ca2+ uptake (which is different from an inhibitory effect of PLB on SERCA2a function) suggests that PS1 regulates SERCA2a independently of PLB.
One previous study has shown that PS1 can act as an activator of SERCA2b [7]. The difference between this and our findings can be explained by different experimental approaches used to measure SERCA function. Green et al. recorded an increase in cytosolic [Ca2+] as an index of SERCA function. Since SERCA Ca2+ transport was measured at resting-low ER Ca2+ loads, the inhibition of SERCA by PS1 at high ER Ca2+ loads could not be detected in that study. Our approach, however, allows studying ER Ca2+ uptake throughout a wide range of ER Ca2+ load: from completely depleted to maximal load. This novel assay identified the complex mechanism of SERCA2a regulation by PS1. First, direct measurement of ER Ca2+ dynamics allows to separate the SERCA-dependent Ca2+ uptake from Ca2+ fluxes mediated by other Ca2+ transporters on the surface membrane and in intracellular organelles. Therefore, we were able to uncover new complex mechanisms of SERCA2a regulation by PS1. In addition, our assay allowed to study the effect of PS1 on the maximum ER Ca2+ load, which is determined by the pump’s catalytic efficiency (i.e. ATPase-Ca2+ transport coupling) and the thermodynamic limit set by the ATP/ADP ratio. We found that PS1 not only inhibits SERCA2a Ca2+ uptake at higher ER Ca2+ loads, but also limits the maximum Ca2+ load. This mechanism might play an important role in preventing SR Ca2+ overload and cardiac arrhythmias during adrenergic receptor stimulation. Moreover, differences between our and Green et al. findings can be explained by differences in regulation of different SERCA isoforms by PS1. As SERCA2b and SERCA2a are characterized by significant structural and functional differences [26], PS1 might have a different functional effect on different SERCA isoforms.
Another mechanism of ER Ca2+ regulation by PS1 involves a clustering of PS1 to function as a low conductance Ca2+ leak channel [17]. Our Ca2+ uptake assay requires similar ER Ca2+ leak (particularly RyR2-independent component) between experimental groups. Increased ER Ca2+ leak in cells overexpressing PS1 could affect ER Ca2+ uptake by offsetting the rate of [Ca2+]ER recovery after full [Ca2+]ER depletion. We found that ER Ca2+ leak was not significantly affected by the overexpression of PS1 in our cell system (Fig 4). However, our results do not rule out a possibility that PS1 can form cation leak channels. PS1-mediated ER Ca2+ leak is likely to be depend on the level of PS1 expression. It seems that the levels of PS1 expression achieved in our cell system were not sufficient to induce opening of PS1 Ca2+ leak channels. It is also possible that experimental conditions (membrane permeabilization, low cytosolic [Ca2+] and high cytosolic [ATP]) used in our studies prevent opening of the PS1 cation leak channels.
5. CONCLUSIONS
This study, conducted in a heterologous cellular environment, reveals PS1 as a novel component of the cardiac Ca2+ regulation that directly interacts with SERCA2a and regulates SR Ca2+ transport. Several clinical studies reported that AD-related PS1 mutations are associated with cardiomyopathies [8] [27] [28]. Since the SERCA2a-PS1 interaction can be beneficial for SR Ca2+ handling, we expect that such PS1 mutations could cause SR Ca2+ mishandling contributing to cardiomyopathies. Therefore, more research is needed to define a functional significance of SERCA2a regulation by PS1 in native environment of cardiomyocytes.
HIGHLIGHTS:
Presenilin 1 directly interacts with SERCA2a.
Direct measurement of ER Ca2+ shows that PS1 directly regulates SERCA2a function.
PS1 regulates SERCA2a with a complex stimulatory/inhibitory profile that is [Ca2+]ER dependent.
AKNOWLEDGMENTS
The authors would like to thank Dr. David Thomas (University of Minnesota, USA) for providing the vector encoding the human hSERCA2a and Dr. Grace B. Stuzmann (Rosalind Franklin University) for providing the vector encoding PS1. The authors also would like to thank Dr. Iino for donating the R-CEPIA1er vector.
SOURCES OF FUNDING
This work was supported by Research Funding Committee Award from Loyola University Chicago (to E.B.), NIH Grants HL151990 (to A.V.Z.), HL092321 and HL143816 (to S.L.R.) and by AHA Grant 20TPA35500032 (to A.V.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
None.
REFERENCES
- [1].Bers DM, Cardiac excitation-contraction coupling, Nature. 415 (2002) 198–205. 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- [2].Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR, Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains, J Biol Chem. 261 (1986) 13333–13341. [PubMed] [Google Scholar]
- [3].Kranias EG, Hajjar RJ, Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome, Circ Res. 110 (2012) 1646–1660. 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, Olson EN, Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides, Sci Signal. 9 (2016) ra119. 10.1126/scisignal.aaj1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].De Strooper B, Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex, Neuron. 38 (2003) 9–12. 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- [6].De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F, Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein, Nature. 391 (1998) 387–390. 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- [7].Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM, SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production, J Cell Biol. 181 (2008) 1107–1116. 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gianni D, Li A, Tesco G, McKay KM, Moore J, Raygor K, Rota M, Gwathmey JK, Dec GW, Aretz T, Leri A, Semigran MJ, Anversa P, Macgillivray TE, Tanzi RE, del Monte F, Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy, Circulation. 121 (2010) 1216–1226. 10.1161/CIRCULATIONAHA.109.879510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bovo E, Nikolaienko R, Bhayani S, Kahn D, Cao Q, Martin JL, Kuo IY, Robia SL, Zima AV, Novel approach for quantification of endoplasmic reticulum Ca2+ transport, Am J Physiol Heart Circ Physiol. 316 (2019) H1323–H1331. 10.1152/ajpheart.00031.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M, Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA, Nat Commun. 5 (2014) 4153. 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bovo E, Nikolaienko R, Cleary SR, Seflova J, Kahn D, Robia SL, Zima AV, Dimerization of SERCA2a Enhances Transport Rate and Improves Energetic Efficiency in Living Cells, Biophys J. 119 (2020) 1456–1465. 10.1016/j.bpj.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nikolaienko R, Bovo E, Rebbeck RT, Kahn D, Thomas DD, Cornea RL, Zima AV, The functional significance of redox-mediated intersubunit cross-linking in regulation of human type 2 ryanodine receptor, Redox Biol. 37 (2020) 101729. 10.1016/j.redox.2020.101729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Singh DR, Dalton MP, Cho EE, Pribadi MP, Zak TJ, Šeflová J, Makarewich CA, Olson EN, Robia SL, Newly Discovered Micropeptide Regulators of SERCA Form Oligomers but Bind to the Pump as Monomers, J Mol Biol. 431 (2019) 4429–4443. 10.1016/j.jmb.2019.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bovo E, Martin JL, Tyryfter J, de Tombe PP, Zima AV, R-CEPIA1er as a new tool to directly measure sarcoplasmic reticulum [Ca] in ventricular myocytes, Am J Physiol Heart Circ Physiol. 311 (2016) H268–275. 10.1152/ajpheart.00175.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS, Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo, Neuron. 17 (1996) 181–190. 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- [16].Stable association of presenilin derivatives and absence of presenilin interactions with APP - PubMed, (n.d.). https://pubmed.ncbi.nlm.nih.gov/9666482/ (accessed March 15, 2021). [DOI] [PubMed]
- [17].Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, Bezprozvanny I, Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations, Cell. 126 (2006) 981–993. 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Supnet C, Bezprozvanny I, The dysregulation of intracellular calcium in Alzheimer disease, Cell Calcium. 47 (2010) 183–189. 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Honarnejad K, Herms J, Presenilins: role in calcium homeostasis, Int J Biochem Cell Biol. 44 (2012) 1983–1986. 10.1016/j.biocel.2012.07.019. [DOI] [PubMed] [Google Scholar]
- [20].Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S, Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1, Nature. 383 (1996) 710–713. 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- [21].Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL, Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease, Proc Natl Acad Sci U S A. 91 (1994) 534–538. 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kasri NN, Kocks SL, Verbert L, Hébert SS, Callewaert G, Parys JB, Missiaen L, De Smedt H, Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knock-out cells, Cell Calcium. 40 (2006) 41–51. 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- [23].Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, Teich AF, Zalk R, Saint N, Arancio O, Bauer C, Duprat F, Briggs CA, Chakroborty S, Stutzmann GE, Shelanski ML, Checler F, Chami M, Marks AR, Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits, Acta Neuropathol. 134 (2017) 749–767. 10.1007/s00401-017-1733-7. [DOI] [PubMed] [Google Scholar]
- [24].Katra RP, Oya T, Hoeker GS, Laurita KR, Ryanodine receptor dysfunction and triggered activity in the heart, Am J Physiol Heart Circ Physiol. 292 (2007) H2144–2151. 10.1152/ajpheart.00924.2006. [DOI] [PubMed] [Google Scholar]
- [25].Venetucci LA, Trafford AW, O’Neill SC, Eisner DA, The sarcoplasmic reticulum and arrhythmogenic calcium release, Cardiovasc Res. 77 (2008) 285–292. 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- [26].Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A, Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses, J Biol Chem. 278 (2003) 47877–47889. 10.1074/jbc.M306784200. [DOI] [PubMed] [Google Scholar]
- [27].Li H, Chen Y, Zhou B, Peng Y, Sheng Y, Rao L, Polymorphisms of presenilin-1 gene associate with dilated cardiomyopathy susceptibility, Mol Cell Biochem. 358 (2011) 31–36. 10.1007/s11010-011-0916-0. [DOI] [PubMed] [Google Scholar]
- [28].Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE, Rare variant mutations identified in pediatric patients with dilated cardiomyopathy, Prog Pediatr Cardiol. 31 (2011) 39–47. 10.1016/j.ppedcard.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]