

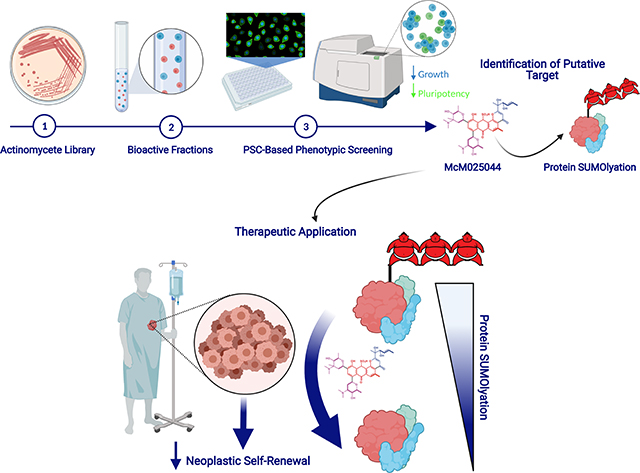
Summary
Natural products (NP) encompass a rich source of bioactive chemical entities. Here, we used human cancer stem cells (CSCs) in a chemical genomics campaign with NP chemical space to interrogate extracts from diverse strains of actinomycete for anti-cancer properties. We identified a compound (McM25044), capable of selectively inhibiting human CSC function vs. normal stem cell (SC) counterparts. Biochemical and molecular studies revealed that McM025044 exerts inhibition on human CSCs through the small ubiquitin-like modifier (SUMO) cascade, found to be hyperactive in human a variety of human cancers. McM025044 impedes SUMOylation pathway via direct targeting of the SAE1/2 complex. Treatment of patient-derived CSCs resulted in reduced levels of SUMOylated proteins and suppression of progenitor and stem cell capacity measured in vitro and in vivo. Our study overcomes a barrier in chemically inhibiting oncogenic SUMOylation activity and uncovers a unique role for SAE2 in the biology of human cancers.
eTOC Blurb
Benoit et al. screened extracts from strains of actinomycete to uncover and isolate a molecule that inhibits SUMOylation in human cancer stem cells. This natural product compound acts via biochemical targeting of a specific motif in a SUMOylation enzyme, SAE2, that regulates self-renewal of a variety of human cancer cells.
Graphical Abstract
Introduction
Natural products (NPs) are enriched for chemical properties that uniquely modulate networks controlling diverse cellular processes(Harvey et al., 2015). Exploitation of distinct NP characteristics is exemplified in cancer treatment, with more than 60% of approved drugs arising from NP inspired molecules(Newman and Cragg, 2012), and the majority of these originating from microorganisms(Harvey et al., 2015; Pye et al., 2017). First generation cancer therapeutics were developed with a focus on targeting proliferating cell that largely comprise of anti-mitotics, antimetabolites, or alkylating agents. While the use of such anti-neoplastic drugs can be effective at achieving cancer remission, they exhibit lack of selectivity for malignant cells, thus affecting all highly proliferative entities including healthy progenitor cells. Rare CSC populations with tumorigenic self-renewal ability capable of de novo initiation of disease have been suggested to elude untargeted antineoplastic therapies, allowing cancer relapse(Bonnet and Dick, 1997; Flemming, 2015; Visvader and Lindeman, 2012). Recent results suggest that the quiescent nature of human CSCs that allows them to avoid chemotherapeutics may not be valid in all cases and is more complex that originally believed(Boyd et al., 2018). The biological processes that govern human CSC survival and self-renewal have been well studied, however, the identification of targetable networks specific to human CSCs dependency vs. healthy counterparts warrants further investigation.
The goal to target specific and rare tumor subpopulations, including popularized CSCs, adds an additional level of complexity for development of anticancer therapies. Natural products (NPs) isolated from plants, microorganisms, and animal tissues have historically proven to be a rich source of unique molecules for new therapeutics(Harvey et al., 2015; Wright, 2017) where the origins of half of FDA-approved drugs over the past 30 years are attributed to NPs(Newman and Cragg, 2016),(Sukuru et al., 2009). As microorganisms represent a diverse and accessible reservoir of bioactive small molecules(Harvey et al., 2015), our group deployed a human SC-based phenotypic testing platform(Sachlos et al., 2012) that is based on transformed vs. normal embryonic pluripotent stem cells, uniquely using crude extracts from soil dwelling actinomycetes to mine this chemical space towards developing lead therapeutics compounds and identifying CSC targets. We identify a natural product compound (McM02544) with selective anti-neoplastic activity on a variety of human cancers.
RESULTS
Activity-guided identification of a natural product with a broad anti-cancer activity
In search of novel biologically active chemical matter capable of selectively targeting human cancer stemness (Bonnet and Dick, 1997; Visvader and Lindeman, 2012), we performed an established multitiered chemical genomics screen (Sachlos et al., 2012) using crude natural product extracts from soil dwelling Actinomycetes as a source of chemical probes. As each strain has been known to produce 30 or more individual molecules, screening crude extracts provided an advantage by intrinsically pooling chemical space that contains compounds not found in curated libraries. A total of 960 distinct crude extracts were prepared from 320 actinomycete strains, where each extract was screened for its ability to inhibit cell growth in a surrogate of human CSCs (transformed H9 human ES cells: t-hESC)(Benoit et al., 2017; Sachlos et al., 2012; Werbowetski-Ogilvie et al., 2009), represented by transformed pluripotent cells previously established to be a powerful tool to identify anti-cancer drugs, that were validated in clinical trials(Aslostovar et al., 2018; Benoit et al., 2017; Sachlos et al., 2012).
The initial screening step allowed separation of active vs. inactive compounds based on growth inhibition assessment in t-hESCs (Figure 1A). Batch-to-batch variation and reproducibility in NP production from microorganisms is well known in this field and limits the efficiency of screening campaigns to identify a suitable number of candidate compounds. Thus, we re-grew cultures of all samples with activity on t-hESCS, as well as a random selection of inactive samples as comparative controls, to generate new batches of extracts for secondary as well as tertiary re-screening (Figure 1B). Using this rigorous approach, we found that 50% of samples that were active in the first batch maintained their activity across the next two batches (Figure 1B). Based on reproducible effects, we identified a group of 12 strains exhibiting consistent bioactivity when isolated from culture conditioned media (Table S1). These strains were subjected to activity-guided fractionation over Diaion HP20 followed by elution using varied concentrations of methanol. Maximum potency for t-hESC growth inhibition was obtained from 90% methanol extraction in 10 strains out of 12 (Figure S1A). Next, we sought to identify extracts causing selective growth inhibition in t-hESC cultures over healthy counterpart PSCs in dose-response experiments (Figure S1B). Extract from strain WAC067 exhibited consistent batch-to-batch activity with the most prominent level of selectivity to preferentially effect t-hESCs vs. low effects in healthy hESCs (Figure 1C, Figure S1B, green shade). To contextualize this result, favourable selectivity for CSC targeting was not observed with chemotherapeutic agents e.g. cytarabine, used to treat several cancers, and forms the standard of treatment in adult myeloid leukemia (Figure S1B, red shade)(Boyd et al., 2018). Using a process of iterative “anti-t-hESC activity” guided purification (LH-20 fractionation followed by IC50 determination, Figure S1C), bioactive extract of WAC067 showing the highest potency was resolved into three distinct peaks using reverse phase chromatography (Figure 1D). These peaks were collected to determine molecular weight by mass spectrometry (Figure S1D). Peak #2 from WAC067 (identified as a strain of Streptomyces maoxianensis by 16S rRNA gene sequence) was found to provide the highest potency and greatest selectivity (green shaded area) to human CSC (Figure 1E) and was thus selected as our lead compound.
Figure 1. Identification of an anti-cancer stem cell lead candidate from crude natural product libraries.
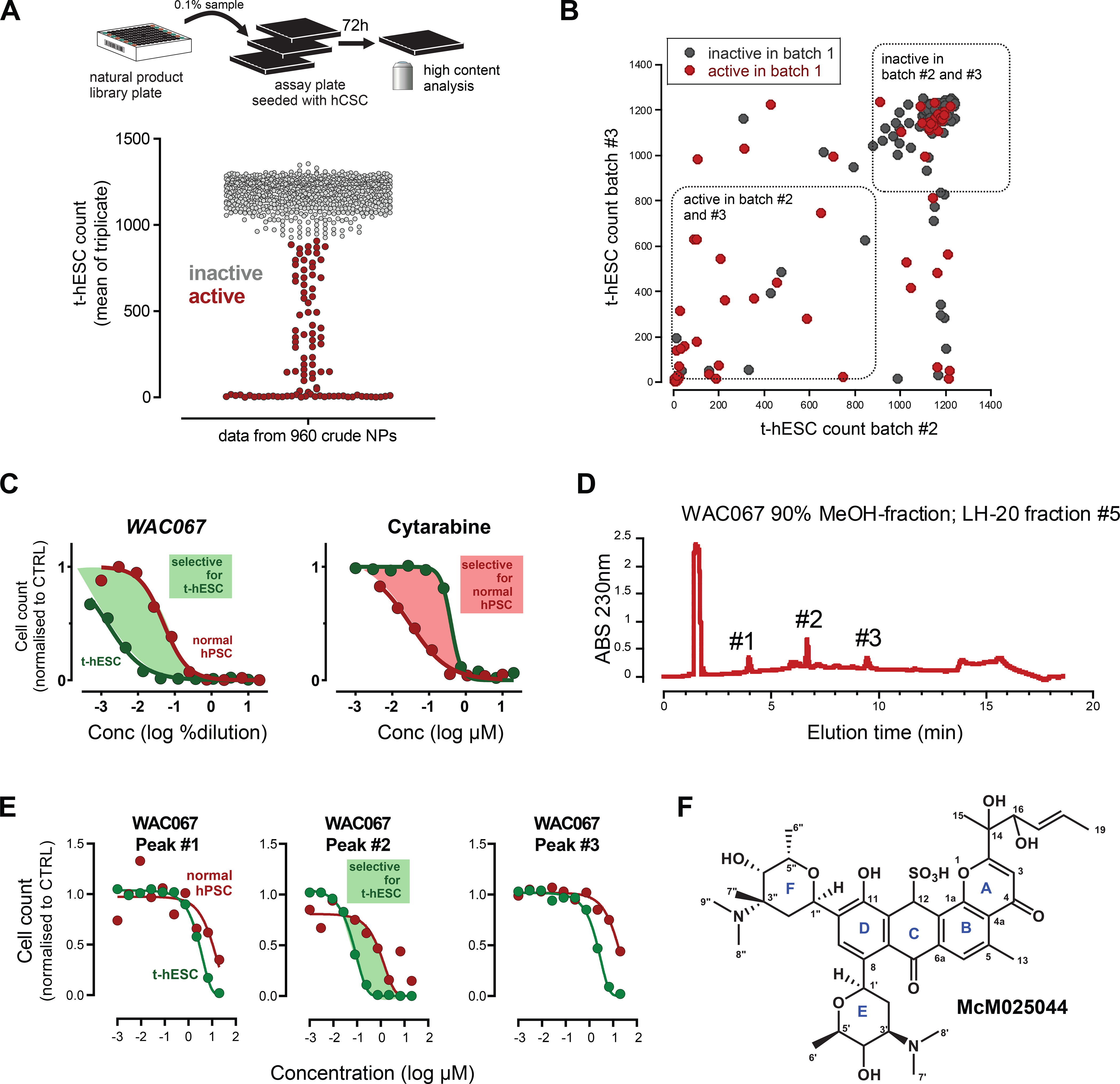
A) High content phenotypic screening assay used to identify CSC bioactive natural products from a unique library of actinomycetes. Transformed human ES cell (t-hESC) counts were used to determine CSC bioactivity from 960 crude natural products derived from actinomycetes cultures. Samples were screened in technical triplicates. Means of triplicates are plotted.
B) To quantify batch-to-batch reproducibility, the active samples and a random subset of inactive samples were regrown from library cultures and the natural product extract regenerated. Half of the samples in batch #1 showed reproducibility across multiple batches (33/67 samples).
C) The cancer-cell selectivity of the 90% methanol fractions was evaluated by comparing the IC50s of t-hESC (green) vs. normal hPSC (red). WAC067 was identified that had cancer-cell selectivity (green shading). In contrast, front line AML chemotherapy Cytarabine has an undesirable higher potency against normal hPSC over t-hESC (red shading).
D) Reverse phase HPLC of LH-20 fraction 5 revealed three distinct peaks for WAC067, LH20 fraction #5. Peaks were isolated, molecular weight determined.
E) Three HPLC separated peaks were isolated and their IC50 for t-hESC and normal hPSC quantified. Peak#2 was found to show cell-selectivity towards t-hESC (shaded green area).
F) Chemical structure of MCM05044. Assignment of stereochemical configuration of rings E and F is based on consistent homology among the related natural products kidamycin, pluramycin A, and hedamycin. Stereochemical configuration at C12, C14, C16 is unknown.
Phenotypic screening has the advantage of being performed in a disease-relevant cell type while remaining agnostic to putative targets. Despite this advantage, subsequent identification of the molecular target of lead candidates identified from phenotyping screens is challenging(Kotz, 2012). This is further complicated by the lack of prior knowledge of compound structure of NPs. To characterize the structure of our lead compound, we employed an approach used previously by our group for novel antibiotic identification e.g. Ibomycin(Robbins et al., 2016). Peak #2 isolated from WAC067 LH-20 Fraction #5 (90%MeOH extraction) was registered as McM025044 and structurally characterised (Figure S1E). HPLC-purified McM025044 was subjected to high-resolution electrospray ionization mass spectrometry (HR-ESI-MS), as well as 1D and 2D nuclear magnetic resonance (NMR) analysis (Figure S1F-J). The molecular formula of McM025044 was determined to be C41H54N2O13S. Two dimensional NMR correlations established a tetracyclic angular anthrapyranone core scaffold with an angolosaminyl C-glycoside substituent at C8 and an N,N-dimethylvancosaminyl C-glycoside group at C10 (Figure S1I, Table S2). The same tetracyclic scaffold, adorned with the same two C-glycosides, is found in multiple members of the pluramycin family of natural products: kidamycin, pluramycin A, hedamycin, and saptomycin B and C1(Hansen and Hurley, 1996). Specifically, McM025044 was found to present a structure highly similar to Saptomycin B (Kitamura et al., 2014). Careful comparison of 1D NMR data of McM025044 to those of Saptomycin B showed that the ketone group (C-12, δ 188.0 ppm) in Saptomycin B was substituted for a sulfonate methine (δ 6.12, 54.4 ppm) in McM025044. The H-14 (δ 2.88) in Saptomycin B was substituted for a hydroxyl group (δ 5.33) in McM025044. The methylene group (C-16, δH 2.50, 2.74 ppm and δC 31.6 ppm) in Saptomycin B was substituted for oxymethine (C-16, δH 4.70 ppm and δC 70.6 ppm) in McM025044. These connections were further confirmed by 1H-1H COSY and HMBC correlations (Suppl. Figure 1E, 1J, Table S2). Unlike all previously described pluramycins, McM025044 contains an sp3-hybridized C12 carbon on its C-ring functionalized with a sulfonic acid moiety, and an unprecedented vicinal diol-containing side chain on the A-ring (Figure 1F), and thus represents a member of this family not reported to date.
We observed that McM025044 is causing significant reduction in proliferation, loss of hallmarks of human stem cell pluripotency, and apoptosis in t-hESCs (Figure 2A-C). Similar effects were observed when McM025044 was tested on different models of human somatic cancers possessing CSC properties, such as breast cancer, colorectal cancer, and leukemia (Figure S2A). In contrast to other pluramycin family members, McM025044 does not induce apoptosis by causing DNA damage (Figure S2B-D). To determine the extent of McM025044 biological effect on neoplastic stem/progenitor activity, we examined the effect of McM025044 in an automated 3D spheroid assays from human colon and breast cancer cells (Figure S2E). McM025044 inhibited sphere-formation capacity (Figure 2D, Figure S2E), and induced growth inhibition across AML cell lines, including OCI-AML3 that is highly enriched for stem/progenitor properties (Mills et al., 2009) (Figure 2D), indicating that targeted biological effectors of MCM025044 can be found in a variety of human somatic lineage-specific cells with CSCs properties. This suggests a broad applicability of this compound in human cancers, including rarer stem/progenitor compartment of heterogeneous human tumor biology.
Figure 2. McM025044 shows anti-neoplastic activity in a broad range of human tumor models.
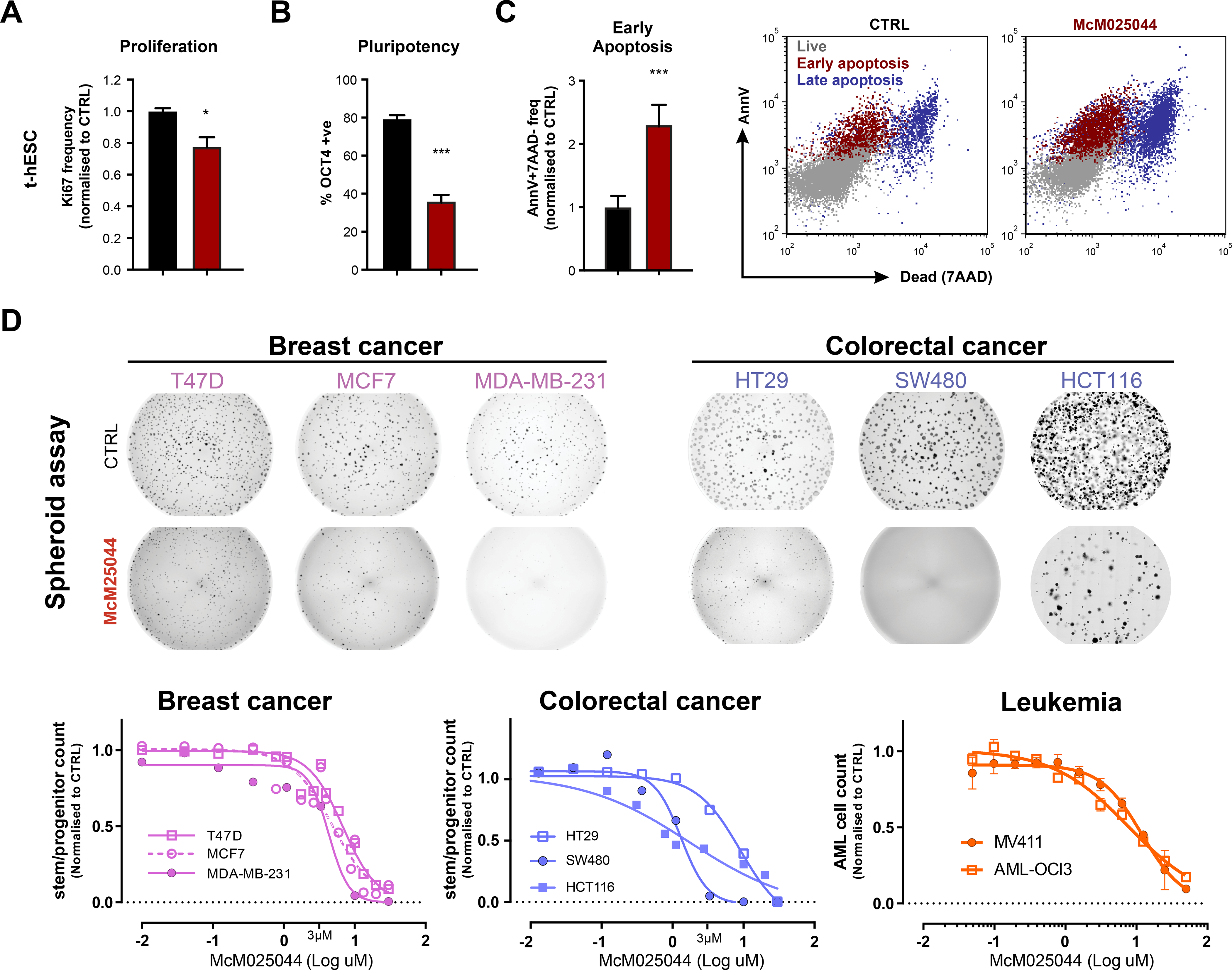
A) McM025044 inhibits proliferation (ki67, n=3), B) induces exit from pluripotency (OCT4, n=12), and C) induces apoptosis (AnnexinV +7AAD−, n=6) in t-hESC, as evaluated by flow cytometry (1uM McM025044 in representative flow plot). Data are represented as mean ± SEM (*: p=0.024, ***: p<0.0001).
D) Sphere forming assays using breast (pink) and colorectal (blue) cell lines, either untreated (DMSO control) or treated with McM025044 (3uM). Full-well images are presented as tiled aggregates of individual micrographs from a same well. McM025044 (3uM shown in image) exhibited a dose-dependent inhibition of spheroid-forming progenitor function across breast, and colon cancer cell lines, as well as MV411 and OCI-AML3 leukemia cell lines enriched in progenitor function. Dose response curves fit through ≥2 independent experiments compared to 0.1% DMSO control.
See also Figure S2
SAE2 regulating the SUMOylation pathway is targeted by the natural product McM025044
To better understand the cellular and molecular targets of McM025044, we appended an alkyne moiety to the C11 phenol of McM025044 to render it suitable for use of “click” cycloaddition-based attachment to azide-coated beads (Figure S3A). The resulting McM025044-alkyne analogue retained biological activity to suppress t-hESCs growth (Figure S3B). Post-assay incubation of protein lysates with azide-agarose beads under copper-catalyzed click-reaction conditions enabled affinity purification of resin-bound protein targets by mass spectrometry (AP-MS)(Tashiro and Imoto, 2012) (Figure S3C). We used human pluripotent cells (t-hESCs vs. normal PSCs) as a model system to further separate CSC-selective McM025044 candidate targets and prioritized those upregulated in AML enriched for neoplastic progenitor activity (Figure S3C, Table S3 and S4)(Mills et al., 2009). AML is among the most studied human neoplasms for CSCs implications and quantitative measurements of these rare cells, and also provides an ideal gateway disease to study novel CSC-targets applicable to other cancers(Benoit et al., 2017; Bonnet and Dick, 1997). Molecular cross referencing this list of putative targets with human interaction networks (ConsensusPathDB database)(Herwig et al., 2016) revealed two members of the small ubiquitin-like modifier or SUMOylation pathway, including the SUMO1 protein itself and the SUMO-activating enzyme subunit 2 (SAE2), acting as a SUMO E1 ligase when forming an heterodimer with SAE1 (Figure S3C)(Lois and Lima, 2005a). Since these putative targets of McM025044 suggested interaction of this compound with the SUMOylation pathway, we deployed transient siRNA knockdown of SUMOylation pathway enzymes SAE2 and UBC9 or the SUMO1 substrate itself. Knockdown of these enzymes caused a reduction in SUMOylation (Figure S3D) and biologically phenocopied the effects of McM025044 treatment on growth reduction of human AML cells (Figure 3A). In addition, these results indicate that human AML is sensitive to modulation of the SUMOylation pathway. As interference with SUMOylation has been shown to be synthetically lethal in conjunction with Myc-hyperactivation across a range of cancers in vitro and in vivo(Kessler et al., 2012), we performed transcriptome analysis of McM025044-treated human AML cells. McM025044 caused transcriptional downregulation of SUMOylation-dependent MYC “switcher” genes (SMS), with loss of expression associated with cell cycle arrest and subsequent apoptosis by GSEA analysis (Figure 3B, Table S4)(Kessler et al., 2012). These observations support recent reports of altered MYC expression upon inhibition of SUMOylation(Li et al., 2019), consistent with biological response of human t-hESCs to McM025044 treatment (Figure 2A-C) and indicate that networks effected by oncogenic SUMOlyation are shared in human CSCs.
Figure 3. McM025044 is targeting the SUMOylation pathway in cancer stem cells through interaction with SAE2.
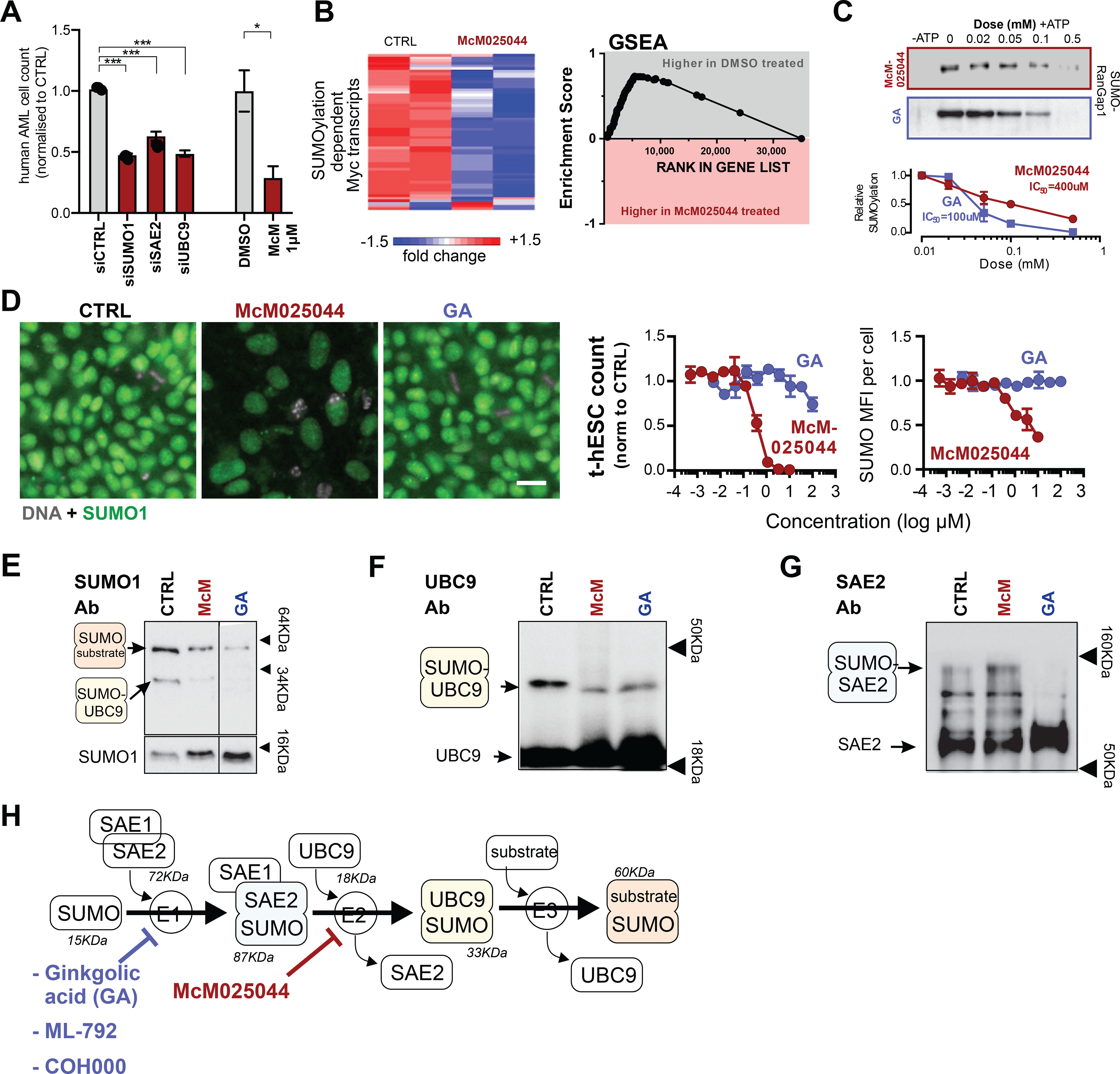
A) Small-interfering RNA (siRNA) knockdowns of SUMOylation machinery members in OCI-AML3 cells results in a reduction in cell count for siSUMO1 (n=15), siSAE2 (n=15), and siUBC9 (n=15), relative to control siRNA (n=14) (***: p<0.0001). McM025044 (1μM, 48hrs) vs. DMSO in OCI-AML3 cells was presented as a reference point (n=3, *: p=0.021). Data are represented as mean ± SEM.
B) Heatmap showing expression levels and GSEA showing enrichment of SUMOylation dependent Myc transcripts(Kessler et al., 2012) after treatment of AML cells with DMSO or McM025044.
C) McM025044 exhibits an ATP-dependent inhibition of SUMOylation in cell-free, biochemical assay (including recombinant SUMO1, SAE1/2, UBC9 and synthetic RanGap1 target protein) with a lower potency than known SUMOylation inhibitor Gingkolic acid (GA) (n=3).
D) McM025044, but not GA inhibits proliferation and reduces cellular SUMO1 levels in t-hESCs. Representative composite images of treated t-hESC (0.1% DMSO (CTRL); 1μM McM025044; 1μM GA, n≥3) immuno-stained for SUMO1 (green) versus nuclei (Hoechst: grey). Scale bar: 20μm.
E) Cell-free biochemical assay shows inhibition of protein target (RanGap1) SUMOylation in the presence of GA and McM25044. Original membrane was cropped to remove irrelevant tracks.
F) Exclusion of the target protein from the reaction (probed with UBC9-Ab) showed a reduction in SUMO1-UBC9 complex formation in the presence of GA and McM025044, demonstrating that neither compound inhibits SUMOylation by inhibiting the transfer of SUMO1 from UBC9 to the target protein.
G) Exclusion of target protein and UBC9 from the reaction (probed with SAE2-Ab) results in a reduction in SUMO1-SAE2 complex formation in GA alone, indicating that McM025044 inhibits the transfer of SUMO1 from SAE2 to UBC9 while GA inhibits the formation of SUMO1-SAE1/2 intermediate.
H) Diagram of the SUMOylation pathway indicating where GA (and other E1 inhibitors ML-792(He et al., 2017) and COH000(Lv et al., 2018)) inhibit the reaction versus McM025044.
To gauge the potential utility of MCM025044 to oncogenic SUMOlyation inhibition, we compared the activity of MCM025044 with ginkgolic acid (GA), that represents a pioneer small molecule in the study of SUMOylation inhibition but requires high concentrations in cell-based assays to exert any effect, thus precluding it for future clinical application(Fukuda et al., 2009). We first compared in vitro SUMOylation assays using human recombinant SAE2, SAE1, and UBC9 transferring SUMO1 to a generic protein target of SUMOylation (partial RanGAP1 fragment). McM025044 exhibited a dose dependent inhibition of SUMOylation in a cell-free in vitro assay for SUMOylation and was less potent than GA in cell-free assays (Figure 3C). However, in human t-hESCs assays, McM025044 demonstrated sub-micromolar activity (IC50 = 300 nM), whereas GA was ineffective, even at doses up to 100,000 nM (Figure 3D). In contrast, inhibition caused by McM025044 in human t-hESCs was accompanied by a reduction in SUMO1 levels and global reduction of protein SUMOylation (Figure 3D, Figure S3E), without affecting total ubiquitination (Figure S3F). We observed that hedamycin can also inhibit SUMOylation in vitro, but its potent toxicity made challenging any further evaluation in human cells (Figure S3G). We next executed stepwise exclusion of key reactional components in vitro (He et al., 2017; Lv et al., 2018) to identify the respective steps in the SUMOylation cascade that McM025044 inhibited (Figure 3E-G). Both GA and McM025044 caused a reduction in SUMO1-UBC9 intermediates (Figure 3E-F) but only GA inhibited the formation of SUMO1-SAE2 thioester bond intermediate (Figure 3G), consistent with its reported inhibition of E1 reaction(Fukuda et al., 2009). In contrast, McM025044 showed persistence of SUMO-SAE1/2 complex (Figure 3G), demonstrating that McM025044 restricts the transfer of the SUMO moiety toward the E2 enzymatic step (as depicted in Figure 3H). This is mechanistically distinct to other SUMOylation inhibitors such as ML-792(He et al., 2017) and COH000(Lv et al., 2018), which block the E1 step via covalent adducts formation and reduction of SAE1/2-SUMO intermediate (Figure 3H). Moreover, we compared the effects of McM025044 with ML-792 in cell growth dose-response experiments, testing small molecules potency and cancer cell-selectivity in t-hESC vs. hPSC. Although the E1 inhibitor ML-792 exhibited a higher potency in both healthy and neoplastic human stem cell models, only McM025044 displayed cancer cell-selectivity over normal stem cells (Figure S3H), supporting potential therapeutic applications for McM025044.
To gain better understanding of McM025044 mechanism of action in intact cells, we overexpressed SAE2 and UBC9 in human t-hESCs and examined the impact on the efficacy of McM025044 treatment. Consistent with biochemical analysis, only overexpression of SAE2 resulted in desensitisation of human t-hESCs response to McM025044, whereas overexpression of UBC9 had no effect on efficacy of McM025044 (Figure 4A, empty vector used as control). Known compounds, such as GA and more recently reported inhibitors COH000 and ML-792, induce their biochemical activity where SAE2 forms a complex with SAE1 to carry out the E1 ligase(He et al., 2017; Li et al., 2019; Lois and Lima, 2005a; Lv et al., 2018). To define the interaction between McM025044 and the SUMO1-SAE1/2 complex, we performed an unbiased approach using SMINA docking(Quiroga and Villarreal, 2016) with all eight possible C12, C14, and C16-stereoisomers of McM025044. The top docking score for McM025044 was in a position that occupied the active adenylation site in SAE1/2 complex (Figure 4B), with the unique sulfonic acid group of McM025044 establishing hydrogen bonds with the side chain of R21 of SAE1 and backbone nitrogen of I384 of SAE2. It also formed several other hydrogen bonds with other residues of SAE1/2 complex including with R59 of SAE2 (Figure 4B). Both R21 and R59 residues are known to be important in the binding of ATP and adenylation reaction(Lois and Lima, 2005a; Lv et al., 2018). To validate this potential mode of binding and biological impact this complex in human CSCs, we introduced a R21A (Arginine to Alanine) mutation in SAE1, as well as a R59A mutation in SAE2 into t-hESCs. Cell-free biochemical assays demonstrated that R21A-SAE1 disrupted SUMOylation reaction while R59A-SAE2 containing complex remained catalytically active (Figure 4C). Independent of McM025044, wild-type (wt) and mutant versions of SAE1 and SAE2 were overexpressed in t-hESCs, and biological role of these proteins was measured in established assays for colony initiating cells (CIC) that rigorously measures clonogenic self-renewal of human t-hESCs (Figure S4A)(Nakanishi et al., 2019; Sachlos et al., 2012; Stewart et al., 2010). Compared to wt SAE1 expression, introduction of R21A-SAE1 mutant drastically depleted clonogenic self-renewal ability in t-hESCs (Figure 4D, Figure S4B). In contrast, overexpression of R59A-SAE2 mutant had no significant impact on CIC frequency compared to wt SAE2 (Figure 4D, Figure S4B). Moreover, global SUMO1 levels were significantly lowered only in R21A-SAE1 transduced t-hESCs vs. other experimental conditions (Figure 4D). This is consistent with in vitro observations where R21A SAE1 suppressed SUMOylation while no impact was noted for R59A-SAE2 (Figure 4C). This also supports the notion that sustained SUMOylation is critical to self-renewal, as we observed a similar pattern of CIC frequency in normal human PSCs expressing SAE1/2 mutants (Figure S4B). Using these SAE2 mutants, we further elucidated the role of SAE2 R59 residue in the context of McM025044 mode of action and evaluated SUMOylation activity in R59A SAE2-containing reactions, in the presence or absence of McM025044 (Figure 4E). McM025044 effectively reduced SUMOylation activity using wt SAE2, whereas inhibitory effects of McM025044 were eliminated upon disruption of the R59 residue of the SAE2 enzyme (Figure 4E). This indicates that R59 represents a critical binding site of McM025044 for its mode of action. To validate the relevance of SAE2 R59 motif in McM025044-dependent inhibition of neoplastic self-renewal, we forced the expression of R59A mutant and wt SAE2 in human colorectal cancer cells (HCT116), and treated with and without McM025044. A GFP reporter system was used to track SAE2-transduced cells. We found that McM025044 significantly reduced the expression of the colorectal cancer self-renewal marker BMI1 in wt SAE2 overexpressing cells (vs. vehicle control treatment), while no effect was observed in cells expresing R59A-SAE2 mutants (Figure 4F). Moreover, HCT116 cells overexpressing R59A-SAE2 mutant displayed no changes in spheroid formation capacity upon McM025044 treatments vs. vehicle, while impaired sphere formation was observed in treated wt SAE2-expressing cells (Figure 4F, Figure S4C). Collectively, these findings reveal MCM025044 as a highly potent SUMOylation inhibitor that uniquely targets SAE2 by altering the SAE1/2 complex dynamics and demonstrates that SUMOylation acting through SAE2 target is essential to self-renewal of human CSC models.
Figure 4. McM025044 binding to SAE2 is inhibiting SUMOylation and stem cell self-renewal.
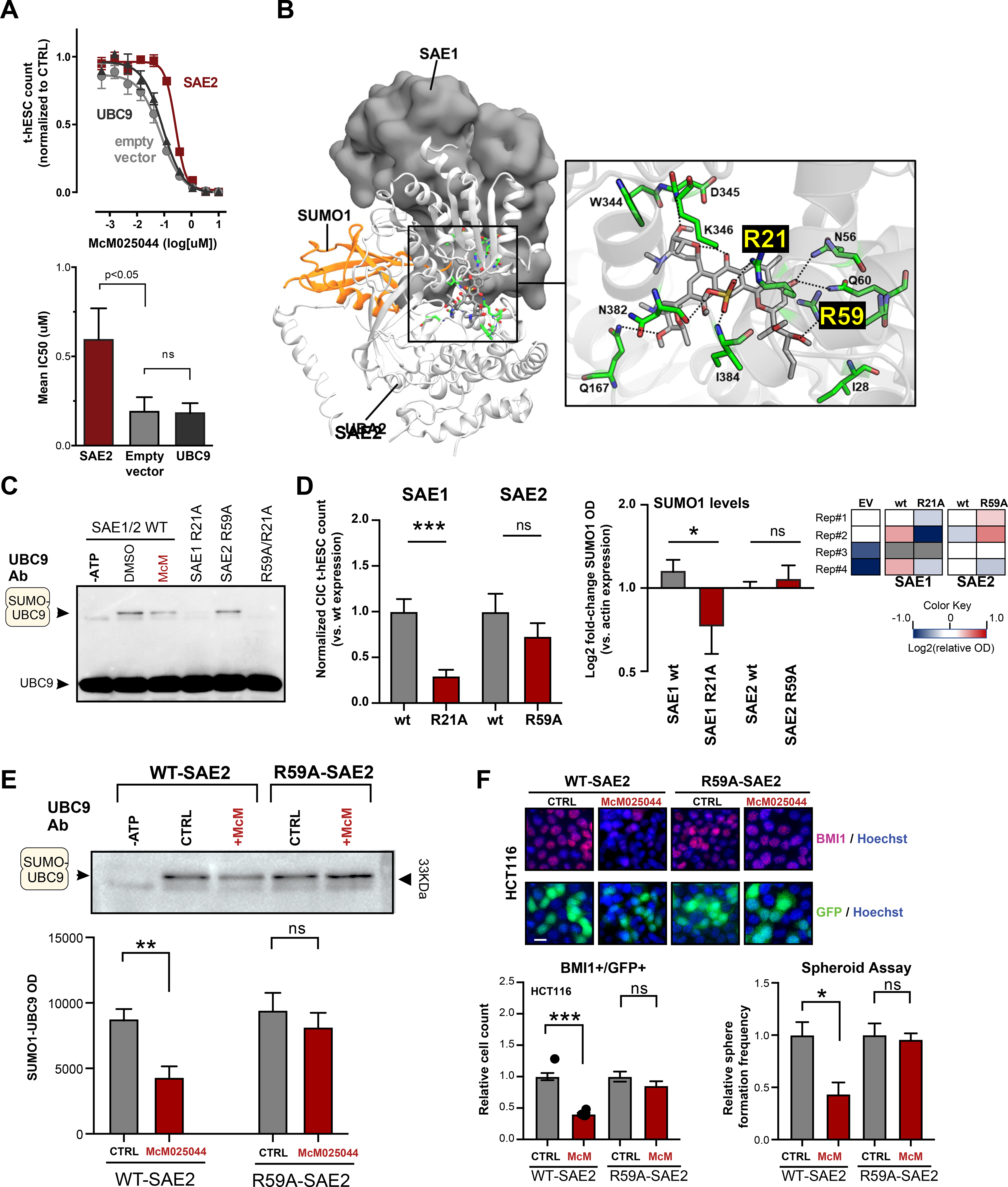
A) Functional testing of McM025044 effect on SUMOylation cascade in t-hESCs through overexpression of SAE2 and UBC9 showed that only cells transduced with SAE2 expression vector exhibited a decrease in sensitivity to McM025044. Data are represented as mean ± SEM (n=3, *: p=0.045, one-tailed t-test).
B) Docked pose of McM025044 (grey) in human SUMO1-SAE1/2 (orange ribbon, gray surface, and white ribbon respectively) complex. Amino acid residues (green stick) highlighting the predicted binding site of McM025044 are shown in an enlarged view. Nitrogen, Oxygen, Sulfur and Hydrogen atoms are shown using blue, red, yellow and white colors respectively. Black dashed line indicates hydrogen bond interaction.
C) Western blot analysis of in vitro SUMO1-UBC9 complex formation using WT SAE1/2 (+/− McM025044) and R21A-SAE1, R59A-SAE2 single mutants, as well as R21A/R59A double mutant. R59A mutation preserved SAE1/2 complex activity, while SAE1-R21A and R21A/R59A double mutant impeded SUMO1-UBC9 complex formation (n=4).
D) Assessment of over-expression of wildtype (wt) SAE1, R21A-SAE1, wt SAE2, or R59A-SAE2 on CIC capacity in t-hESCs (n=9, ***: p<0.001). Total SUMOylation was assessed in t-hESC by SUMO1 immunodetection (represented as heat maps) for each CIC experimental condition (vs. empty vector) (n≥3, *=p:0.039). Data are represented as mean ± SEM.
E) Western blot analysis of SUMO1-UBC9 complex catalyzed by WT-SAE2 and R59A-SAE2 in the presence of McM025044 vs. DMSO control. Quantitative analysis demonstrates that R59A mutation in SAE2 restricts the capacity of McM025044 to inhibit SUMO1-UBC9 intermediate formation. Data are represented as mean ± SEM (n≥5, ***: p=0.01, n.s.: p=0.4).
F) High-content imaging assessment of self-renewal marker BMI1 expression in WT-SAE2 and R59A-SAE2 expressing HCT116 cells (GFP-positive), upon McM025044 (5μM, 48hrs) or DMSO treatments (n=22, ***: p<0.0001). WT-SAE2 and R59A-SAE2 expressing HCT116 cells were also used in 3D sphere formation assays, where GFP+ sphere count was measured following vehicle or McM025044 (5μM) treatments (n=3, *: p=0.0278). Bar graph data are represented as mean ± SEM. Representative micrographs are composites of BMI1 (red), GFP (green), and Hoechst (blue) channels. Scale bar: 20μm.
See also Figure S4
AML patient derived cells exhibit elevated SUMOylation and response to McM025044
McM025044 may represent a viable candidate to chemically probe biological effects of putative CSCs derived directly from patients, with future therapeutic value. Consistent with previous suggestions using other cancer models(Benoit et al., 2017; Bogachek et al., 2014; Du et al., 2016), we demonstrated that pluripotent human CSCs exhibit elevated SUMOylation levels compared to normal pluripotent SCs (Figure S3E). To determine if this differential expression was consistent and could be exploited beyond laboratory model systems, hematopoietic tissue was obtained from healthy as well as AML patient donors to measure SUMOylation levels and sensitivity to McM025044 inhibition. Established assays for stem and progenitor cells, as well as the ability to easily obtain normal human SC counterparts for comparative studies are characteristics of malignant hematological disease, which provides an ideal system for clinical validations. AML patient cells had higher levels of SUMOylation in comparison to healthy controls (Figure S5A, B) with the primitive sub-fraction expressing cell surface antigen CD34 (CD34+) enriched for leukemic stem and progenitor activity(Bhatia et al., 1997) expressed twice the levels of SUMO1 (Figure 5A, Figure S5C). Using the ratio of SUMO1 levels in CD34+ vs. MNCs, a total of 12 AML patients exhibiting the highest levels of SUMOylation in the stem cell compartment were associated with the most severe “adverse risk” category (based on European LeukemiaNet (ELN) classification(Döhner et al., 2017)) (Figure 5B, Figure S5C), underscoring a clinical relevance for targeting SUMOylation in AML patients. To determine whether stemness properties of AML were sensitive to SUMOylation inhibition, we first determined the initial baseline frequency of stem and progenitor cells in patients by functional readout prior to treatment with McM025044 to allow quantitative before and after analysis (Figure S5D). Treatment with McM025044 was sufficient to inhibit progenitor capacity using a 6-fold less concentration of the compound (IC50’s) compared to response of healthy progenitors (Figure 5C-E, Figure S5E, F), revealing a highly selective therapeutic index for McM025044 in this broad cohort of AML patients tested. Notably, McM025044 produces no consistent lineage bias in treated healthy progenitors, as examined by CFU colony type (Figure S5G).
Figure 5. McM025044 selectively inhibits leukemia stem/progenitor function over healthy hematopoietic cells.
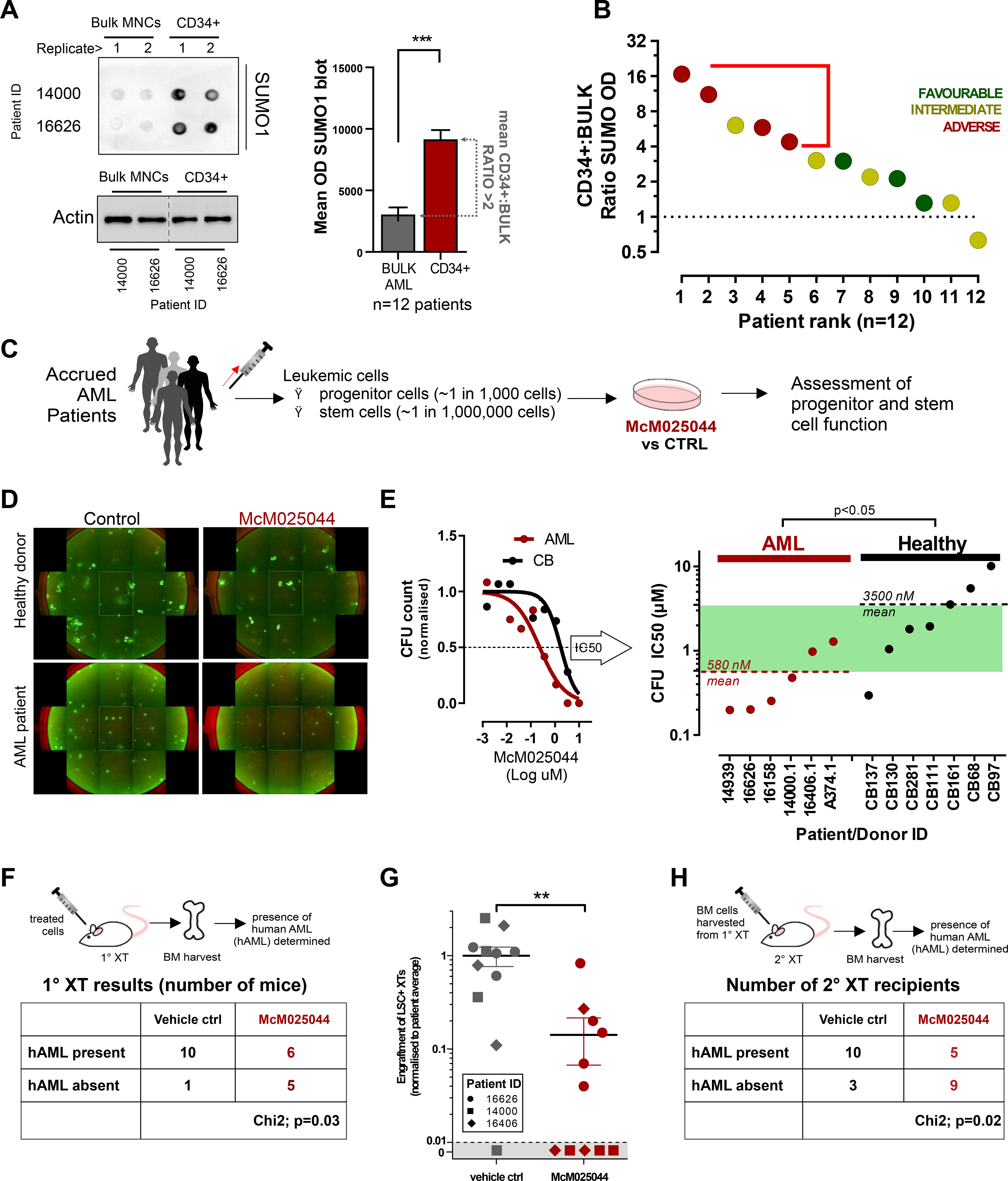
A) Protein Dot blot detection of total SUMO1 levels in bulk and CD34+ sorted MNC populations from primary AML patient samples shows that global SUMOylation is significantly higher in the stem-cell/progenitor compartment (CD34+ cells) in comparison to bulk cells. Actin was used as loading control and original membrane was cropped to remove irrelevant tracks. Quantitative analysis was presented in the histogram where data are represented as mean ± SEM (n=12, ***: p<0.0001).
B) Global SUMOylation was higher in the CD34+ cells relative to bulk cells across AML ELN risk stratification.
C) Diagram illustrating the evaluation of 24h McM025044 treatment of patient samples on stem-cell and progenitor function.
D) Progenitor function was evaluated through quantification of colony forming units (CFU) in semi-solid media. 7d after treatment with McM25044 (1uM shown) with a range of concentrations (0.1% DMSO as vehicle control), colonies in 24 well plates were stained with Calcein AM and quantified though high content imaging. Full-well images are presented as tiled aggregates of individual micrographs from a same well.
E) Representative IC50 curve for one AML patient sample (#16626) vs. one healthy donor (CB #111), determined from >5 concentrations of McM025044 (see Figure S5E and F). The IC50s for AML samples were on average 580 nM and the IC50 for healthy was 3.5 μM indicating a cell selectivity (green shade) for AML progenitors (n=6) over healthy progenitors (n=7) compared to control treatment of 0.1% DMSO (p<0.05).
F) Treated (1μM McM025044 or 0.1% DMSO as vehicle control; 24h) AML patient samples (n=3 individual patients) were each transplanted into ≥3 irradiated immunocompromised recipient mice to determine effect of McM025044 on leukemic initiation cells. Samples retained LSC after treatment (i.e. LSC+) if human engraftment was detected at 8 weeks post-transplant. Treatment with McM025044 (n=12) significantly inhibited LSC function compared to vehicle control (n=11, Chi square: p=0.03).
G-H). Samples that were LSC+ had significantly reduced levels of human engraftment (n=11, mean ± SEM, **: p<0.001) and had reduced ability to engraft secondary recipient mice (Chi square: P=0.02).
To determine whether McM025044 could effectively target rarer AML CSCs with disease initiating capacity in vivo, termed leukemia initiating cells (LICs), we treated patient cells with McM025044 and transplanted treated vs. treated vehicle control cells into primary recipient NOD/SCID mice (Figure 5F). Recipient xenografted mice were sacrificed, and bone marrow (BM) chimerism for human AML disease was assessed 8–10 weeks post-transplant using human specific pan-leukocyte marker CD45 and leukemic marker CD33 (CD45+CD33+). Limiting dilution analysis was used to quantify human AML CSCs determined by absence or presence of leukemia in recipients (Chi-Square Variance). McM025044 treatment reduced global SUMOylation as well as CSC activity in AML samples compared to vehicle-treated controls (Figure 5F, Figure S5H, Table S5). In rarer recipient mice that achieved a level of leukemic engraftment after McM025044 treatment, we found the disease burden (AML chimerism) was significantly reduced (Figure 5G, Table S5). However, no consistent changes in phenotypic marker expression, such as CD34, CD14, and CD11b (progenitor vs. lineage commitment) were observed in the McM025044-treated group vs. controls (Figure S5I). Human AML cells were harvested and purified from these primary recipients, and intravenously transplanted into secondary recipient mice as a further rigorous method to measure disease sustainability. Analysis of these secondary mice revealed McM025044 was able to suppress AML CSC activity in 64% of secondary recipients (Figure 5H, Figure S5J).
Collectively, these data indicate that McM025044 targets clinically relevant primitive leukemic cells measured by in vitro progenitor assays or serial transplantation assessment of CSC activity in vivo. The identification of McM02044 demonstrates that higher SUMOylation activity in CSCs and vs. healthy SCs can be chemically exploited for selective therapeutic targeting.
DISCUSSION
Cellular SUMOylation of proteins has been associated with non-oncogene addiction of human cancers and thus represents a potential basis of anti-cancer therapeutics (Bogachek et al., 2016; He et al., 2017; Kessler et al., 2012; Li et al., 2019; Licciardello and Kubicek, 2016; Lv et al., 2018). Using a chemical genomics approach with novel chemical space, our study identifies a NP based compound in McM025044, that represents a molecular inhibitor of the SUMOylation cascade that functionally reduces growth of colon, breast, and myeloid human cancers. Molecular characterization of McM025044 structure revealed this compound as an undescribed member of the tetracyclic pluramycin family with distinct chemical and biological features. From a sequence of biochemical and multi-omics analyses, we identified the SUMO-activating enzyme SAE1/2 complex as the main target of MCM025044, distinguishing it from pluramycins that exclusively target DNA. Although proteomic profiling of affinity pull down assays revealed other potential McM025044 interactors, these are either SUMOylation targets (APEX1, DDX3X), or known to form complexes with SUMO substrates (EWSR1) (Figure S3C)(Oughtred et al., 2019). Transcriptional changes induced by McM025044 in human AML cells strongly reflected the effects of SAE2 knockdown on a described subset of MYC switcher genes in human breast epithelial cells(Kessler et al., 2012). Nontheless, we cannot fully exclude that McM025044 can target additional pathways in specific contexts.
Our results provide evidence for the biological dependency of human CSCs on SAE2 using McM025044 as chemical probe of this previously unappreciated role of SUMOylation biochemistry in self-renewal of human cancers. Previous characterization of a loss of SAE2 expression in the context of MYC oncogenesis is marked by reduced proliferation and apoptosis induction(Kessler et al., 2012). This observation is similar to our results in response to McM025044-treated human CSCs. McM025044 uniquely targets SAE2 and disrupts a unique pocket within the SAE1/2 complex to inhibit SUMOylation processing of proteins. Our findings indicate that SUMOylation activity is higher in human CSCs compared to healthy SC counterparts, and therefore inhibition of this pathway selectively affects human CSCs to a larger extent. This selectivity provides a potential therapeutic window for further development and application to human cancers where the CSC compartment is responsible for relapse or resistance to standard therapies. In addition to McM025044 biological activity directly correlating to self-renewal capacity of CSCs, genetic mutation of SAE2 at residues critical to the binding site of McM025044, was able to reduce CSC sensitivity to treatments. The ability of SAE2 R59A mutation to neutralize the effects of McM025044 removing this unique motif of SAE2 provides evidence for the specific role of this protein in CSC self-renewal that warrants further study. For instance, stochastic R59 mutations in human tumors could hinder McM025044 efficacy. Thus, developing and testing new molecules taking advantage of the same binding pocket in the SAE1/2 complex could shed light on such a potential resistance mechanism.
CSCs constitute the root of the disease, actively contributing to tumor growth, relapse, and metastasis(Flemming, 2015), therefore representing a highly relevant target toward effective and prolonged cancer remission. Recent studies suggest activation of the SUMOylation pathway as a key factor in sustaining CSC growth and non-oncogenic addiction of human cancers(Benoit et al., 2017; Bogachek et al., 2014; Bossis et al., 2014). Accordingly, targeting the SUMOylation cascade represents a promising therapeutic for cancer treatment. Other chemical inhibitors targeting SUMOylation have been previously developed via in silico approaches or identified through cell-free screening, including GA, anachardic acid, and kerriamycin B(Fukuda et al., 2009; Hirohama et al., 2013; Kumar et al., 2013). However, most of these compounds exhibited poor potency in complex eukaryotic systems and remain far from clinical applications. Similar to McM025044, molecules that serve as chemical probes of SUMOylation such as ML-792 and COH000(He et al., 2017; Li et al., 2019; Lv et al., 2018) demonstrated efficacy at blocking SUMOylation E1 step via modes of action distinct from McM025044, involving the formation of covalent adducts with SAE1/2. Importantly, such an irreversible pharmacological approach to inhibit SUMOylation failed to demonstrate neoplastic cell-selective response (ML-792: Figure S3H). In contrast, McM025044 distinct mode of action enables selective antineoplastic functions, through targeting of SUMOylation machinery in patient-derived human CSCs. Given that overexpression of R21A-mutated SAE1 in hPSC substantially altered CIC activity, whereas McM025044 had no impact on hPSC biology further supports a potential non-covalent/reversible mode of action for this SUMOylation inhibitor. McM025044 reduces CSC activity in a serial xenotransplantation model, the gold standard in vivo assay for pre-clinical validation of CSC-targeting agents.
Significance
Our results represent a key proof of concept for the importance of the SUMO pathway vs. antimitotic drug therapies to combat rare human CSCs through the development of drugs that target oncogenic SUMOylation of human cancers. How SAE2 and inhibition of SUMOylation in CSCs collaborates with self-renewal machinery, and if this process and biochemical interaction can be extended to other cancers or is associated with chemotherapeutic resistance and cancer relapse(Boyd et al., 2018) will be further evaluated through alternative and recently developed in vivo xenotransplantation models in our program.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Mickie Bhatia (mbhatia@mcmaster.ca).
Materials availability
Reagents generated in this study are available from the Lead Contact under Materials Transfer Agreements.
Data and code availability
Transcriptomics data are available at GSE160551
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Pluripotent Stem Cell Culture
H9 (hPSCs) and transformed H9 (t-hESCs) were cultured on Matrigel-coated (BD Biosciences 353234) tissue culture plates in mouse embryonic fibroblast-conditioned (MEFCM) medium supplemented with 8 ng/ml bFGF (Thermo Fisher 13256–029) at 37°C 5% CO2. MEFCM is composed of KO-DMEM (Thermo Fisher 10829–018), 20% KO-Serum Replacement (Thermo Fisher 10828–028), 1% Non-Essential Amino Acids (Thermo Fisher 11140–050), 1mM L-Glutamine (Thermo Fisher 25030081), and 0.1mM β-mercaptoethanol (Sigma Aldrich M7522). H9 cell cultures were passaged every 7 days using Collagenase IV 100units/mL (Thermo Fisher 17104–019) and mechanical scraping of cell clumps at a ratio of 1:2.5). Transformed H9 cell cultures were passaged every 3 days at a ratio of 1:4, as previously described(Werbowetski-Ogilvie et al., 2009).
OCI-AML3 Cell Culture
Leukemia cell line OCI-AML3 (male) was obtained from the German collection of microorganisms and cell cultures (DSMZ). OCI-AML3 cells were cultured in Alpha MEM (Gibco) supplemented with 20% FBS (Hyclone) at 37°C 5% CO2. Original cultures were seeded at 500k per mL of culture medium in ultra-low attachment tissue culture plates (Corning) and passaged every 3 days at a ratio of 1 to 3.
SW480 Cell Culture
SW480 (Male) were cultured in Leibovitz’s L-15 medium (Gibco) supplemented with 10% FBS (Hyclone) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 5 days at a ratio of 1 to 4.
HT29 Cell Culture
HT29 (Female) were cultured in McCoys 5A medium (Gibco) supplemented with 10% FBS (Hyclone), 1mM L-Glutamine (Thermo Fischer) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 5 days at a ratio of 1 to 5.
HCT116 Cell Culture
HCT116 (Male) were cultured in McCoys 5A medium (Gibco) supplemented with 10% FBS (Hyclone) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 5 days at a ratio of 1 to 10.
MCF7 Cell Culture
MCF7 (Female) were cultured in Alpha MEM (Gibco) supplemented with 10% FBS (Hyclone), 0.01mg/ml Insulin (Sigma) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 3 days at a ratio of 1 to 4.
MDA-MB-231 Cell Culture
MDA-MB-231 (Female) were cultured in DMEM (Gibco) supplemented with 10% FBS (Hyclone) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 3 days at a ratio of 1 to 4.
T-47D Cell Culture
T-47D (Female) cells were cultured in RPMI-1640 (Thermo Fisher Scientific, MA, USA) supplemented with 10% FBS (Hyclone) at 37°C 5% CO2. Original cultures were seeded at 500k per 25cm2 surface area in TC-treated flasks (Corning) and passaged every 3 days at a ratio of 1 to 4.
Breast cancer cell lines and colon cancer cell lines were obtained from the American Type Culture Collection (ATCC, Virginia, USA). All cell lines were cultured according to the suppliers’ instructions, authenticated and frequently tested for mycoplasma contamination.
293-FT Cell Culture
293FT cells were cultured in DMEM 10%FBS, 1mM non-essential amino acids, 1mM L-Glut, 1mM sodium pyruvate at 37°C 5% CO2. Original cultures were seeded at 500k per 75cm2 TC-treated flasks and passaged every 3 days at a ratio of 1 to 6.
Primary donor and patient samples
Primary AML samples of peripheral blood (PB) and/or bone marrow (BM) were collected and characterized at the time of clinical presentation (Key Resources Table and Table S5). Patient disease risk classification (Favourable, Intermediate, Adverse) was determined based on European LeukemiaNet (ELN) categories(Döhner et al., 2017). Umbilical cord blood (CB) was collected after delivery in heparinized bags. All samples were obtained after informed consent, according to the Research Ethics Board approved protocols at McMaster University, and mononuclear cells (MNC) were immediately isolated as previously described(Bhatia et al., 1997). Healthy progenitors were enriched from CB using lineage depletion by negative antibody-based selection using a StemSep system (Stemcell Technologies).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| PE-conjugated Ki 67 antibody | BD Biosciences | Cat# 556027 RRID: AB_2266296 |
| FITC-conjugated Oct¾ antibody Clone 40/Oct-3 | BD Biosciences | Cat# 560217 RRID: AB_1645305 |
| FITC Annexin V | BD biosciences | Cat#556419 |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | Cat# 16–193, RRID: AB_310795 |
| SUMO-1 (D-11) antibody | Santa Cruz Biotechnology | Cat# sc-5308 RRID: AB_628300 |
| SUMO-1 (human) (CT) polyclonal antibody | Enzo lifesciences | Cat# BML-PW9460 |
| Anti-SAE2 / UBA2 antibody [EPR14880] | Abcam | Cat# ab185955 |
| Anti-UBE2I / UBC9 antibody [EP2938Y] | Abcam | Cat# ab75854 |
| Anti-Ubiquitin Antibody (FL-76) | Santa Cruz Biotech | Cat# sc-9133; RRID: AB_2180553 |
| Anti-BMI1 antibody (JJ093–3) | ThermoFisher | Cat# MA5–32486; RRID: AB_2809763 |
| PARP antibody | Cell Signaling | Cat# 9542; RRID: AB_2160739 |
| Anti-Actin Antibody, clone C4 | Millipore | Cat# MAB1501; RRID: AB_2223041 |
| Alexa Fluor® 647 Mouse anti-Human TRA-1–60 Antigen | BD biosciences | Cat#560850 AB_10565983 |
| FITC-conjugated Mouse Anti-CD33 Monoclonal Antibody, Clone P67.6 | BD Biosciences | Cat# 340533 RRID: AB_400047 |
| APC-conjugated Mouse Anti-CD34 Monoclonal Antibody, Clone 581 | BD Biosciences | Cat# 555824 RRID: AB_398614 |
| APC-conjugated anti-CD11b | BD Biosciences | Cat# 550019 RRID: AB_398456 |
| FITC-conjugated Mouse Anti-CD14 Monoclonal Antibody, Clone M phi P9 | BD Biosciences | Cat# 347493; RRID: AB_400311 |
| APC-conjugated Mouse Anti-Human CD45 Clone 2D1 | BD Biosciences | Cat#340943 |
|
| ||
| Biological Samples | ||
|
| ||
| AML patient sample | This paper | #11040 |
| AML patient sample | This paper | #11040.1 |
| AML patient sample | This paper | #12526 |
| AML patient sample | This paper | #13051.1 |
| AML patient sample | This paper | #13814.1 |
| AML patient sample | This paper | #13814.2 |
| AML patient sample | This paper | #13963.1 |
| AML patient sample | This paper | #14000.1 |
| AML patient sample | This paper | #14939 |
| AML patient sample | This paper | #16150 |
| AML patient sample | This paper | #16150.1 |
| AML patient sample | This paper | #16158.2 |
| AML patient sample | This paper | #16406.1 |
| AML patient sample | This paper | #16626 |
| AML patient sample | This paper | #17223 |
| AML patient sample | This paper | #A374.1 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| McM025044 | This paper | N/A |
| Cytarabine | Sigma-Aldrich | Cat# C6645 |
| Hedamycin | Boc Sciences | Cat# 11048–97-8 |
| Bleomycin | Millipore Sigma | Cat# B8416 |
| 7-aminoactinomycin (7-AAD) | ThermoFisher | Cat# A1310 |
| Ginkgolic acid (GA) | Tocris | Cat# 6326 |
| Calphostin C | Tocris | Cat# 1626 |
| N-Ethylmaleimide | Millipore Sigma | Cat# E3876 |
| ML-792 | MedChemExpress | Cat# HY-108702 |
| Recombinant R59A SAE2 | This paper | N/A |
| Recombinant R21A SAE1 | This paper | N/A |
| bFGF | BD biosciences | Cat# 359282 |
| Matrigel | BD Biosciences | Cat#353234 |
| KO Serum Replacement | ThermoFisher | Cat#10828–028 |
| Fetal Bovine Serum | Hyclone | Lot#AXG45146 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| SUMOylation Kit® | ENZO Life Sciences | Cat# BML-UW8955–0001 |
| OxiSelect™ Comet Assay Slides | Cell Biolabs Inc. | Cat# STA-353 |
| EdU Staining Proliferation Kit (iFluor-647) | Abcam | Cat# ab222421 |
| CellEvent™ Caspase-3/7 Green Detection Reagent | ThermoFisher | Cat# C10423 |
| Click-iT™ Protein Reaction Buffer Kit | ThermoFisher | Cat# C10276 |
| Azide agarose beads | Jena Bioscience | Cat# CLK-1038–2 |
| BD Fixation Buffer | BD Bioscience | Cat#554655 |
| BD Cytofix/Cytoperm kit | BD Bioscience | Cat#554714 |
| InFusion HD Cloning | Clontech | Cat# 638910 |
| qScrip cDNA SuperMix | Quanta | Cat# 95048–100 |
| Total RNA isolation kit | Norgen | Cat# 37500 |
| Immobilon Western Chemiluminescent HRP Substrate | Millipore | WBKLS0500 |
|
| ||
| Deposited Data | ||
|
| ||
| OCI-AML3 Vehicle vs. MCM025044-treated | This paper | GSE160551 |
| SUMOylation-dependent MYC “switcher” genes (SMS) | (Kessler et al., 2012) | GSE34055 |
| Human SUMO E1 complex with a SUMO1-AMP mimic | (Olsen et al., 2010) | PDB:3KYC |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| H9 PSCs | Wicell | N/A |
| Transformed H9 PSCs | M.Bhatia lab | PMID:19122652 |
| OCI-AML3 | DSMZ | ACC 582 |
| MDA-MB-231 | ATCC ® | HTB-26™ |
| MCF7 | ATCC® | HTB-22™ |
| HT29 | ATCC® | HTB-38™ |
| HCT116 | ATCC® | CCL-247™ |
| SW480 | ATCC® | CCL-228™ |
| T-47D | ATCC® | HTB-133™ |
| 293FT | Thermo Fisher | Cat#R70007 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| See Table S1 | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV-mCherry | Pantelis Tsoulfas (unpublished) | RRID:Addgene_36084 |
| pHIV-eGFP | (Welm et al., 2008) | RRID:Addgene_21373 |
| pET-11c-hSEA1 | GenScript Biotech | N/A |
| pET-28b(+)-hSAE2 | GenScript Biotech | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Control siRNA-A (scramble) | Santa Cruz Biotechnology | Cat# sc-37007 |
| SUMO1 siRNA (human) | Santa Cruz Biotechnology | Cat# sc-29498 |
| SAE2/UBA2 siRNA (human) | Santa Cruz Biotechnology | Cat# sc-61740 |
| UBC9 siRNA (human) | Santa Cruz Biotechnology | Cat# sc-36773 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Acapella Script Collection | PerkinElmer | http://www.perkinelmer.com/content/relatedmaterials/brochures/bro_acapellasoftware.pdf |
| FlowJo™ v10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| Partek Genomic Suite 6.6 | Partek Inc | http://www.partek.com/pgs |
| micrOTOF control version 3.0 | Bruker Daltonics | https://www.bruker.com/service/support-upgrades/software-downloads/mass-spectrometry.html |
| Sequest version 1.4.0.288 | ThermoFisher | https://www.thermofisher.com/ca/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| X! Tandem, version CYCLONE 2010.12.01.1 | GMP | https://www.thegpm.org |
| AutoDock Tools | The Scripps Research Institute | http://mgltools.scripps.edu/ |
METHOD DETAILS
High-content screening using t-hESC and counter-screening with hPSC
Screening of extracts from the WAC library was performed on neoplastic hPSC (t-hESC) and normal hPSC as previously described by our group(Sachlos et al., 2012). Briefly, t-hESCs were seeded at 5,000 and cells per well, in black optical 96-well plates (Falcon) coated with Matrigel basement membrane extract. Human pluripotent stem cells (hPSCs) were mechanically dissociated at confluence and plated in Matrigel-coated black optical 96-well plates at a ratio 1 confluent 6-well for 1 full 96-well plate. hPSCs and t-hESCs were cultured in mouse embryonic fibroblast conditioned media (MEFCM) supplemented with 8 ng/ml bFGF. Twenty-four hours post-plating, the media was exchanged for MEFCM containing 0.1% of each extract from the WAC library for single-dose cell count experiments or containing 0.001% to 20% of extracts for the 10-point dose-response curve experiments. t-hESC were incubated with natural product extracts for 48 hr then exchanged with fresh media with compound for a further 24 hr (total compound treatment time 72 hr). Treatment of hPSCs was conducted over 5 days, with daily fresh extract-containing media exchange. After the treatments, t-hESCs and hPSCs plates were fixed with 2% formaldehyde and washed 3 times with Perm/Wash buffer prior to nuclei staining using Hoechst 33342. Plates were scanned using an Operetta High Content Imaging System (Perkin Elmer). Images were analyzed using custom Acapella scripts (PerkinElmer).
Preparation of natural product crude extracts from actinomycetes strains for chemical screening
A loop of spores from strains of the Wright Actinomycete Collection (WAC) library(Thaker et al., 2013) was streaked on Bennett’s or TSB agar, and incubated for 6 days. Then, Bennett’s media was inoculated from solid cultures and incubated for 6 days. Liquid Cultures were spun down, the supernatant was filter sterilized and diluted to 0.1% for screens. Extracts were made from either Conditioned Media (CM; the sterile filtered supernatant from liquid cultures); culture pellets (the concentrated extract of pellets from CM preparations) resuspended in DMSO and diluted to 0.1% for screens; or obtained from the concentrated extract of solid media culture (SM). The culture and agar were pulverized and extracted with methanol. Agar and bacterial debris were removed by filtration. The methanol was evaporated. Dried samples were resuspended in DMSO and diluted to 0.1% for screening. For Large-scale fermentation of actinomycetes strains, a loop of spores from relevant WAC strains was inoculated into Bennett liquid medium (3 mL/tube) in 60 test tubes and incubated for 3 days at 30°C. The seed culture (3 mL x 60) was inoculated into ten 1 L Bennett liquid medium in 2.8 L flasks and cultivated for 7 days at 30°C on a shaker at 250 rpm.
Isolation and Purification of CSC-bioactive fractions
Supernatant from CSC-bioactive strain WAC067 (60L) was extracted with 2% (W/V) HP-20 (Diaion) resin. The resin was eluted with H2O (1L), 10% methanol (1 L), 90% methanol (2 L), and 100% methanol (1L) to yield four fractions WAC067–1~4. The active fraction WAC067–3 was applied to reverse-phase CombiFlash ISCO (RediSep Rf C18, Teledyne) and eluted with a Water-Acetonitrile linear gradient system (0–100% acetonitrile) to give 60 sub-fractions (WAC067–3-1~60). The active sub-fractions WAC067–3-41 to 55 were combined and passed through a Sephadex LH-20 column (100 mL), eluting with 90% MeOH, to yield 9 sub-fractions. The most bioactive sub-fraction out of 9 (WAC067–3-41~55–5) was purified by semi-preparative HPLC (gradient elution 12–18% acetonitrile, 0.1% Formic acid, 4.0 mL/min). The active supernatant from WAC308 (6L) was extracted with 2% (W/V) HP-20 (Diaion) resin. The resin was eluted with H2O (200mL), 10% methanol (200mL), 90% methanol (1L), and 100% methanol (500mL) to yield four fractions WAC308–1~4. The active fraction WAC308–3 was applied to a Sephadex LH-20 column (35 ml), eluting with 50% methanol, to yield 9 sub-fractions WAC308–3-1~9. The active sub-fractions WAC308–3-5 were purified by semi-preparative HPLC (gradient elution 5–70% acetonitrile, 0.1% formic acid, 4.0 mL/min) to afford MCM025044 (2mg). HRESI mass spectrometry, 1H-1H COSY and HMBC NMR analysis were subsequently performed as detailed below.
Mass spectrometry analysis of NP purified peaks
The mass spectrometer was a Bruker Daltonics micrOTOF II, equipped with electrospray (ESI) ion source. Compound spectra were obtained in the positive mode with direct infusion from an external syringe at a rate of 180 μL/hour. Nitrogen was used both as a nebulizer gas (pressure 0.4 bar) and a drying gas (gas flow 4.0 L/min). The drying temperature was 180 °C. The applied capillary voltage was 4500 V, the capillary exit was 150.0 V and skimmer 1 was set to 50.0 V. The transfer time of the ions from hexapole to orthogonal acceleration was 45 μs and hexapole radio frequency was 800.0 Vpp. Mass spectral data were collected from m/z 200 to 2000. TOF-MS was operated with micrOTOF control version 3.0 by Bruker Daltonics.
Structure characterization of CSC-bioactive natural products
McM025044 was isolated as pale-yellow powder. Its molecular formula was determined as C41H54N2O13S based on HRESI-MS at m/z 815.3466 [M+H]+ (calcd 815.3425). The UV spectrum of McM025044 (maxima at 222, 276 and 343 nm) suggested the present of a conjugated aromatic system in McM025044. The 1H- and 13C-NMR spectra of McM025044 displayed signals for two carbonyl, thirteen quarternary carbons, fourteen methines, two methylenes, and ten methyl groups (Table S2). Careful analysis of 1H-1H COSY NMR data of McM025044 revealed three proton sequences form oxymethine H-16 (δ 4.70 ppm) to methyl H-19 (δ 1.59 ppm), from oxymethine H-1’ (δ 5.10 ppm) to methyl H-6’ (δ 1.29 ppm), and from oxymethine H-1” (δ 5.41 ppm) to methyl H-6” (δ 1.28 ppm). These three sequences were extended to structure unit A, C, and D according to HMBC correlations from H-15 (δ 1.51 ppm) to C-2 (δ 170.9 ppm), 14 (δ 75.9 ppm) and 16 (δ 70.6 ppm), from H-3 (δ 6.37 ppm) to C-2 (δ 170.9 ppm) and C-4 (δ 179.2 ppm), from H-1’ (δ 5.10 ppm) to C-5’ (δ 76.5 ppm), from H-7’ (δ 2.86 ppm) and H-8’ (δ 2.78 ppm) to C-3’ (δ 66.9 ppm), from H-1” (δ 5.41 ppm) to C-5” (δ 68.7 ppm), from H-7” (δ 1.11 ppm) to C-2” (δ 34.5 ppm), C-3” (δ 64.8 ppm) and C-4” (δ 67.9 ppm), and from H-8” (δ 2.67 ppm) and H-9” (δ 2.60 ppm) to C-3” (δ 64.8 ppm). Structure unit B was established base on HMBC correlations from 11-OH (δ 9.72 ppm) to C-10 (δ 136.6 ppm), C-11 (δ 150.0 ppm) and C-11a (δ 126.8 ppm), from H-1’ (δ 5.10 ppm) to C-7a (δ 128.8 ppm), C-8 (δ 135.3 ppm) and C-9 (δ 123.1 ppm), from H-1” (δ 5.41 ppm) to C-9 (δ 123.1 ppm), C-10 (δ 136.6 ppm) and C-11 (δ 150.0 ppm), from H-12 (δ 6.12 ppm) to C-6a (δ 136.7 ppm), C-7a (δ 128.8 ppm), C-11a (δ 126.8 ppm) and C-12a (δ 124.8 ppm), from H-13 (δ 2.82 ppm) to C-4a (δ 123.3 ppm), C-5 (δ 139.0 ppm) and C-6 (δ 123.3 ppm), and from H-6 (δ 7.68 ppm) to C-4a (δ 123.3 ppm) and C-12a (δ 124.8 ppm). (Figure S1E, Table S2). These four structure units were connected according to the HMBC correlations form H-3 (δ 6.37 ppm) to C-4a (δ 123.3 ppm), H-13 (δ 2.82 ppm) to C-4 (δ 179.2 ppm), H-1’ (δ 5.10 ppm) to C-7a (δ 128.8 ppm), C-8 (δ 135.3 ppm) and C-9 (δ 123.1 ppm), and H-1” (δ 5.41 ppm) to C-9 (δ 123.1 ppm), C-10 (δ 136.6 ppm) and C-11 (δ 150.0 ppm), (Figure S1E, Table S2).
DNA damage assays
Drug induced DNA strand breaks was evaluated in t-hESC using single cell gel electrophoresis also known as the COMET assay(Tice et al., 2000). Treated cells (approx. 20,000) were resuspended in 75 μL of molten agarose, laid on glass slides (Cell Biolabs) and let cool down for 15 min at 4°C. Cell membrane lysis was performed on the embedded cells for 30 min at 4°C. DNA was relaxed and denatured in a cold alkaline solution (300 mM NaOH; 1mM EDTA; pH=13) for 30 min at 4°C. Samples were electrophoresed at 1 V/cm in a horizontal chamber in alkaline solution. DNA was stained using Vista Green DNA dye (OxySelect Comet assay, Cell Biolabs). DNA in damaged cells migrated further than intact condensed DNA producing a “comet tail” shape. Hedamycin was used as positive control. The % of DNA in the tail was quantified using custom Acapella scripts (PerkinElmer). Gamma phosphorylation of histone H2A.X was used as a marker of induction of repair mechanisms of DNA double-strand breaks(Bonner et al., 2008). The intensity of γH2A.X nuclear staining (Key Resources Table) was quantified in treated human CSCs using high-content image analysis. Bleomycin and hedamycin were used as positive controls.
Flow cytometry
All antibodies used for flow cytometry (Key Resources Table) were titrated in order to generate signal-based populations consistent with those demonstrated by the antibody manufacturer. All extracellular staining was performed in PBS 3%FBS 0.5mM EDTA (PEF) where 100K/ml cells of interest were incubated with antibodies for 1hr at 4°, washed with 10 volumes of PEF and then stained with 7-amino actinomycin D (7-AAD) to eliminate dead cells prior to analysis. Surface marker expression and 7-AAD staining were analyzed using LSRII (BD Biosciences) and FlowJo software. Cell sorting experiments were performed using a FACSAria II (Becton Dickinson). Human primary AML MNCs were live stained for CD34 expression to enrich progenitor population. Doublet discrimination was used for all cell-sorting experiments.
EdU incorporation assays
Proliferation rate of cancer cells was determined using a 5-ethynyl-2’-deoxyuridine (EdU) Staining Proliferation kit (iFluor 647) (Abcam), according to the manufacturer’s instructions. Briefly, 5,000 cells per well of MDA-MB-231 and HT29 cultures were plated in 96-well plates. 24 hours post-seeding, cells were treated with McM025044 for 48 hours vs. DMSO control. Two hours before the end of the drug incubation period, a pulse of EdU was added to all cultures, according to manufacturer’s protocol. Then, cells were formalin-fixed (2% v/v) and EdU-positive cells were fluorescently labeled following a click chemistry reaction. Nuclei were counterstained with Hoechst 33342. The same procedure was applied to OCI-AML3 cells, which were grown and treated in ultra-low adhesions round bottom 96-well plates (Corning), and immobilized under a sealed glass coverslip in viscous fluorescence mounting medium (Vector). Image acquisition and EdU-positive cell scoring was done by high-content imaging, as described above.
Apoptosis detection assay
Apoptosis levels in DMSO and McM025044-treated cancer cells were determined using CellEvent caspase-3/7 green detection reagent (ThermoFisher Scientific, no. C10423), according to the manufacturer’s recommendations. MDA-MB-231 and HT29 cells were plated at 5,000 cells per well in 96-well plates and maintained for 24 hours in standard growth media. Then, cells were treated with DMSO or McM025044 for 48 hours. As a positive control, each cell line was treated with 1 μM of staurosporine for 6 h. Next, 5μM of the CellEvent™ Caspase 3/7 Green labeling reagent was added to each well and incubated for 30 min at 37 °C. Cells were washed twice with PBS and formalin-fixed (2% v/v) for 30 min. Hoechst 33342 was used to stain nuclei. Image acquisition and activated Caspase-3/7-positive cell scoring was done by high-content imaging, as described above.
High-throughput progenitor assays
For OCI-AML 3 colony forming unit (CFU) assays, serially diluted cells were seeded in semi-solid media at 3 cell densities (H4434, Stem Cell Technologies) and incubated for up to 14 days at 37°C and 5% CO2 prior to staining with 10uM Calcein Green AM for 40 min at room temperature (Thermo Fisher Scientific, MA, USA) to score robust colonies. For spheroid-formation assays (breast and colorectal), cells were resuspended in 0.5% methylcellulose DMEM/F12 (2%B-27 supplement + 20ng/ml EGF + 20ng/ml bFGF) and seeded at 1500 cells per well in ULA 96-well flat-bottom plates (VWR, Pennsylvania, USA). Serial dilutions of McM025044 or DMSO controls were generated in the same media and added to the plates using automated liquid handlers. After treatment, plates were incubated at 37°C and 5% CO2 for 7 days before staining with 10uM Calcein Green AM for 40 min at room temperature. Plates were then imaged with an Operetta High-Content Imaging System (Perkin Elmer, MA, USA). Images were analyzed using in-house custom analysis scripts (Acapella, Perkin Elmer, MA, USA) to quantify the number of discrete spheroids per well.
Alkyne conjugation of McM025044
In order to perform affinity pull down experiments on CSC cell lysates and identify McM025044 target, the reactive phenolic hydroxyl group in C-11 of the McM025044 was selected for alkyne conjugation via an ether bond (Figure S3A). An excess of 80% propargyl bromide (50μl) was added to a suspension of McM025044 (5mg, 0.006mmol) in anhydrous acetonitrile. Reaction was then carried out for 24 h at room temperature. The modified McM025044 alkyne was purified using semipreparative HPLC on XSelect C18 column (Waters, 5μm, 10–100mm).
1H NMR (700 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.94 (t, J = 5.3 Hz, 1H), 7.76 (d, J = 35.4 Hz, 2H), 6.38 (s, 1H), 6.27 – 6.10 (m, 2H), 6.01 (d, J = 7.2 Hz, 1H), 5.89 (d, J = 10.3 Hz, 1H), 5.56 (s, 1H), 5.44 (t, J = 6.8 Hz, 2H), 5.35 (s, 1H), 4.97 (d, J = 6.4 Hz, 1H), 4.65 (d, J = 5.0 Hz, 1H), 4.07 (dd, J = 6.5, 2.4 Hz, 1H), 3.85 (t, J = 3.4 Hz, 1H), 3.77 – 3.55 (m, 2H), 3.02 (s, 3H), 2.83 (s, 3H), 2.66 (dd, J = 26.6, 4.9 Hz, 7H), 2.24 (dd, J = 13.5, 6.6 Hz, 1H), 2.13 (d, J = 12.0 Hz, 1H), 1.62 (s, 4H), 1.44 (d, J = 5.4 Hz, 3H), 1.38 (d, J = 5.6 Hz, 3H), 1.26 (d, J = 6.3 Hz, 3H), 1.08 (s, 3H). 13C NMR (176 MHz, DMSO) δ 186.31, 179.16, 172.02, 154.31, 150.39, 138.71, 134.27, 130.05, 128.58, 125.68, 125.16, 123.65, 123.19, 109.17, 76.33, 72.38, 71.09, 70.45, 64.86, 54.71, 54.22, 51.24, 47.65, 40.02, 36.90, 35.86, 22.94, 22.53, 18.28, 16.36, 14.27, 13.19. HRMS calculated for C44H57N2O13S [M+H]+ expected 853.3581, found: 853.364 (Figure S3A). McM025044-alkyne maintained biological activity against neoplastic ES cells (Figure S3B).
Affinity-Purification Mass Spectrometry analysis
10 mg of total protein were harvested from around 8 millions of normal and neoplastic PSC, using 1 mL of lysis buffer (200 mM TRIS, 4% CHAPS, 1 M NaCl, 8 M urea, pH 8.0, 20mM N-ethylmaleimide) supplemented with 1X Halt protease inhibitor cocktail (Pierce Technologies) and lysate protein contents were quantified. Alkyne-conjugated McM025044 was synthesised as above-described. hPSC and t-hESC lysates were incubated with 10 μM of alkyne-conjugated or unconjugated McM025044. Samples were incubated for 2h at room temperature under agitation. Then, 0.2 ml of Azide-agarose beads (Jena Bioscience) was incubated with 8 mg (0.8 mL) of protein lysates and 1 mL of Copper-catalyzed click-reaction reagents from the Click Chemistry Protein Reaction Buffer Kit, according to manufacturer’s recommendations (Click Chemistry Tools). After click reaction, resin bound proteins were reduced with dithiothreitol (10 μM), alkylated with iodoacetamide (40 mM), and washed sequentially with 100 mM TRIS, 1% SDS, 250 mM NaCl, 5 mM EDTA, pH 8.0 buffer, 8 M urea/100 mM TRIS (pH 8.0), and 20% acetonitrile. Resin-bound proteins were digested for 16 h at 37°C using trypsin (5 μg/mL) diluted in 0.2 mL of a digestion buffer (100 mM TRIS, 2 mM CaCl2, 10% acetonitrile). Digested peptides were desalted using a solid-phase extraction column (C-18), eluted in 0.7 mL of 50% acetonitrile/0.1% trifluoroacetic acid. Samples were analyzed by liquid chromatography tandem mass spectrometry (LC/MS) using a linear ion-trap instrument (LTQ, ThermoFisher). Most common contaminant peptides in H. sapiens, as documented by Hodge et al. were filtered out (Table S3)(Hodge et al., 2013).
MS/MS spectral analysis and drug target identification
Tandem mass spectra were extracted and analyzed using Sequest (Thermo Fisher Scientific, San Jose, CA, USA; version 1.4.0.288) and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Sequest was set up to search Human-UniProt_Mar_26_2014.fasta (unknown version, 69011 entries) assuming the digestion enzyme trypsin. X! Tandem was set up to search the Uniprot-Human_May13–2014 database (unknown version, 46448 entries) also assuming trypsin. Sequest and X! Tandem were searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 2.0 Da. Carbamidomethyl of cysteine was specified in Sequest and X! Tandem as a fixed modification. Deamidation of asparagine and glutamine and oxidation of methionine were specified in Sequest as variable modifications. Glu->pyro-Glu of the n-terminus, ammonia-loss of the n-terminus, gln->pyro-Glu of the n-terminus, deamidated of asparagine and glutamine and oxidation of methionine were specified in X! Tandem as variable modifications. Scaffold (version Scaffold_4.3.4, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 97.0% probability to achieve a false-discovery rate (FDR) lower than 0.1%. Peptide Probabilities from Sequest were assigned by the Scaffold Local FDR algorithm. Peptide Probabilities from X! Tandem were assigned by the Peptide Prophet algorithm (Keller et al., 2002) with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 95.0% probability and contained at least 1 identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm(Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters.
Small-interfering RNA (siRNA) silencing
Human leukemia OCI-AML3 cells were cultured in Alpha MEM (Gibco) supplemented with 20% FBS (Hyclone), in ultra-low attachment plates (Corning). OCI-AML3 leukemia cell line was seeded at 25K cells/well in 96 well ultra-low-attachment round bottom plates (Corning). 0.1μg siRNA against SUMO1, SAE2, UBC9, or scramble control (siCTRL) (Santa Cruz Biotechnology Gene Silencers) was diluted in 10μL OptiMEM and mixed with 0.1μL RNAiMAX diluted in 10μL optiMEM. The siRNA mixture was vortexed for 5 seconds and incubated at room temperature for 20min. Prior to addition of the transfection mixture, cells were washed with 100μL of OptiMEM, re-suspended in 30μL of fresh OptiMEM and then mixed and incubated with the siRNA-containing transfection mixture for 6 hours. Reactions were diluted in 100μL of culture media and replaced with pure culture media after an additional 18 hours. Knockdown was evaluated by western blot and subsequent experimentation was performed 48hr post transfection.
Transcriptome profiling analysis
OCI/AML3 cells were treated with 1μM of McM025044 or vehicle control for 72h and total RNA was isolated using a commercial RNA purification kit according to the manufacturer’s instructions (Norgen Biotek). Purified RNA was quantified on a Nanodrop 2000 Spectrophotometer (Thermo Scientific), and RNA integrity was assessed by a 2100 Bioanalyser (Agilent Technologies). RNA hybridization was performed using Affymetrix Gene Chip Human Gene 1.0 ST arrays (London Regional Genomics Centre). Data were normalized using the Robust Multichip Averaging algorithm with Partek Genomics Suite software (version 6.6). For GSEA performed on SMS genes, previously profiled by Kessler et al.(Kessler et al., 2012), all samples were RMA normalized using Partek GS and mean of probsets for the same gene were calculated to collapse data to gene symbols. Myc-activated HMEC samples were compared to Control HMEC and 474 genes were found to be significantly different (p<=0.05, FC>=1.2), 308 of which were upregulated in Myc activated HMEC samples. Next, we compared HMEC SAE2 depleted (shSAE2) Myc-activated samples to HMEC shSAE2 alone, and 68 genes out of 308 Myc-induced transcripts where not induced or became repressed in response to Myc when SAE2 was depleted. The 68 gene list was applied to McM025044-modulated gene list (vs. DMSO-treated) in OCI-AML3 cells for a GSEA analysis, which resulted in high enrichment score (NES = 3.11 and p<0.001).
Immunoblotting analysis
Western analyses were performed on SDS-PAGE gels under denaturing conditions. Total protein samples were prepared in Laemmli Sample Buffer (20mM N-ethylmaleimide, 60mM Tris-HCL pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue), sonicated and thermo-reduced/denatured (5 min, 95°C) prior to electrophoresis on 12% polyacrylamide gels, as previously described(Benoit et al., 2010). For dot blot experiments, equal amounts of protein content were spotted on nitrocellulose membranes (5μl/cm2) and air-dried for 30 min. Membranes were blocked in PBS containing 5% skim milk and 0.1% TWEEN 20. Primary antibodies were diluted (Key Resources Table) in the blocking solution, and incubated overnight at 4 °C. After 3 sequential washing steps (10 min each) with PBS, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Goat anti-Mouse HRP-conj., Cat#1721011 and Goat anti-Rabbit HRP-conj., Cat#1706515, BioRad), washed 3 times (10 mins) in PBS-Tween, and developed using the Immobilon Western Kit (Millipore, WBKLS0100). Blot images were acquired using a ChemiDoc™ XRS+ System with Image Lab™ Software (Bio Rad). Quantitative optical densitometry signal analysis was performed using Image J software (National Institutes of Health).
In vitro SUMOylation assays
In vitro SUMOylation reaction were performed using a SUMOylation kit according to the Manufacturer’s instructions (ENZO Life Sciences). Increasing doses (0.02 to 0.5 mM) of Ginkgolic acid (GA) and McM025044 were used to validate SUMOylation inhibition effects. Reaction products were detected by western blot analysis (Key Resources Table). Reaction intermediate formation was evaluated by excluding subsequent substrate/enzymatic components from mixtures. β-mercaptoethanol was omitted from Laemmli Sample Buffer to preserve thioester bonds integrity.
Immunofluorescence analysis
HCT116 and t-hESCs were plated, cultured, and treated as above described, prior to fixation with 2% formalin and incubated in Perm/Wash buffer (BD Biosciences) at 4°C for 15–30 min prior to immunostaining(Benoit et al., 2017). Anti-SUMO1 and anti-BMI1 primary antibodies (Key Resources Table) were diluted in 1% BSA-PBS solution and incubated overnight at 4°C. Secondary antibodies were used at 1:500 in a 1% BSA-PBS solution. Nuclei were stained with Hoechst 33342, and images were acquired using an Operetta High Content Imaging System (Perkin Elmer). Images were analyzed using custom Acapella scripts (PerkinElmer).
E1 Ligase Cloning, Expression and Purification
Human SAE1 and SAE2 genes were codon-optimized for expression in Escherichia coli, commercially synthesized and sub-cloned into pET-11c and pET-28b(+) respectively (GenScript). Mutant SAE1 (R21A) and SAE2 (R59A) genes were created by site-directed mutagenesis using the site-saturation mutagenesis protocol(Zheng et al., 2004). Expression and purification of SAE1 and SAE2 plasmids was carried out as described previously(Lois and Lima, 2005b), with minor changes. In brief, SAE1 and SAE2 plasmids were co-transformed into Escherichia coli BL21-CodonPlus (DE3) (Agilent). 10L cultures were fermented in Terrific Broth at 37°C to an OD600 = 1 and then induced with IPTG at 30°C for 4 hours. Cells were harvested by centrifugation and lysed by sonication in 50mM Tris pH 8.0, 20% sucrose, 350mM NaCl, 0.1% NP-40, 0.1% β-mercaptoethanol, 1mM PMSF and 10ug/mL DNase I (NEB). After clarification by centrifugation, soluble SAE1/2 was purified by nickel-affinity chromatography, with elution of SAE1/2 in 50mM Tris pH 8.0, 200mM NaCl, 250mM imidazole and 0.1% β-mercaptoethanol. Further purification was carried out by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 column (GE Healthcare) in 20mM Tris pH 8.0, 350mM NaCl and 0.1% β-mercaptoethanol and anion exchange chromatography on a 5mL HiTrap Q HP (GE Healthcare) in 20mM Tris pH 8.0 and 0.1% β-mercaptoethanol, with elution over a gradient of 0–1M NaCl. A final purification step occurred via cation exchange chromatography on a 5mL HiTrap SP HP column (GE Healthcare) using the same buffers as anion exchange, where the SAE1/2 complex did not bind to the column, while the remaining contaminants did. After purification, the SAE1/2 complex was exchanged into 10mM Tris pH 9.0, 87mM NaCl and 1mM DTT before being flash frozen with liquid nitrogen and stored at −80°C.
Generation of overexpression plasmids and lentiviral transduction
cDNA synthesis was performed from total RNA extracted from t-hESC and primed with oligo(dT), using SuperScript III First Strand Synthesis System (Thermo Fisher Scientific). Wild type SAE2 and UBC9 ORF were amplified by RT-PCR using Phusion Hot Start II DNA polymerase (Thermo Fisher Scientific) and subcloned into pUC19 vector using InFusion HD Cloning Kit (Takara Bio USA). Both, SAE2 and UBC9 ORFs were then PCR amplified and subcloned into pLV-mCherry plasmid (Addgene #36084). All PCR steps described above have been performed using Phusion Hot Start II DNA polymerase (Thermo Fisher Scientific). All subcloning steps have been performed in OneShot Stbl3 Chemically Competent E. coli cells (Thermo Fisher Scientific). Lentiviral expression plasmids were co-transfected with pMD2.G (Addgene #12259) and psPAX2 (Addgene #12260) as previously described(Benoit et al., 2017). Medium was collected, centrifuged to remove cells and debris, filtered through a 0.45um filter and then ultracentrifuged at 20K x g for 2hr at 4°. Concentrated lentivirus was re-suspended in 0.5ml of PBS per 75cm2 flask of 293-FT cells worth of supernatant. Individual lentivirus preps were titrated on the desired cell line to be transduced in order to optimize expression but minimize transduction induced cell death.
Molecular docking modelling
To predict the binding site of McM025044 in human SUMO E1 complex, we performed an unbiased docking calculation using SMINA docking software(Quiroga and Villarreal, 2016). 3-D X-ray crystallographic structure of human SUMO E1 complex with a SUMO1-AMP mimic downloaded from the Protein Data Bank (PDB) Web site (http://www.rcsb.org) using PDB ID 3KYC. The structure file was edited to remove all but the two interacting chains SAE1 and SAE2. All missing atoms in the PDB files were added using WHAT IF web server (https://swift.cmbi.umcn.nl/servers/html/index.html). Finally, the clean structure file with two chains was converted to the PDBQT format using AutoDock Tools (http://mgltools.scripps.edu/). We treated all the bonds in a protein complex as rigid, i.e. no flexible residues were selected. McM025044 was sketched in 2-D using ChemDraw tool of ChemBio office package (http://www.cambridgesoft.com). 2-D structure was converted to 3-D using Chem3D tool. Ligand geometry was then optimized by performing an energy minimization run using MMFF94 force-field(Halgren Thomas, 1996). Lastly, optimized 3-D structure of McM025044 was converted to the PDBQT format using AutoDock Tools (http://mgltools.scripps.edu/) with the default identification of rotatable bonds. Ligand docking was carried out by creating a grid box of size 60 Å × 60 Å × 92 Å, centered at the geometric center of the SAE1/2 complex, with a grid spacing of 1 Å. No region of the complex protein complex was excluded in the search for the most favorable interactions of McM025044 i.e. the ligand was allowed to explore its binding pocket in an unbiased manner. The input exhaustiveness parameter for the docking was set to 1000. The number of top docking orientations with high docking scores was fixed to 15. Top docking pose was then analyzed to study the interaction of McM025044 with SAE1/2 complex.
Colony initiating cell (CIC) assays
t-hESCs and normal hPSCs were cultured on Matrigel coated plates (Corning) in MEF conditioned media supplemented with 20 ng/uL bFGF and passed every 5–7 days as needed. The day after passing, the cells were transduced with lentivirus based on the virus titre. t-hESCs and hPSCs were fed with fresh MEF-conditioned media + bFGF 24- and 48-hours post-transduction. 72 hrs post-transduction, and washed with warm DPBS (Wisent) prior to dissociation using cell dissociation buffer (Gibco). hPSCs were treated with Collagenase IV (ThermoFisher Scientific) before the dissociation step. Cells were collected by centrifugation at 1500 rpm for 5 minutes, filtered and counted. Dissociated cells were re-suspended in 200 uL of sorting buffer (200 mL DPBS (Gibco), 1 mM EDTA (Invitrogen), 25 mM HEPES (ThermoFisher Scientific), 0.2% BSA (ThermoFisher Scientific)) and stained for fluorescent activated cell sorting (FACS). Cells were sorted based on GFP (expression vector reporter system), viability (7AAD, Beckman Coulter, 1:50) and TRA-1–60 expression (BD Biosciences, 1:100, 1hr at 4°C). After staining, cells were sorted directly into prepared 6-well plates. Single cells were seeded at 1000 cells (t-hESC) or 2500 cells (hPSC) per well into 3 wells of a 6-well plate, pre-coated with Matrigel (Corning) and pre-filled with 2.5 mL MEF-conditioned media + bFGF. Wells hosting hPSC were previously seeded with 200,000 irradiated mouse embryonic fibroblasts (iMEFs). After seeding, t-hESCs and hPSCs were fed every other day and colonies were counted by phase contrast microscopy on day 7 (t-hESC) or day 9 (hPSC) post-seeding.
Functional evaluation of AML primary patient samples
For control vs. McM025044 treatments, primary AML and normal hematopoietic cells were maintained in serum-free media (StemSpan #09600, StemCell Technologies) supplemented with stem cell factor (SCF) (100 ng/mL), FLT3 ligand (100 ng/mL) and thrombopoietin (TPO) (20 ng/mL) at a density of 250,000 cells/mL. For functional analysis of hematopoietic clonogenic progenitors in colony/cluster forming units (CFU) assays, primary cells were treated with McM025044 (doses ranging from 0.0015 to 10 μM) or vehicle control for 24h and seeded (25,000 MNC AML and 500 lineage depleted CB) in methylcellulose-containing media (Methocult H4434, StemCell Technologies) in 12-well plates as previously reported(Hess et al., 2002). After 14 days, hematopoietic colonies were scored manually under a 10X magnification. IC50 curves were fit with a four-parameter logistic curve function (GraphPad Prism). The R squared values for all of the curves were >0.5 except for cord blood #68 and #97 where the IC50 was difficult to define because McM025044 only impacted CFU counts at the highest concentration tested.
Xenotransplantation assays
AML primary cells were cultured in serum-free StemSpan media supplemented with SCF, FLT3 ligand, and TPO at a density of 250,000 cells/mL and were treated with 1 μM McM025044 or vehicle control for 24h. NOD.SCID adult mice were sub-lethally irradiated with 325 rads, 24 hours before transplantation. 1 × 106 live AML MNCs (McM025044 or vehicle treated) were injected via tail vein. After 8 weeks, animals were culled, and the BMs were harvested and the presence of cells expressing human (CD45) leukemic myeloid marker (CD33), (hCD45+/CD33+) was quantified by flow cytometry (LSRII, BD Biosciences). For secondary transplants, unsorted BM engrafted human AML cells were transplanted IV in adult irradiated NOD.SCID mice as described for primary transplants. Presence of human chimerism was evaluated in secondary recipients, 8 weeks post-transplantation as described for primary mice. In all cases, the threshold for engraftment detection was set at ≥0.1% human chimerism, as previously described(Boyd et al., 2018). Outlier values were removed from presented data by applying a maximum normed residual test (Grubbs’ test). The McMaster University Animal Care Council approved all in vivo procedures and protocols. AML samples # 11040, 13051.1, and 13814.1 were used (Key Resources Table and Table S5).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data is represented as the mean ± SEM. P-values ≤ 0.05 were considered significant. “n” denotes the number of times the data was replicated. Specific “n” number for each experiment and scale bar length are indicated in figure legends. Significant differences between groups were determined by 2-way ANOVA test and unpaired one or two-tailed Student t-test, using GraphPad Prism software. Grubbs’ test was applied to determine outlier’s exclusion. Fisher’s exact test was used to determine statistical significance between two classifications.
Supplementary Material
Table S1. Unique actinomycetes strains showing CSC bioactivity across all three batches of crude samples, related to Figure 1.
Table S2. 1H and 13C NMR data of compound Peak-1 and McM025044, related to Figure 1.
Table S3. List of peptides isolated by McM025044 affinity-purification in human CSCs, related to Figure 3.
Table S4. Lists of genes upregulated in AML patient samples (Mills et al., 2009) and SMS genes, related to Figure 3.
Table S5. AML patient samples information and engraftment level in xenotransplantation experiments, related to Figure 5.
Highlights.
Natural product compound (McM025044) has selective anti-neoplastic activity
McM025044 binds to SAE2 to reduce SUMOylation of intracellular proteins
SUMOylation inhibition by McM025044 impairs stem cell self-renewal
McM025044 restricts in vivo activity of SUMO-dependent leukemia initiating cells
Acknowledgments
This work was supported by grants from the Ontario Research Fund (OCRiT) led by MB and the David Braley Foundation, work in this study related to human leukemia was funded and supported by CCS Impact Grant #706694, whereas results and studies using human pluripotent stem cells was supported by Canadian Institute of Health Research (CIHR) foundation grant FRN 159925 to Mickie Bhatia approved by the Stem Cell Oversight Committee in Canada, and finally the Canada Research Chair in human stem cell biology (M.B.) CIHR for molecular studies of antibiotics (G.D.W.). Y.D.B. is supported by the NSERC (RGPIN-2018–06521), the Cancer Research Society (#24039) and the CIHR (PJT- 173541). J.S.P. was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM104420. Computer resources were provided by the Institute for Bioinformatics and Evolutionary Studies Computational Resources Core sponsored by the National Institutes of Health (P30 GM103324). We acknowledge the Centre for Microbial Chemical Biology (CMCB) core facility and staff members Susan McCusker and Nikki Henriquez for assistance with protein production and mass spectrometry respectively.
Footnotes
Declaration of Interests
The authors declare no competing financial interests. However, some authors are inventors of a provisional patent related to unique targeting of human cancer cells using SAE2 and related probe compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aslostovar L, Boyd AL, Almakadi M, Collins TJ, Leong DP, Tirona RG, Kim RB, Julian JA, Xenocostas A, Leber B, et al. (2018). A phase 1 trial evaluating thioridazine in combination with cytarabine in patients with acute myeloid leukemia. Blood advances 2, 1935–1945. 10.1182/bloodadvances.2018015677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Mitchell RR, Risueño RM, Orlando L, Tanasijevic B, Boyd AL, Aslostovar L, Salci KR, Shapovalova Z, Russell J, et al. (2017). Sam68 Allows Selective Targeting of Human Cancer Stem Cells. Cell Chem Biol 24, 833–844.e839. 10.1016/j.chembiol.2017.05.026. [DOI] [PubMed] [Google Scholar]
- Benoit YD, Pare F, Francoeur C, Jean D, Tremblay E, Boudreau F, Escaffit F, and Beaulieu JF (2010). Cooperation between HNF-1alpha, Cdx2, and GATA-4 in initiating an enterocytic differentiation program in a normal human intestinal epithelial progenitor cell line. Am J Physiol Gastrointest Liver Physiol 298, G504–517. ajpgi.00265.2009 [pii] 10.1152/ajpgi.00265.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Wang JC, Kapp U, Bonnet D, and Dick JE (1997). Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A 94, 5320–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogachek MV, Chen Y, Kulak MV, Woodfield GW, Cyr AR, Park JM, Spanheimer PM, Li Y, Li T, and Weigel RJ (2014). Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell 25, 748–761. 10.1016/j.ccr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogachek MV, Park JM, De Andrade JP, Lorenzen AW, Kulak MV, White JR, Gu VW, Wu VT, and Weigel RJ (2016). Inhibiting the SUMO Pathway Represses the Cancer Stem Cell Population in Breast and Colorectal Carcinomas. Stem Cell Reports 7, 1140–1151. 10.1016/j.stemcr.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, and Pommier Y (2008). GammaH2AX and cancer. Nat Rev Cancer 8, 957–967. 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, and Dick JE (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3, 730–737. [DOI] [PubMed] [Google Scholar]
- Bossis G, Sarry JE, Kifagi C, Ristic M, Saland E, Vergez F, Salem T, Boutzen H, Baik H, Brockly F, et al. (2014). The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep 7, 1815–1823. 10.1016/j.celrep.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Boyd AL, Aslostovar L, Reid J, Ye W, Tanasijevic B, Porras DP, Shapovalova Z, Almakadi M, Foley R, Leber B, et al. (2018). Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell 34, 483–498.e485. 10.1016/j.ccell.2018.08.007. [DOI] [PubMed] [Google Scholar]
- Du L, Li YJ, Fakih M, Wiatrek RL, Duldulao M, Chen Z, Chu P, Garcia-Aguilar J, and Chen Y (2016). Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nature communications 7, 12326. 10.1038/ncomms12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447. 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming A (2015). Cancer stem cells: Targeting the root of cancer relapse. Nat Rev Drug Discov 14, 165. 10.1038/nrd4560. [DOI] [PubMed] [Google Scholar]
- Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, Kimura K, Sodeoka M, and Yoshida M (2009). Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol 16, 133–140. 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Halgren Thomas A (1996). Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry 17, 490–519. ;2-P. [DOI] [Google Scholar]
- Hansen MR, and Hurley LH (1996). Pluramycins. Old drugs having modern friends in structural biology. Acc. Chem. Res 29, 249–258. [Google Scholar]
- Harvey AL, Edrada-Ebel R, and Quinn RJ (2015). The re-emergence of natural products for drug discovery in the genomics era. 14, 111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, Bernard H, Yang X, Liao H, Rabino C, et al. (2017). Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol 13, 1164–1171. 10.1038/nchembio.2463. [DOI] [PubMed] [Google Scholar]
- Herwig R, Hardt C, Lienhard M, and Kamburov A (2016). Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11, 1889–1907. 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- Hess DA, Levac KD, Karanu FN, Rosu-Myles M, White MJ, Gallacher L, Murdoch B, Keeney M, Ottowski P, Foley R, et al. (2002). Functional analysis of human hematopoietic repopulating cells mobilized with granulocyte colony-stimulating factor alone versus granulocyte colony-stimulating factor in combination with stem cell factor. Blood 100, 869–878. [DOI] [PubMed] [Google Scholar]
- Hirohama M, Kumar A, Fukuda I, Matsuoka S, Igarashi Y, Saitoh H, Takagi M, Shin-ya K, Honda K, Kondoh Y, et al. (2013). Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS chemical biology 8, 2635–2642. 10.1021/cb400630z. [DOI] [PubMed] [Google Scholar]
- Hodge K, Have ST, Hutton L, and Lamond AI (2013). Cleaning up the masses: exclusion lists to reduce contamination with HPLC-MS/MS. J Proteomics 88, 92–103. 10.1016/j.jprot.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. (2012). A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353. 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Maezawa Y, Ando Y, Kusumi T, Matsumoto T, and Suzuki K (2014). Synthesis of the pluramycins 2: total synthesis and structure assignment of saptomycin B. Angew Chem Int Ed Engl 53, 1262–1265. 10.1002/anie.201308017. [DOI] [PubMed] [Google Scholar]
- Kotz J (2012). Phenotypic screening, take two. Science-Business eXchange 5, 380–380. 10.1038/scibx.2012.380. [DOI] [Google Scholar]
- Kumar A, Ito A, Hirohama M, Yoshida M, and Zhang KY (2013). Identification of sumoylation activating enzyme 1 inhibitors by structure-based virtual screening. J Chem Inf Model 53, 809–820. 10.1021/ci300618e. [DOI] [PubMed] [Google Scholar]
- Li YJ, Du L, Wang J, Vega R, Lee TD, Miao Y, Aldana-Masangkay G, Samuels ER, Li B, Ouyang SX, et al. (2019). Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem Biol 26, 278–288.e276. 10.1016/j.chembiol.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciardello MP, and Kubicek S (2016). Pharmacological treats for SUMO addicts. Pharmacol Res 107, 390–397. 10.1016/j.phrs.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Lois LM, and Lima CD (2005a). Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. The EMBO Journal 24, 439–451. 10.1038/sj.emboj.7600552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois LM, and Lima CD (2005b). Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. Embo j 24, 439–451. 10.1038/sj.emboj.7600552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z, Yuan L, Atkison JH, Williams KM, Vega R, Sessions EH, Divlianska DB, Davies C, Chen Y, and Olsen SK (2018). Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat Commun 9, 5145. 10.1038/s41467-018-07015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, Wei W, Bowen DT, Loeffler H, Hernandez JM, et al. (2009). Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 114, 1063–1072. 10.1182/blood-2008-10-187203. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Mitchell RR, Benoit YD, Orlando L, Reid JC, Shimada K, Davidson KC, Shapovalova Z, Collins TJ, Nagy A, and Bhatia M (2019). Human Pluripotency Is Initiated and Preserved by a Unique Subset of Founder Cells. Cell. 10.1016/j.cell.2019.03.013. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Newman DJ, and Cragg GM (2012). Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75, 311–335. 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DJ, and Cragg GM (2016). Natural Products as Sources of New Drugs from 1981 to 2014. Journal of natural products 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Olsen SK, Capili AD, Lu X, Tan DS, and Lima CD (2010). Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463, 906–912. 10.1038/nature08765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, et al. (2019). The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541. 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye CR, Bertin MJ, Lokey RS, Gerwick WH, and Linington RG (2017). Retrospective analysis of natural products provides insights for future discovery trends. Proc Natl Acad Sci U S A 114, 5601–5606. 10.1073/pnas.1614680114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga R, and Villarreal MA (2016). Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PloS one 11, e0155183. 10.1371/journal.pone.0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Spitzer M, Wang W, Waglechner N, Patel DJ, O’Brien JS, Ejim L, Ejim O, Tyers M, and Wright GD (2016). Discovery of Ibomycin, a Complex Macrolactone that Exerts Antifungal Activity by Impeding Endocytic Trafficking and Membrane Function. Cell Chem Biol 23, 1383–1394. 10.1016/j.chembiol.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al. (2012). Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 149, 1284–1297. 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- Stewart MH, Bendall SC, Levadoux-Martin M, and Bhatia M (2010). Clonal tracking of hESCs reveals differential contribution to functional assays. Nat Methods 7, 917–922. 10.1038/nmeth.1519. [DOI] [PubMed] [Google Scholar]
- Sukuru SC, Jenkins JL, Beckwith RE, Scheiber J, Bender A, Mikhailov D, Davies JW, and Glick M (2009). Plate-based diversity selection based on empirical HTS data to enhance the number of hits and their chemical diversity. J Biomol Screen 14, 690–699. 10.1177/1087057109335678. [DOI] [PubMed] [Google Scholar]
- Tashiro E, and Imoto M (2012). Target identification of bioactive compounds. Bioorg Med Chem 20, 1910–1921. 10.1016/j.bmc.2011.10.081. [DOI] [PubMed] [Google Scholar]
- Thaker MN, Wang W, Spanogiannopoulos P, Waglechner N, King AM, Medina R, and Wright GD (2013). Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat Biotechnol 31, 922–927. 10.1038/nbt.2685. [DOI] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, and Sasaki YF (2000). Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35, 206–221. [DOI] [PubMed] [Google Scholar]
- Visvader JE, and Lindeman GJ (2012). Cancer stem cells: current status and evolving complexities. Cell Stem Cell 10, 717–728. 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, and Werb Z (2008). Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2, 90–102. 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbowetski-Ogilvie TE, Bosse M, Stewart M, Schnerch A, Ramos-Mejia V, Rouleau A, Wynder T, Smith MJ, Dingwall S, Carter T, et al. (2009). Characterization of human embryonic stem cells with features of neoplastic progression. Nature biotechnology 27, 91–97. nbt.1516 [pii] 10.1038/nbt.1516. [DOI] [PubMed] [Google Scholar]
- Wright GD (2017). Opportunities for natural products in 21. Nat Prod Rep 34, 694–701. 10.1039/c7np00019g. [DOI] [PubMed] [Google Scholar]
- Zheng L, Baumann U, and Reymond J-L (2004). An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Research 32, e115–e115. 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Unique actinomycetes strains showing CSC bioactivity across all three batches of crude samples, related to Figure 1.
Table S2. 1H and 13C NMR data of compound Peak-1 and McM025044, related to Figure 1.
Table S3. List of peptides isolated by McM025044 affinity-purification in human CSCs, related to Figure 3.
Table S4. Lists of genes upregulated in AML patient samples (Mills et al., 2009) and SMS genes, related to Figure 3.
Table S5. AML patient samples information and engraftment level in xenotransplantation experiments, related to Figure 5.
Data Availability Statement
Transcriptomics data are available at GSE160551