
Figure 2. Integrative genome‐scale metabolic modeling identifies altered metabolic pathways in residual cells compared with normal cells.
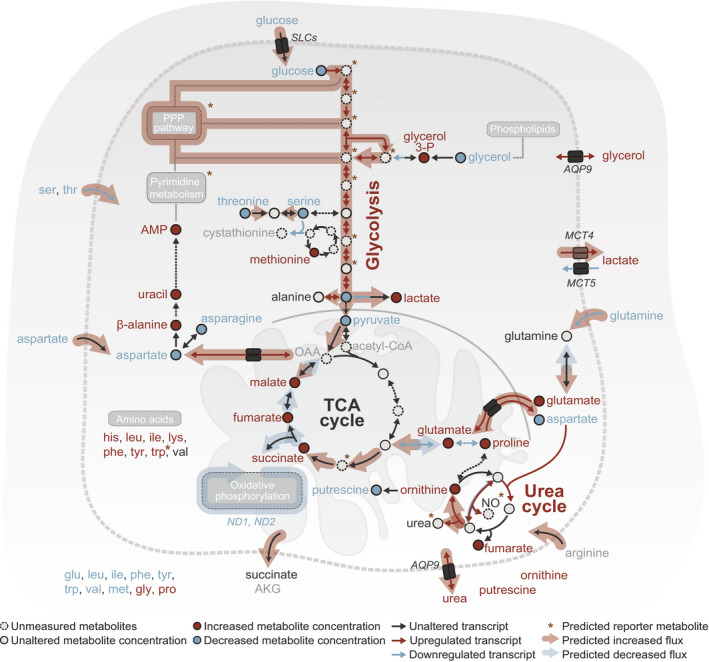
A selection of genes with significantly altered expression (Bonferroni‐adjusted P < 0.1; normal, n = 8; tumor and residual, n = 4), targeted metabolites with significantly altered levels (Benjamini–Hochberg‐adjusted P ≤ 0.05; normal, tumor, and residual, n = 8; WT n = 2), and significant reporter metabolites (top 5% with P < 0.1) of core metabolic processes are presented. A two‐sided Wald test with a Negative Binomial GLM (Love et al, 2014) was used as test statistics for gene expression. For metabolite levels, statistics were calculated using the limma package (Ritchie et al, 2015) in R with the significance threshold corresponding to a Benjamini–Hochberg‐adjusted P‐value ≤ 0.05 (residual compared to normal). For the reporter metabolites, a gene set enrichment analysis was performed from a theoretical null distribution using the reporter method (Varemo et al, 2013). Bonferroni‐adjusted P‐values of the expression analysis were used as gene‐level statistics. Metabolite‐gene sets were derived from a genome‐wide human metabolic model (HMR2), with genes mapped to mouse orthologs (Mardinoglu et al, 2014). Flux balance analysis predicted metabolic fluxes, which are altered in the corresponding (overlaid) pathways. Significantly altered gene expressions and metabolites were used to inform the predictions. Number of replicates corresponds to individual mice used to derive organoids.