

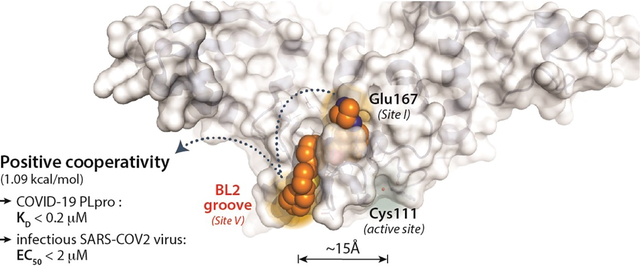
Abstract
Antiviral agents that complement vaccination are urgently needed to end the COVID-19 pandemic. The SARS-CoV-2 papain-like protease (PLpro), one of only two essential cysteine proteases that regulate viral replication, also dysregulates host immune sensing by binding and deubiquitination of host protein substrates. PLpro is a promising therapeutic target, albeit challenging owing to featureless P1 and P2 sites recognizing glycine. To overcome this challenge, we leveraged the cooperativity of multiple shallow binding sites on the PLpro surface yielding novel 2-phenylthiophenes with nanomolar inhibitory potency. New co-crystal structures confirmed that ligand binding induces new interactions with PLpro: by closing of the BL2 loop of PLpro forming a novel “BL2 groove”; and by mimicking the binding interaction of ubiquitin with Glu167 of PLpro. Together, this binding cooperativity translates to the most potent PLpro inhibitors reported to date, with slow off-rates, improved binding affinities, and low micromolar antiviral potency in SARS-CoV-2-infected human cells.
Keywords: COVID-19, cysteine protease, non-covalent inhibitor, PLpro, antiviral agent
Graphical Abstract
INTRODUCTION
The COVID-19 pandemic, caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),1 has brought about profound socioeconomic challenges for humankind. Currently approved antiviral agents have not effectively addressed the COVID-19 pandemic, and we are learning belatedly that it is essential to proactively create new antiviral agents for future outbreaks of this and other zoonotic viruses. The expedited approval and administration of the first vaccines is one important step in ending the pandemic. However, there are questions about the long-term effects of immunoprotection from: the resurgence of COVID-19 in a population with high seroprevalence in Manaus, Brazil;2 and recent evidence suggesting those who previously infected with Alpha, Beta, and Gamma variants are more susceptible to the rapidly spreading Delta variant.3 With the evolution and spread of new variants, there exists an urgent need to develop small molecule antiviral agents to treat patients who do not respond or cannot tolerate vaccines and to address future outbreaks.
The early sequencing of the SARS-CoV-2 genome has allowed comparisons with other coronaviruses including the Middle East Respiratory Syndrome CoV (MERS-CoV) and the earlier SARS-CoV, which like SARS-CoV-2 uses the angiotensin-converting enzyme 2 (ACE2) receptor to enter host cells.4 SARS-CoV-2 shares 86% overall amino acid sequence identity with SARS-CoV and ~50% identity with MERS-CoV.1 The high homology of SARS-CoV-2 to other coronaviruses has allowed the rapid understanding of its viral biology, from particle attachment, entry, replication and primary translation (polyprotein processing), assembly, maturation, to release and shedding.5 The SARS-CoV-2 spike protein recognizes and attaches to ACE2 and utilizes the cell surface serine protease TMPRSS2 to promote viral entry. 4, 6 Following entry, viral RNA is translated by the host ribosome to yield two large overlapping polyproteins, pp1a and pp1ab. Two viral cysteine proteases, the coronavirus main protease (3CLpro; nsp5) and the papain-like protease (PLpro; nsp3), proteolytically process these two viral polyproteins to yield individual non-structural proteins (nsps) that then assemble into complexes with host membrane components.7 3CLpro cleaves at 11 polyprotein sites to release itself and 11 other nsps, including the RNA-dependent RNA polymerase, the molecular target of FDA-approved COVID-19 treatment remdesivir.8 PLpro, which recognizes the P4–P1 sequence LxGG, cleaves at three sites within the polyprotein to release nsps 1–3. Nsp3 (1922aa, 215 kDa) incorporates PLpro itself (residues 1602–1855) and is the largest component of the replication and transcription complex.9, 10 The catalytic activities of 3CLpro and PLpro are essential for viral replication, making inhibition of these enzymes a compelling strategy for antiviral therapy.
PLpro supports viral replication beyond the role of viral polyprotein processing by disrupting the host innate immune response. Specifically, PLpro removes ubiquitin (Ub) and ubiquitin-like proteins (UbL), such as interferon-stimulated gene product 15 (ISG15), from host proteins.11–17 Post translational modification by Ub and UbL can regulate host protein cellular localization, stability, or involvement in specialized responses such as antiviral immunity. PLpro recognizes and cleaves the C-terminal RLRGG sequence of many UbLs, acting as a deubiquitinase (DUB) towards Ub- and UbL-modified proteins. PLpro DUBactivity is hypothesized to cause dysregulation of both the initial inflammatory and subsequent interferon responses. Substantial SARS-CoV-2-related mortality is associated with cytokine storms that arise from dysregulated inflammatory responses.18 Thus, targeting PLpro is an attractive strategy to inhibit viral replication and to prevent disruption of the host immune response to viral infection.
Despite the lack of tangible success from repurposing approaches to the earlier SARS-CoV and MERS-CoV outbreaks, there have been many publications on drug repurposing for COVID-19. Of the two essential cysteine proteases of SARS-CoV-2, 3CLpro (or Mpro) is inhibited by many known cysteine protease inhibitors, the majority of which act via covalent modification of the active site cysteine and 3CLpro would seem to be a more amenable target for drug repurposing. The promiscuity of many human cysteine protease inhibitors has slowed the progress of these agents into clinical use; however, off-target inhibition by calpain-1 inhibitors of cathepsin-L and 3CLpro may be opportunistically exploited, since cathepsin-L also facilitates viral entry. 19, 20 Discovery of PF-008352313 as a covalent active-site-directed inhibitor of SARS-CoV 3CLpro in 2003 allowed the relatively rapid translation of this agent into clinical trials for SARS-CoV-2 in 2020.21 Like 3CLpro, PLpro from SARS-CoV-2 has 100% active site homology with the enzyme from SARS-CoV. In contrast to 3CLpro, there are very few potent inhibitors of SARS-CoV PLpro with experimentally validated efficacy; therefore, targeting PLpro with repurposed drugs is problematic.22–25 A key reason for the lack of potent PLpro inhibitors is the restricted binding pockets at the P1 and P2 substrate binding sites (Gly-Gly recognition). This presents severe challenges for inhibitor design and precludes a rapid drug discovery strategy.26
The resolution of crystal structures of SARS-CoV PLpro by Ratia et al. demonstrated a conformationally flexible BL2 loop, remote from the active site cysteine, which could be stabilized by small molecule SARS-CoV PLpro inhibitors.22 Although some of these inhibitors had reported poor metabolic stability, GRL0617 (1) attained 14.5 μM potency in inhibition of host cell death to infectious SARS-CoV. 23, 24, 27 Recently, the efficacy of GRL0617 (1) in a SARS-CoV-2 viral plaque reduction assay (EC50 27.6 μM) was confirmed;16 and several simple derivatives were shown to retain activity (e.g. rac5c at 33 μM, the maximum soluble dose: 60% antiviral activity compared to remdesivir).28 Peptide-based covalent inhibitors of PLpro have also been reported.29 The two best inhibitors, VIR250 and VIR251, showed weak potency with IC50s of 50 μM in biochemical assays, with no cellular antiviral data reported; highlighting again the challenge in developing potent PLpro inhibitors.
To address the relatively featureless active site, we hypothesized that the development of potent PLpro inhibitors would require exploiting binding cooperativity to leverage multiple shallow binding sites on the PLpro surface. Positive cooperativity occurs when multiple binding interactions lead to a ligand with a binding affinity better than the sum of the individual interactions.30, 31 Therefore, to improve inhibitor potency, we explored binding cooperativity at potential binding sites distal to the active site cysteine. Two such binding sites were apparent: one created by Glu167, important in the binding of ubiquitin by PLpro; and a second novel binding site that we termed the “BL2 groove”. The BL2 groove is positioned between the β8 and β9 strands, adjacent to the BL2 loop, and is not engaged by any reported PLpro inhibitors or substrates. Inhibitors that bound to these ancillary sites were observed to have improved potency and slower off-rates. Out of almost 100 compounds synthesized, those that engaged the “BL2 groove” represent, to our knowledge, the most potent PLpro inhibitors yet to be reported, displaying low nanomolar potency against SARS-CoV-2 PLpro. Two of these compounds also displayed promising, low micromolar potency against viral infection in human lung epithelial A549 cells expressing hACE2, the human receptor by which SARS-CoV-2 gains viral entry, showing potency improved greatly over GRL0617 (1). The two lead compounds showed both good microsomal stability and satisfactory bioavailability after i.p. injection. The demonstration that a non-covalent, non-catalytic-site PLpro inhibitor can be rationally designed is significant, because such an inhibitor in combination with antivirals blocking other viral targets (such as remdesivir) could have a major impact on therapy of COVID-19 and future coronavirus outbreaks.
RESULTS
Design and Optimization
To select a starting scaffold for structure-based PLpro inhibitor design, we first carried out high-throughput screening (HTS) to identify SARS-CoV-2 PLpro inhibitors, the details of which will be published elsewhere. The hit rate of this HTS campaign was low, in accord with contemporary reports.28, 32 Consequently, we selected the naphthalenyl benzamide of GRL0617 as a starting scaffold, based on: i) the availability of a SARS GRL0617:PLpro co-crystal structure from Ratia et al. (PDB: 3E9S)33; ii) the sequence identity of the SARS PLpro to SARS-CoV-2 PLpro (83% overall and 100% at the active site); and iii) the superior potency of GRL0617 to all other identified hits from HTS. Optimization was driven by measurement of potency for SARS-CoV-2 PLpro inhibition and affinity for PLpro measured using SPR (Figure S1)
Initial structure-based design leveraged the GRL0617:PLpro co-crystal structure (PDB: 3E9S): the design rationale is summarized in Figure 1. The benzamide of GRL0617 forms two key hydrogen bonding interactions with the mainchain nitrogen of Gln269 and sidechain of Asp164 in PLpro, thereby closing the BL2 loop (Figure 1A). Replacement of benzamide with benzylamine or benzyl sulfonamide bioisosteres (DY2-64 (17) and DY3-63 (18)) led to a sharp decline in potency, therefore the benzamide was conserved moving forward (Table S1).
Figure 1. Structure-guided design of SARS-CoV-2 PLpro inhibitors to explore druggable binding sites.
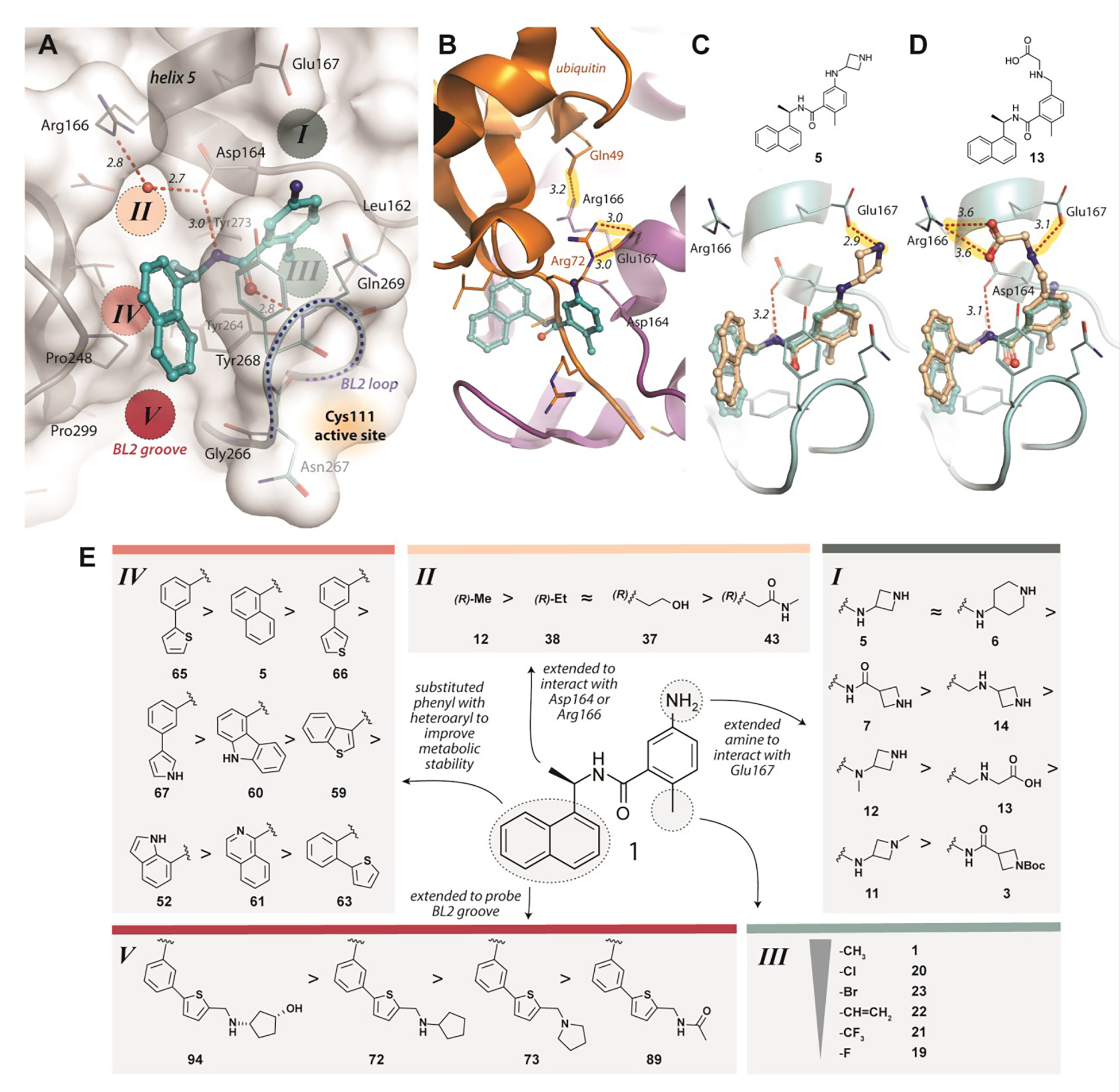
A) Identification of potential ligand binding Sites I-V (PDB: 3E9S). Key hydrogen bonds are shown as red, dashed lines, with distances (Å) labeled in italics. B) A superposition of GRL0617 (cyan; PDB 3E9S) onto the PLpro-ubiquitin structure (orange/magenta; PDB 4MM3) shows that Glu167 of PLpro (magenta) interacts with Arg72 of ubiquitin (orange) in Site I and Arg166 interacts with Gln49 of ubiquitin in Site II. New compounds were designed to mimic these two key interactions to improve binding affinity and to engage Sites I and II. C) Modeling of ZN-2-184 (5) (wheat) bound to PLpro, superimposed with PLpro-GRL0617 (cyan, PDB 3E9S), with the azetidine ring capturing the electrostatic interaction with Glu167 in Site I; D) Modeling of ZN-3-56 (13) (wheat) bound to PLpro, superimposed with PLpro-GRL0617 (cyan, PDB 3E9S; showing the glycine sidechain of ZN-3-56 (13) forming electrostatic interactions with Glu167 and Arg166. E) Summary of structure activity relationships of selected compounds designed to engage with Sites I-V of PLpro (Table 1 details potency and affinity for the selected compounds and full SAR is provided in Tables S1–S5).
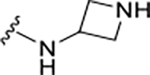
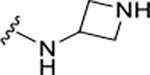
A detailed analysis of PLpro residues interacting with GRL0617 and those providing recognition for ubiquitin/ISG15 substrates revealed four potential binding sites (Sites I-IV) (Figure 1A). We hypothesized that engaging these sites could increase inhibitor affinity and potency. In addition, we identified a potential binding site formed by closure of the BL2 loop and not exploited by ubiquitin substrates nor known inhibitors (Site V) (Figure 1A,B). Engaging Site I appeared straightforward by extending from the benzamide group towards Glu167. This residue forms electrostatic contacts with Arg72 of ubiquitin in the Ub:PLpro SARS-CoV co-crystal structure (PDB: 4MM3) (Figure 1A,B).14 We envisioned that a basic amine appended to the aniline group would capture this interaction to improve binding affinity. A library of 16 compounds was synthesized to identify suitable basic side chains (Figure 1E Pane-I). The azetidine-substituted ZN-2-184 (5) yielded a two-fold improvement in affinity relative to GRL0617, consistent with engagement of Glu167 (Figure 1D). The increase in affinity was accompanied by improved potency for PLpro inhibition (Figure 2; Table 1).
Figure 2. PLpro inhibition and binding affinity.
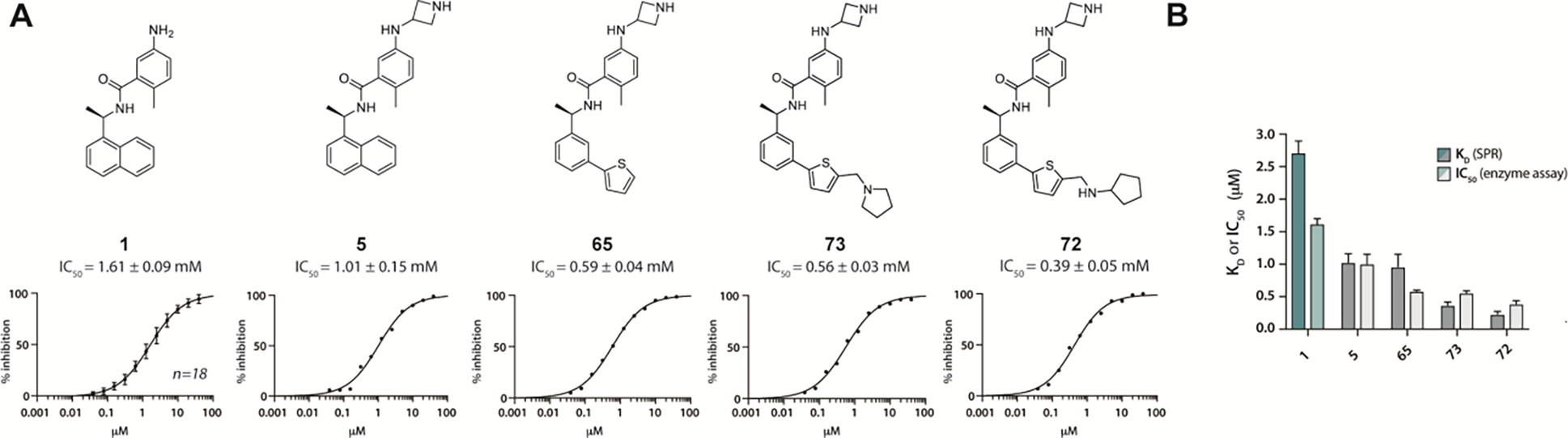
A) Chemical structures and dose response of the most potent PLpro inhibitors in enzymatic assays: GRL0617 (1), ZN-2-184 (5), ZN-3-80 (65), XR8-24 (73), XR8-23 (72). B) Comparison of KD measured by SPR with IC50 measured in enzyme inhibition assay. Also see Figure S1.
Table 1.
Structures, potency, and affinity for compounds explicitly discussed in the text.
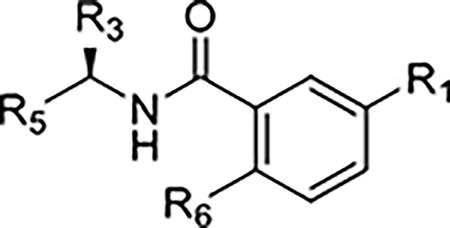
| |||||||
|---|---|---|---|---|---|---|---|
| Compound Code | No. | R1 | R3 | R5 | R6 | Enzyme inhibition IC50 (μM) | SPR binding assays KD (μM) |
| GRL0167 | 1 |
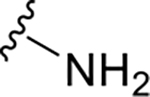
|
CH3 |
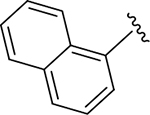
|
CH3 | 1.61 | 2.70 |
| ZN-2-182 | 3 |
|
CH3 |
|
CH3 | 5.5 | 32.6 |
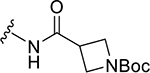
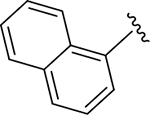
| ZN-2-184 | 5 |
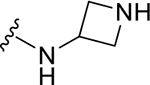
|
CH3 |
|
CH3 | 1.01 | 1.03 |
| ZN-2-185 | 6 |
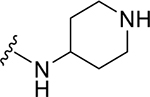
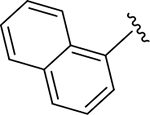
|
CH3 |
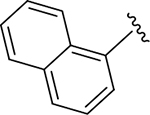
|
CH3 | 0.6 | 1.8 |
| ZN-2-186 | 7 |
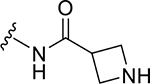
|
CH3 |
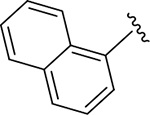
|
CH3 | 1.2 | 3.1 |
| ZN-2-188-2 | 11 |
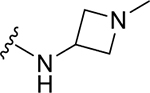
|
CH3 |
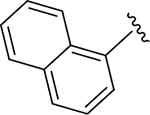
|
CH3 | 4.3 | 3.4 |
| ZN-2-197 | 12 |
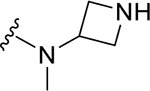
|
CH3 |
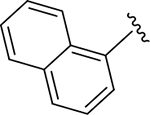
|
CH3 | 2.4 | 2.8 |
| ZN-3-56 | 13 |
|
CH3 |
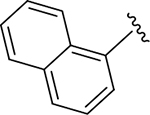
|
CH3 | 3.9 | 26.5 |
| DY2-144 | 14 |
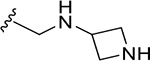
|
CH3 |
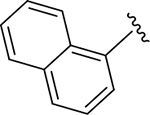
|
CH3 | 1.3 | 6.0 |
| ZN-2-190 | 19 |
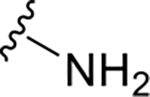
|
CH3 |
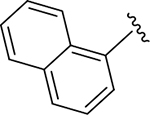
|
F | >>100 | >1000 |
| ZN-2-192 | 20 |
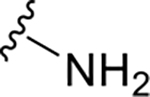
|
CH3 |
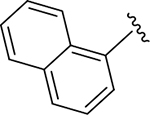
|
Cl | 4.8 | 2.0 |
| ZN-2-193 | 21 |
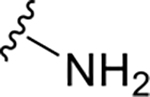
|
CH3 |
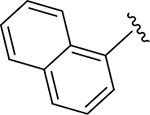
|
CF3 | >10 | 454 |
| ZN-3-3 | 22 |
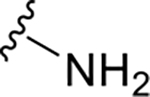
|
CH3 |
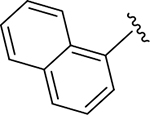
|
|
>10 | 54.6 |
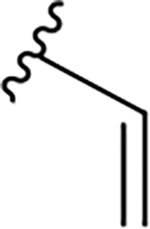
| DY2-109 | 23 |
|
CH3 |
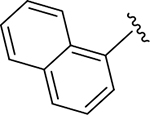
|
Br | 21 | 83.0 |
| DY2-97 | 37 |
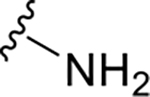
|
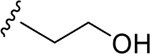
|
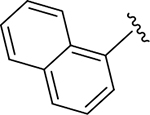
|
CH3 | ~100 | 721.5 |
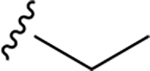
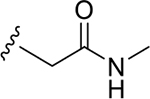
| ZN-3-61 | 38 |
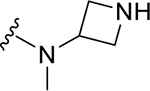
|
|
|
CH3 | >>10 | 281.5 |
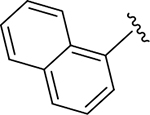
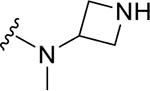
| DY2-116 | 43 |
|
|
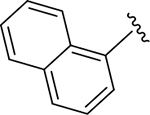
|
CH3 | NI | >1000 |
| ZN-3-45 | 52 |
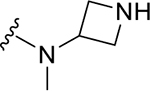
|
(R/S)-CH3 |
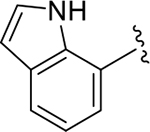
|
CH3 | 5.7 | 18.8 |
| ZN-3-79 | 59 |
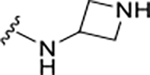
|
CH3 |
|
CH3 | 1.9 | 8.4 |
| DY-2-153 | 60 |
|
CH3 |
|
CH3 | 1.8 | 3.9 |
| ZN-3-36 | 61 |
|
CH3 |
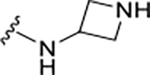
|
CH3 | 56 | 19.6 |
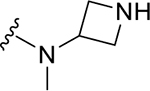
| DY2-139 | 63 |
|
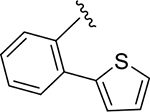
CH3 |
|
CH3 | >40 | NA |
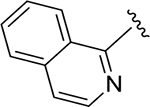
| ZN-3-80 | 65 |
|
CH3 |
|
CH3 | 0.59 | 0.963 |
| XR8-8 | 66 |
|
CH3 |
|
CH3 | 1.3 | 1.39 |
| XR8-9 | 67 |
|
CH3 |
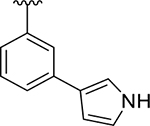
|
CH3 | 1.8 | 2.89 |
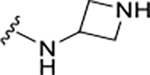
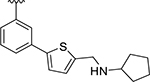
| XR8-23 | 72 |
|
CH3 |
|
CH3 | 0.39 | 0.235 |
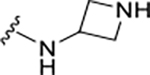
| XR8-24 | 73 |
|
CH3 |
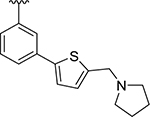
|
CH3 | 0.56 | 0.372 |
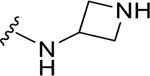
| XR8-69 | 89 |
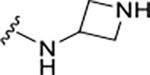
|
CH3 |
|
CH3 | 0.37 | NA |
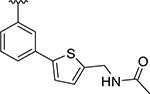
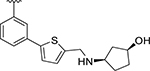
| XR8-83 | 92 |
|
CH3 |
|
CH3 | 0.21 | 0.337 |
| XR8-89 | 94 |
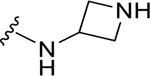
|
CH3 |
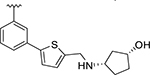
|
CH3 | 0.113 | 0.113 |
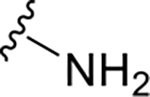
| XR8-106 | 100 |
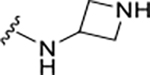
|
NH2 |
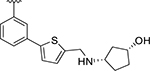
|
CH3 | 1.4 | 1.88 |
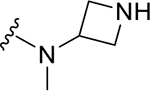
Site II is positioned adjacent to the charged side chains of Arg166 and Asp164 (Figure 1A). Arg166 forms an electrostatic interaction with Asp164 via its charged guanidino group, leaving the other guanidine nitrogens available for hydrogen bonding interactions. In the Ub:PLpro complex (PDB 4MM3), Arg166 and Asp164 are captured by hydrogen bonding with Gln49 and Arg72 of ubiquitin, respectively (Figure 1B). Engaging Site II in tandem with Site I is more complicated than Site I alone, because of the risk in disruption of the benzamide hydrogen bonding network. To engage Arg166 at Site II, modifications were made to: 1) α-methyl; 2) 2-napthalene; and 3) aniline nitrogen. The simplest approaches to extend from the α-methyl position led to loss of potency: even a minor ethyl modification led to a significant decrease in potency (ZN3-61) and further modification resulted in almost completely inactive compounds such as DY2-97 (37) and DY2-116 (43) (Figure 1E pane-II; Tables 1, S3). As an example of the second strategy to target Site II, the 2-isoquinoline ZN3-36 (61) (Figure 1E pane-II; Table 1) was designed to engage with a structurally conserved water molecule between Asp164 and Arg166 (Figure 1A); however, ZN3-36 (61) (IC50 = 56 μM) and all related approaches led to significant loss of activity (Tables 1, S4). We propose that the dihedral angle between the plane of the naphthalene ring and amide of GRL-0617 (81.7°, PDB: 7JRN) is important to maintain the benzamide hydrogen bonding network. In ZN3-36 (61) the comparable angle is 27.9° (low energy conformation from B3LYP/6-31G* calculation with a polarizable continuum model for aqueous solvation) (Figure S2). The third approach, to extend from the aniline towards Site II (Figure 1E Pane-III) resulted in only one compound (ZN-3-56 (13)) with improved potency over GRL0617. The proposed binding model of ZN-3-56 (13) predicts electrostatic interaction with both Arg 166 and Glu167 (Figure 1C). Further exploration of Site II interactions was terminated.
Site III is located at the P3 site of the substrate-binding channel, which is formed by the BL2 loop, helix 5, and neighboring hydrophobic residues Tyr264, Tyr273, and Leu162 (Figure 1A). Small hydrophobic moieties such as a halide or trifluoromethyl were introduced to probe the hydrophobic interaction at this site (Figure 1E Pane-III). Interestingly, small substitutions such as methyl to fluorine at Site III led to a dramatic decrease in potency. Only bromo- and chloro-substituents did not significantly decrease potency. Attempts to make fused-ring indole analogs to replace the aniline also did not lead to any improvement in potency (Table S2).
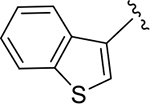
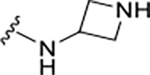
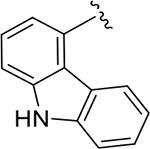
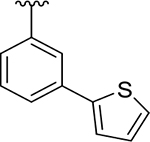
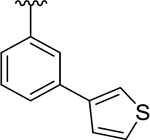
To probe Site IV (Figure 1A), we explored scaffolds to replace the naphthalene of GRL0617. Retaining the essential geometry between the benzamide and naphthalene rings should be possible using heteroaryl or bi-aryl group replacements (Figure S2), and replacement of the naphthalene ring was anticipated to improve metabolic stability.23, 25 Modeling indicated that the preferred torsional geometry between the planes of the amide and the naphthalene rings could be maintained using aryl group replacements (Figure S2). Fused heteroaryls such as benzothiophene, indole, and carbazole with various linkages were prepared and tested (Table S4); however, most modifications led to a loss in activity. Only the 3-benzothiophene (ZN-3-79 (59)) and the carbazole-based (DY2-153 (60)) analogues showed reasonable potency (IC50 = 1.9 μM and 1.8 μM, respectively; Tables 1, S4). However, the biaryl analogues showed significantly improved potency: 2-phenylthiophene (ZN-3-80 (65); IC50 = 0.59 μM) and 3-phenylthiophene (XR8-8; (66); IC50 = 1.3 μM) (Figure 2A; Tables 1, S5). ZN-3-80 (65), the most potent analog in this subset, was also found to be more stable than GRL0617 in human liver microsome stability assays (Table S6).
Identification and engagement of the BL2 groove
Examination of available crystal structures identified a potential ligand binding site, coined the “BL2 groove”, positioned at the N-terminal side of the BL2 loop, between strands β8 and β9 and on top of β13 (Site V, Figure 1A). The BL2 groove features hydrophobic residues such as Pro248 and Pro299 and potential hydrogen bonding partners such as the backbone amide of Gly266. We explored derivatization of the 2-phenylthiophene scaffold (ZN-3-80 (65)) to exploit interactions with the BL2 groove. Of 22 derivatives synthesized, nine showed significantly improved potency (IC50 < 500 nM; Figure 2A; Table S5).
To confirm engagement of the BL2 groove, we co-crystalized potent inhibitors with SARS-CoV-2 PLpro. In contrast to the identical active sites, there are differences in amino acids proximal to the BL2 loop between SARS and SARS-CoV-2 PLpro; therefore, it was important to obtain new co-crystal structures to confirm our binding hypotheses for this specific PLpro. The superimposed structures of SARS-CoV-2 PLpro complexed with XR8-24 (73), XR8-65 (86), XR8-69 (89), and XR8-83 (92) show all inhibitors enforcing the same binding mode with the closed BL2 loop (Figure 3, Supplemental Data 2), confirming our drug design hypotheses.
Figure 3. Superposition of four novel SARS-CoV-2 PLpro:inhibitor crystal structures.
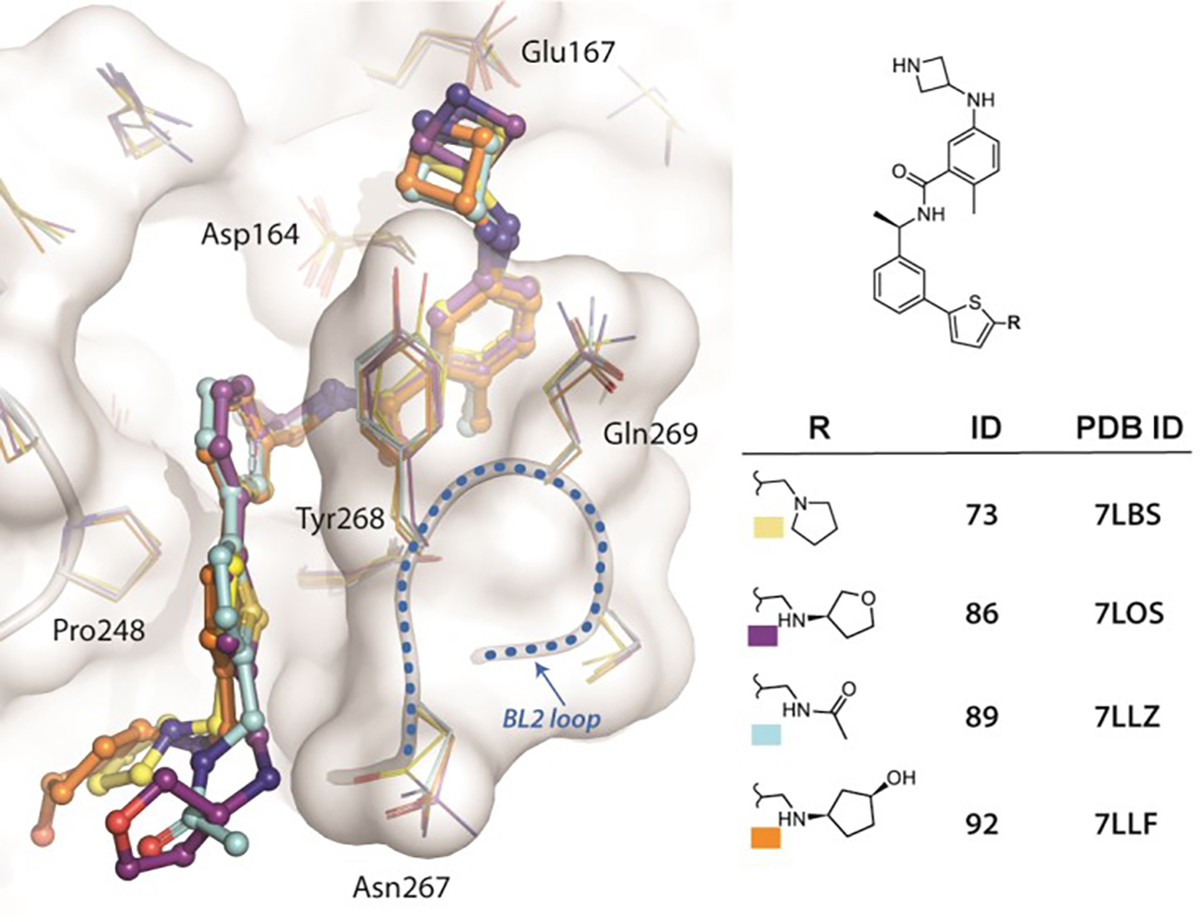
The chemical structures of inhibitors, their IDs, and associated pdb codes are listed at right, with colored boxes corresponding to the coloring used in the structures at left: XR8-24 (73), XR8-65 (86), XR8-69 (89), XR8-83 (92). The statistics for the crystal data processing and refinement, as well as Fo-Fc maps, are included in Supplemental Data 2.
Superposition of XR8-24 (73) with GRL0617, bound to SARS-CoV-2 PLpro, clearly demonstrates that XR8-24 (73) has gained the additional binding interactions that we had targeted (Figure 4A); thus, the azetidine ring extends into Site I to within 3 Å of Glu167, gaining the intended electrostatic stabilization (Figure 4A,B). The benzamide hydrogen-bonding network, involving the mainchain of Gln269 on the BL2 loop, is retained; however, there is a shift in the biaryl ring of the new inhibitors relative to the naphthalene ring of GRL0617 (Figure 4A). This shift places the thiophene ring firmly in the BL2 groove (Site V), where it takes part in van der Waals interactions with residues surrounding the cavity (Pro248, Tyr264, Tyr268; Figure 4A,B).
Figure 4. Structural characterization of SARS-CoV-2 PLpro inhibitors.
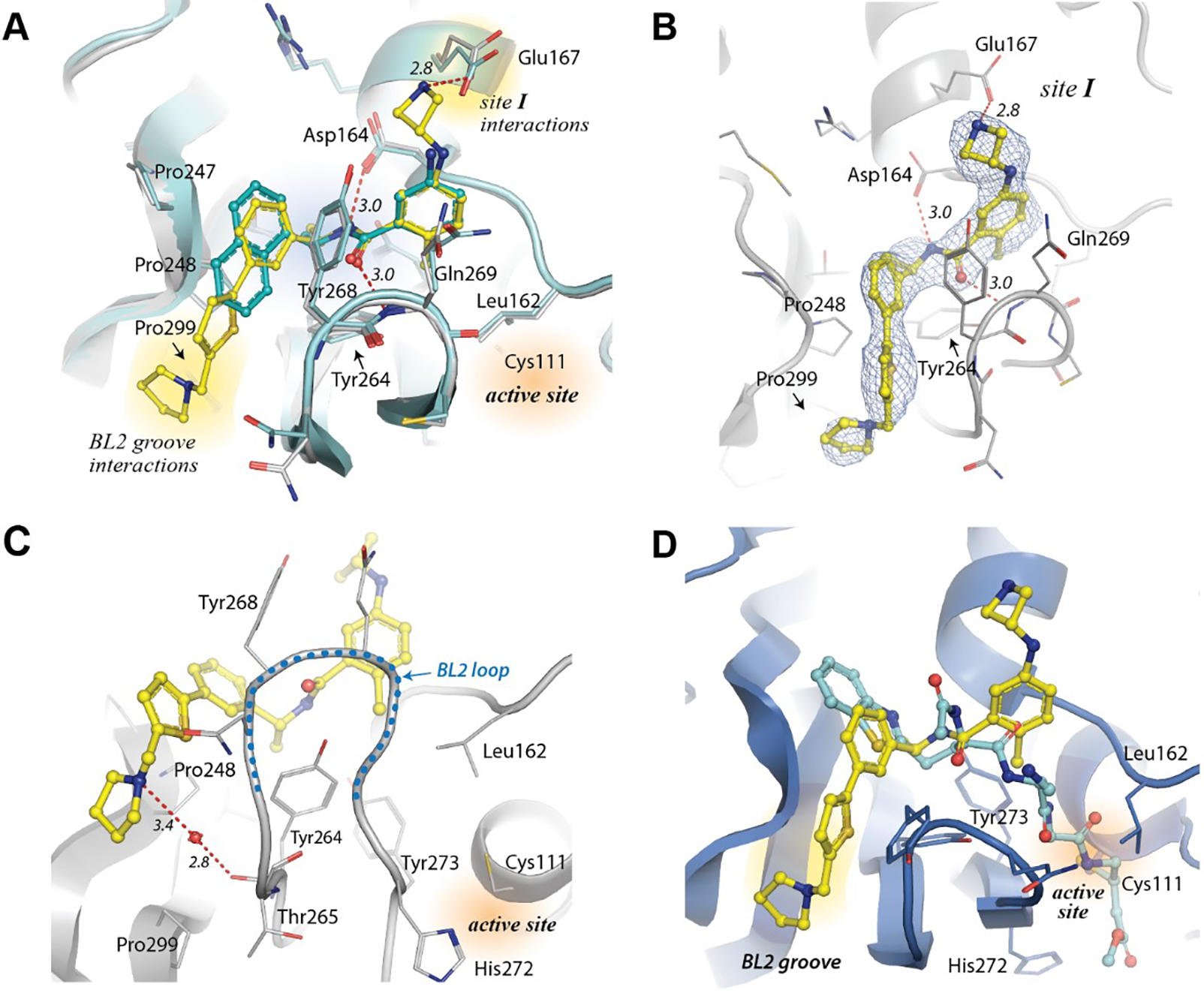
A) XR8-24 (73)-bound PLpro structure (yellow) superimposed with GRL-0617-bound (cyan) PLpro (PDB:7JRN). The extended structure of XR-8-24 (73) allows: 1) an additional electrostatic interaction with Glu167; and 2) occupies the BL2 groove. B) 2Fo-Fc electron density map of XR8-24 (73). The map is shown as blue mesh and is contoured at 1 sigma around the inhibitor (PDB: 7LBS). C) Details of the water-mediated interaction of XR8-24 (73) (yellow) with PLpro. D) Superposition of XR8-24 (73) (yellow) onto PLpro (blue) complexed with a covalent peptide-based inhibitor (cyan), VIR250 (PDB: 6WUU).
The alicyclic “tail” of XR8-24 (73) sits perpendicular to the thiophene and adjacent to the body of the protein near Pro248 and Pro299 (Figure 4). The tail of XR8-24 (73) is well-defined (Figure 4B), with the pyrrolidine ring forming a putative water-mediated hydrogen bond to the mainchain carbonyl oxygen of Tyr264 (Figure 4C), which may account for its superior potency. The co-crystal structures of XR8-65 (86), XR8-69 (89) and XR8-83 (92) show disorder in the “tail”, which may be due this being a solvent-exposed region, or to crystal packing forces (a second symmetry-related monomer is found adjacent to this region). Superposition of the XR8-24(73):PLpro structure with that of a peptide inhibitor, VIR250, covalently bound to the active site cysteine illustrates the spatial relationship of the BL2 groove with the active site in SARS CoV-2 PLpro (Figure 4D). Clearly, the new 2-phenylthiophene non-covalent inhibitors do not access the active site. These inhibitors engage the BL2 groove enforcing a blockade of the active site.
Loop reorganization leads to lower off-rates and binding cooperativity
Binding of host and viral protein substrates is controlled by the flexible β-hairpin BL2 loop, containing an unusual beta-turn formed by Tyr268 and Gln269. Superposition of the XR8-24 co-crystal structure with that of the apo-enzyme (PDB: 7CJD) vividly illustrates the conformational flexibility of the BL2 loop (Figure 5A). Binding of XR8-24 (73) induces closing of the loop, with the repositioning of Gln269 and Tyr268 graphically demonstrating the substantial loop reorganization. Substrate access to the PLpro catalytic site is controlled by the BL2 loop as shown by the XR8-24 (73) co-crystal structure superposed with those of ubiquitin (PDB: 6XAA) and ISG15 (PDB: 6YVA) bound to PLpro (Figure 5B).
Figure 5. PLpro inhibition by loop reorganization and distal blocking of substrate access to the active site.
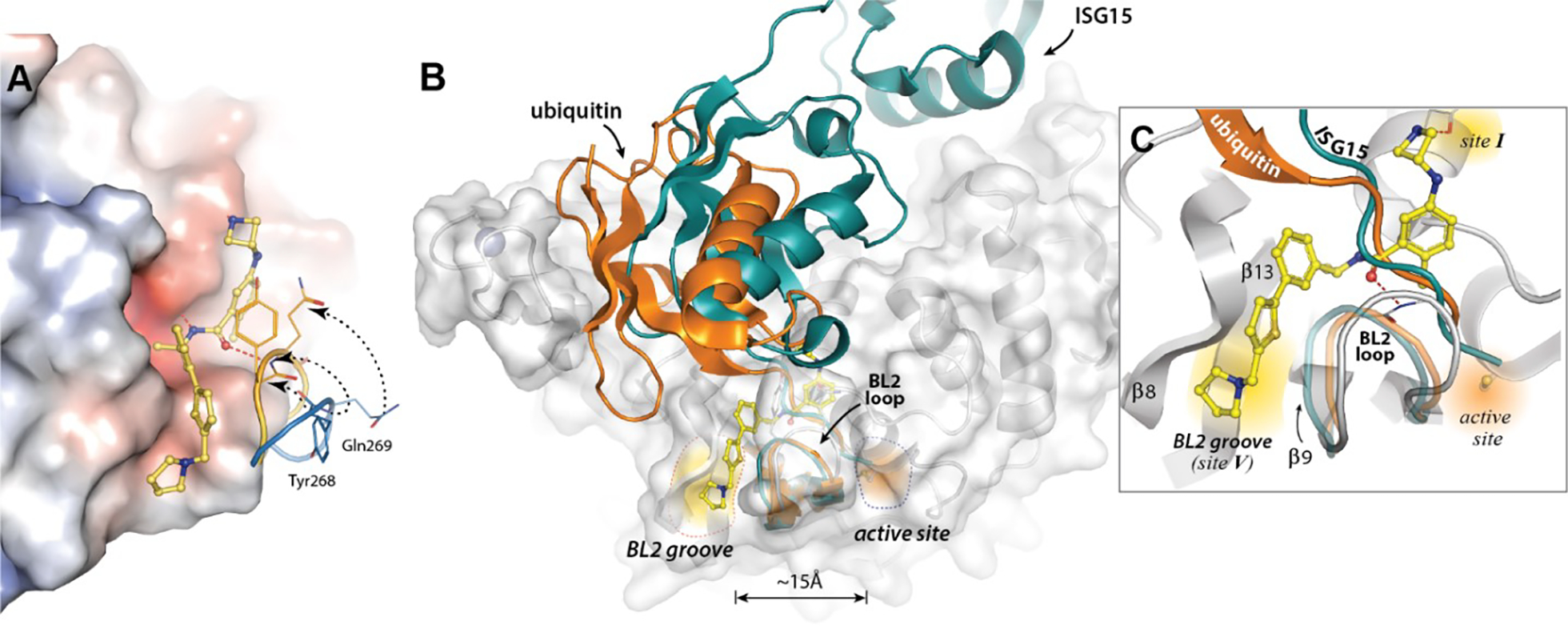
A) An overlay of the XR8-24 (73):SARS CoV-2 PLpro structure with that of the apoenzyme structure (PDB: 7CJD), highlighting the BL2 loop reoragnization as the Gln269 mainchain residue in the BL2 loop is closed to form a hydrogen bond interaction with XR8-24 (73). B) The structure of the XR8-24(73)-bound PLpro structure superimposed with Ub-bound PLpro (PDB: 6XAA, orange) and ISG15-bound Plpro structures (PDB: 6YVA, teal). C) XR8-24 (73) extends into a novel binding site, the BL2 groove, which is positioned between the β8 and β9 strands, adjacent to the BL2 loop. The BL2 groove is approximately 15Å from the active site. Binding of XR8-24 (73) blocks the tails of ubiquitin (orange) or ISG15 (teal) from accessing the active site channel.
A closer look at the channel by which substrates enter the active site (Figure 5C) emphasizes that both the closed loop and the inhibitor itself occupy the channel used by substrates to access the active site. It is reasonable to postulate that binding by induced fit couples inhibitor binding to the substantial reorganization of the tertiary structure of the PLpro BL2 loop. Possible sequelae would be variable ligand off-rates and slow ligand dissociation. Intuitively, dissociation rates would be expected to inversely correlate with the number of binding interactions and binding affinity and the correlation between protein conformational reorganization and slow off-rates has previously been noted.34, 35 PLpro association and dissociation rates were measured by SPR (Figure 6A,B). The measured association rates were significantly slower than the 1×109 M−1s−1 diffusion-controlled rate. Moreover, the extended ligands, designed to engage the BL2 groove, showed slower dissociation rates than GRL0617 (Figure 6B). XR8-23 (72) contains a basic amine side chain extending from the thiophene scaffold of ZN-3-80 (65), which resulted in slower off-rates compared to ZN-3-80 (65) and 4-fold slower dissociation rates compared to GRL0617: an inverse correlation between inhibitor potency and off-rate was observed (Figure S3; Table S7).
Figure 6. Association and dissociation rates and binding cooperativity.
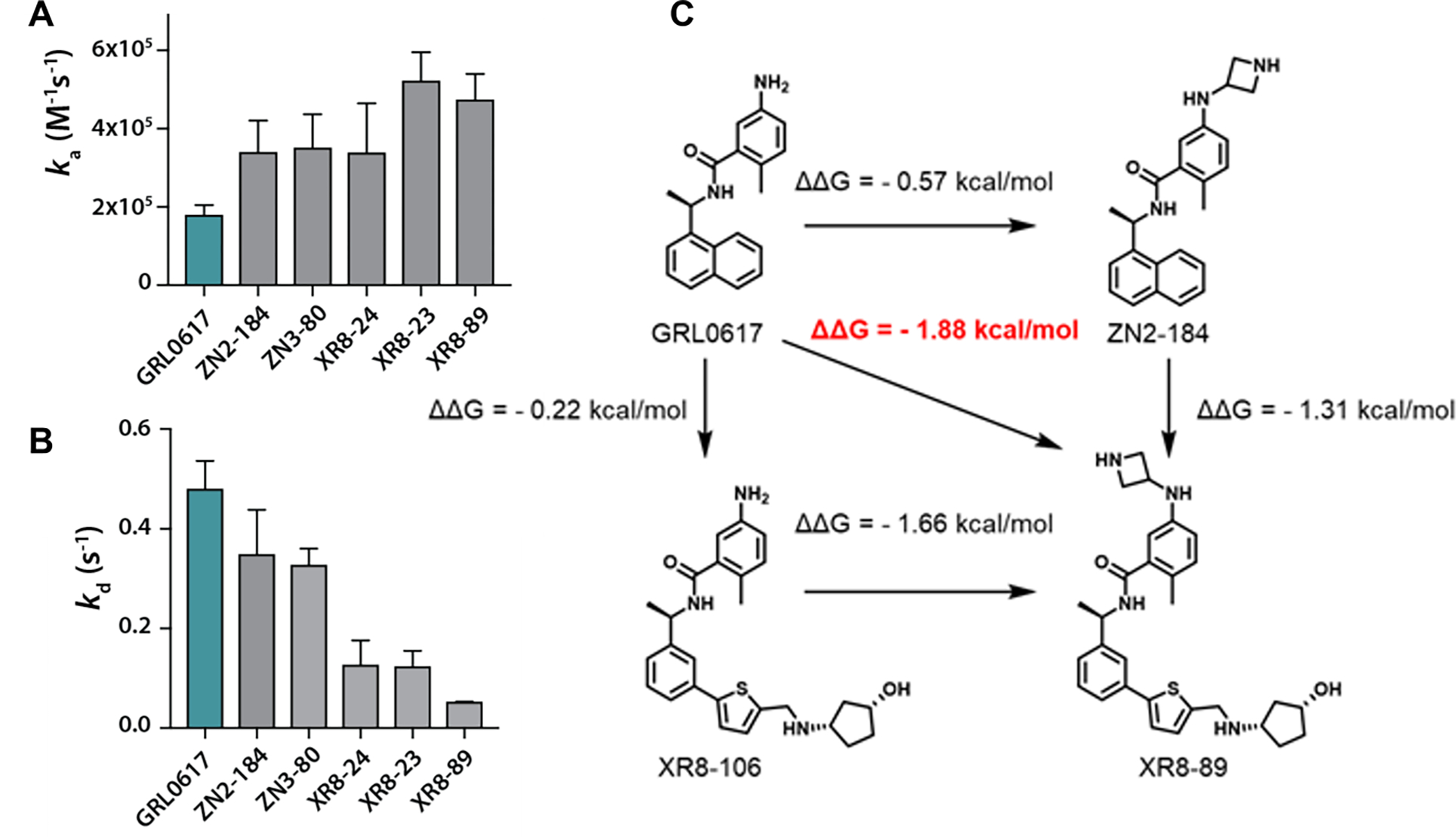
SPR was used to measure (A) association rates and (B) dissociation rates for PLpro inhibitors: GRL0617 (1), ZN-2-184 (5), ZN-3-80 (65), XR8-24 (73), XR8-23 (72), XR8-89 (94). (C) Binding affinity (KD determined by SPR) was used to demonstrate potential cooperativity by engaging multiple weak interactions across multiple binding sites.
PLpro Site I is engaged by ubiquitin and ISG substrates, as introduced above; whereas, Site V (the BL2 groove) is 15 Å from the active site and not a known binding site for any substrates or inhibitors (Figures 4, 5). Our novel 2-phenylthiophene inhibitors engage Sites I, IV, and V, in addition to the benzamide hydrogen bonding network engaged by GRL0617. Using the binding affinity data from SPR (Table 1, S7): engaging Site I with the azetedine ring gains 0.57 kcal/mol stabilization (ZN2-184 (5)); while enaging with the BL2 groove increases affinity by 0.22 kcal/mol (XR8-106 (100)) (Figure 6C). Engaging both sites (XR8-89 (94)) leads to an additional stabilization of 0.8 kcal/mol, which represents positive cooperativity due to binding at multiple sites in and around the BL2 loop, including the novel BL2 groove.
Inhibition of DUB activity is specific for PLpro
PLpro disrupts the host innate immune response by cleaving the isopeptide bond that ligates Ub and ISG-15 to lysine sidechains of host proteins.15, 17, 36–40 Using Ub-AMC and ISG-15-AMC as substrates, we observed complete ablation of the DUB activity of PLpro by 2-phenylthiophene inhibitors (30 μM) (Figure 7). Comparing with GRL0617, at the approximate IC50 concentration, all novel inhibitors gave greater inhibition of DUB activity. Our novel PLpro inhibitors effectively block deubiquitination and deISGylation catalyzed by SARS-CoV-2 PLpro-mediated in biochemical assays; which is compatible with the structural biology data on these inhibitors (Figures 4, 5). We tested our most potent novel PLpro inhibitors against the closest human, structural homolog of PLpro, the catalytic domain of human USP7: no inhibition of USP7-catalyzed Ub-AMC hydrolysis was observed at ≤ 30 μM (Figure 7).
Figure 7. Inhibition PLpro DUB activity.
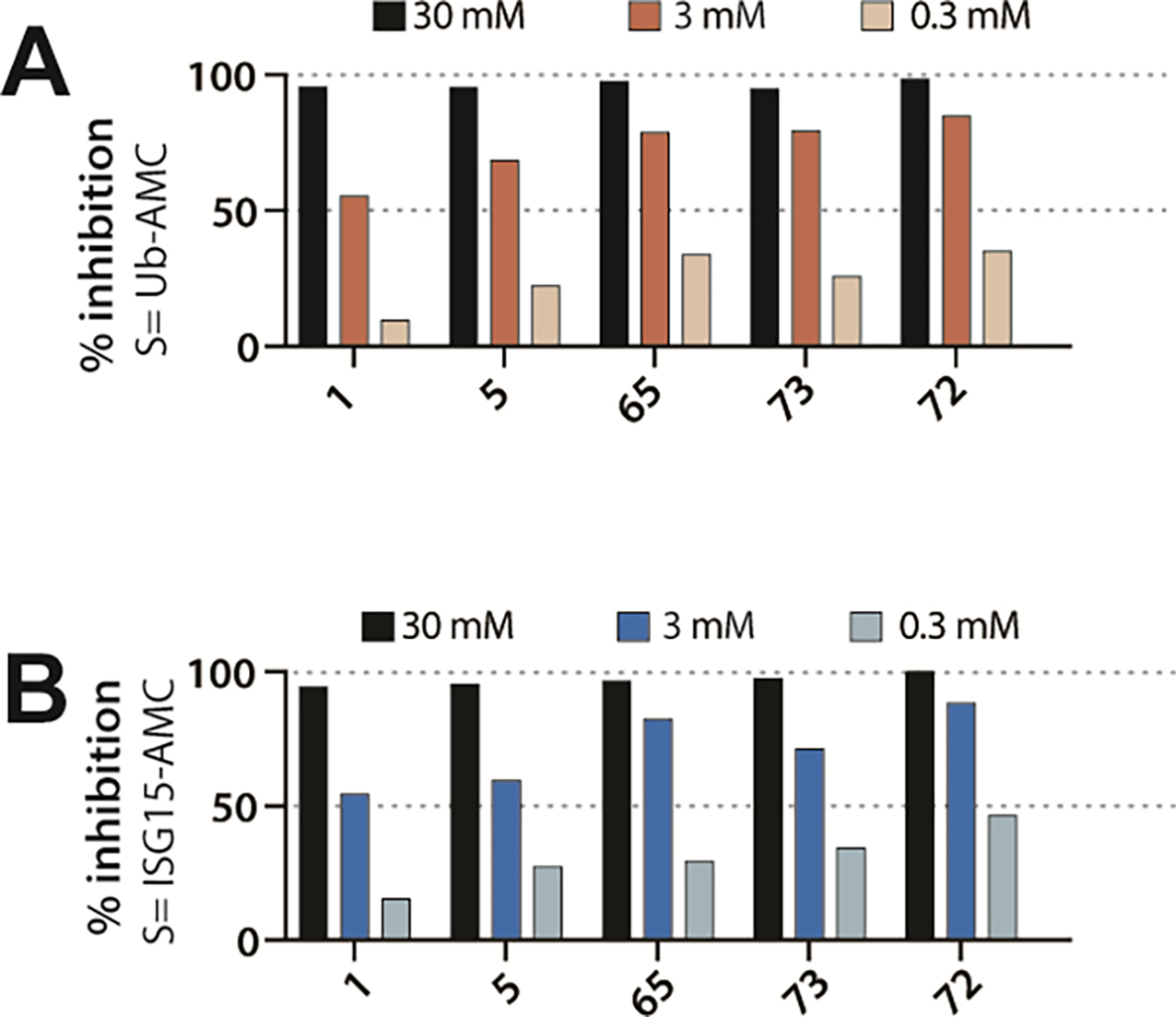
Inhibition of (A) deubiquitinating and (B) de-ISGylating activities of Plpro inhibitors: GRL0617 (1), ZN-2-184 (5), ZN-3-80 (65), XR8-24 (73), XR8-23 (72).
Preclinical Efficay and Bioavailability
Two of the most potent 2-phenylthiophene PLpro inhibitors, XR8-23 (72) and XR8-24 (73), were tested in human lung epithelial A549 cells, stably overexpressing the human ACE2 receptor. This represents a preclinical model relevant to the severe, acute respiratory tract infection that is a feature of COVID-19. Although inhibitor 94 showed high binding affinity and low dissociation rate, this compound was not effective in preliminary antiviral studies, compared to 72 and 73.41 Viral RNA was measured by RT-qPCR as a measure of replication of infectious SARS-CoV-2 USA/WA1/2020. Inhibitor cytotoxicity was not observed under the assay conditions at < 30 μM, although at 100 μM, 72 and 73 were more cytotoxic than 1 (Figure S4). The antiviral activity of XR8-23 (72) and XR8-24 (73) in this model system was compared to GRL0617 and the clinical SARS-CoV-2 antiviral agent, remdesivir (10 μM) as a positive control (Figure 8). GRL0617 was ineffective at preventing viral replication in A549 cells (IC50 > 20 μM). By unpaired nonparametric t-test: 1) the effect of treatment with XR8-23 (72) and XR8-24 (73) (1.3 μM) was significantly different from vehicle control; and 2) the effect of treatment with XR8-24 (73) (20 μM) was not significantly different from that of remdesivir (10 μM).
Figure 8. Improved PLpro inhibitors show potent antiviral efficacy.
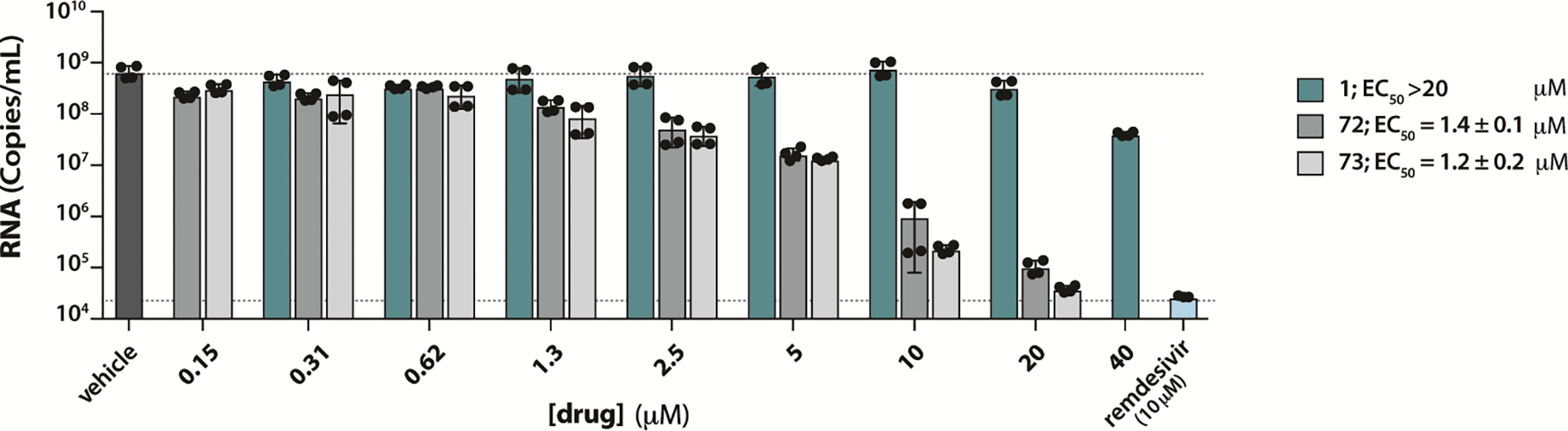
To measure reduction in virus yield, A549-hACE2 cells were infected with MOI = 0.01 of SARS-CoV-2 cultured in Vero E6 cells with and without various concentrations GRL0617, XR8-23 (72), or XR8-24 (73) (cytotoxicity was not observed under the assay conditions at < 50 μM for XR8-24 (72) and < 10 μM for XR8-23 (73). After 48 hours, supernatants were harvested, and RNA was isolated and quantified by RT-qPCR. The data show mean ± S.D.
To estimate the ability of these inhibitors to reach plasma concentrations compatible with the observed antiviral potency, XR8-23 (72) and XR8-24 (73) were administered to male C57BL/6 mice (50 mg/kg i.p.). The Cmax measurements (6,130 ng/mL for XR8-23 (72); 6,403 ng/mL for XR8-24 (73); Figure S5) correspond to 12–13 μM plasma concentrations. Although these inhibitors have not been optimized for in vivo antiviral activity, the superior potency to all other PLpro inhibitors in both biochemical enzyme assays and cell-based antiviral assays, combined with early indications if in vivo bioavailability are supportive of these inhibitors representing leads for further development.
Chemistry
The convergent synthesis of PLpro inhibitors was based mainly on reductive amination, amine coupling, and Suzuki–Miyaura cross-coupIling reactions. A general synthesis is summarized in Scheme 1. The commercially available substituted 5-amino-2-methylbenzoic acids were coupled with (R)-1-(1-naphthyl)ethylamine through HATU condensation reactions to afford the compounds GRL0167, 3, 5-7, 19-23, using reductive amination or amine coupling with Boc protected cyclic amines and then deprotection with HCl solutions.
Scheme 1.
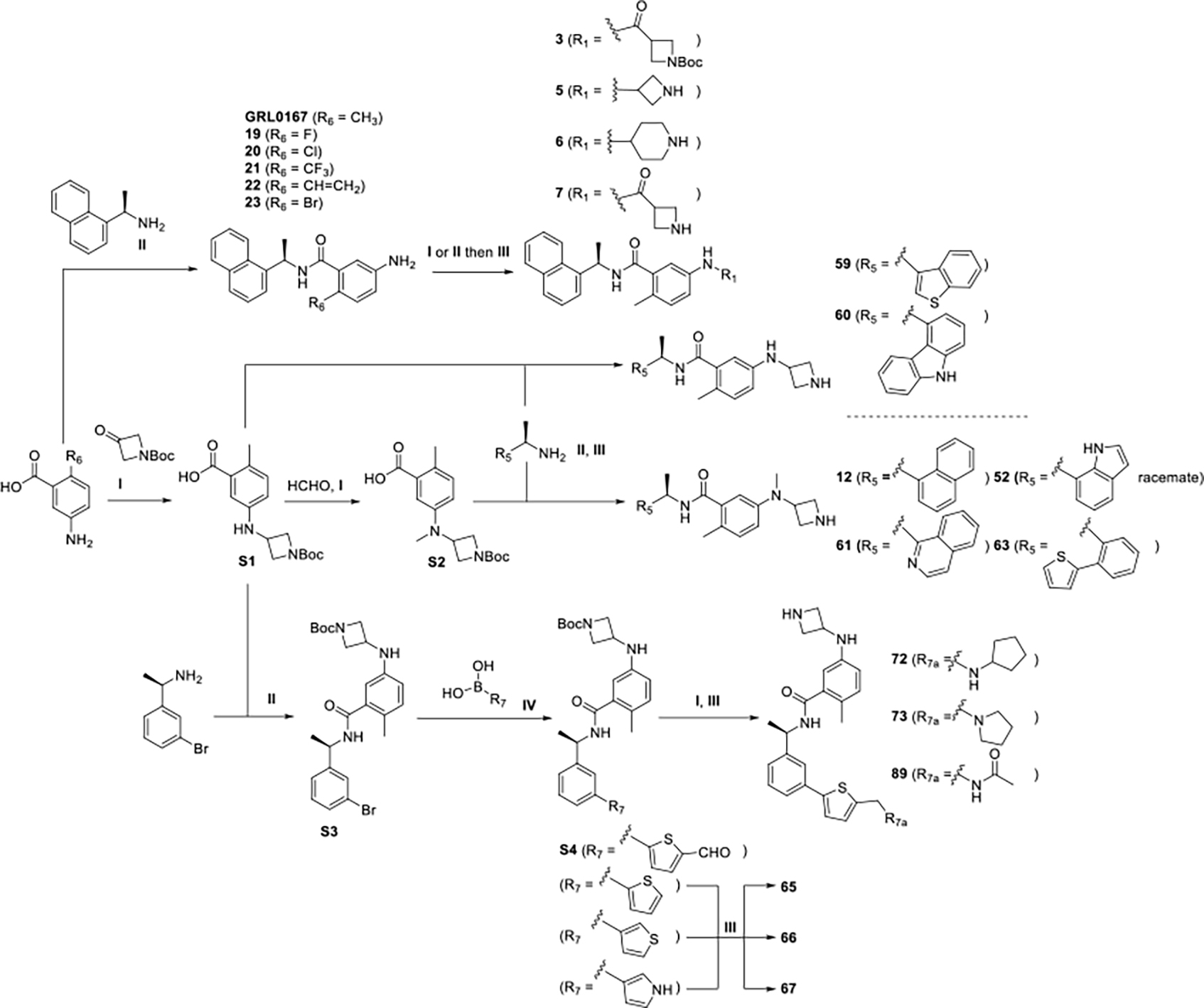
Reagents and conditions: I. (Amines, aldehydes or ketones), HOAc, NaBH3CN, MeOH; II. (Amines or carboxylic acids), HATU, DMAP, DMF, rt; III. HCl (4M in dioxane), DCM; IV. XPhos Pd G2, K3PO4, DMF/EtOH/H2O, 95 °C.
For synthesis of the azetidine derivatives, synthons S1, S2, and S3 were key in preparation of the common synthons with structure S4 (Scheme 1). We synthesized the S1 via 5-amino-2-methylbenzoic acid and 1-Boc-3-azetidinone, attaching a methyl group using formaldehyde solution to afford S2. After coupling the S1 or S2 synthons with benzylamines in the presence of HATU and deprotection, we readily generated the coumpounds 52, 59, 60, 61, 63. Synthon S4 and the meta-substituted intermediates were prepared through a sequence of reactions starting from the condensation of S1 and (1R)-1-(3-bromophenyl)ethanamine to give S3, followed by Suzuki–Miyaura coupling with 3-thienylboronic acid using XPhos Pd G2 as the catalyst. Deprotection of the meta-substituted intermediates gave 65-67. The aldehyde synthon S4 was readily reacted with amines through a reductive amination, followed by Boc deprotection using HCl (4M in dioxane) to afford compounds 72, 73, 89 with good yield.
For amines not available commercially, the chiral benzylamine could be synthesized through Ellman’s sulfinamide; for example as shown in Scheme 2. The substituted acetophenone was reacted with (R)-2-methyl-2-propanesulfinamide using the Lewis acid Ti(OEt)4, followed by NaBH4 reduction to yield the two diastereomeric synthons, S12 and S13, with the R configuration as the major product. Removing the sulfinyl group from the diastereomers after separation with concentrated HCl gave the amine synthons S14 and S15, respectively. After amine coupling and deprotection as described above, the desired compounds was obtained.
Scheme 2.
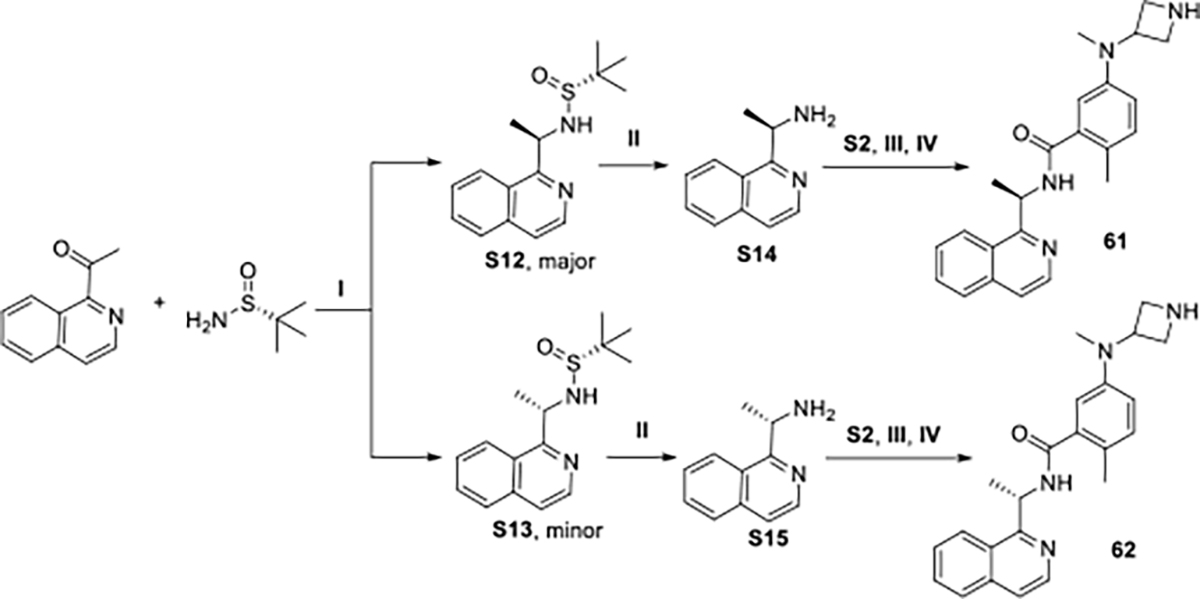
Reagents and conditions: I.Ti(OEt)4, NaBH4, THF, −78 °C to rt; II. HCl (conc. aq.), dioxane; III. HATU, DMAP, DMF, rt; IV. HCl (4M in dioxane), DCM
DISCUSSION & CONCLUSIONS
The essential SARS-CoV-2 cysteine proteases, 3CLpro and PLpro, are excellent therapeutic targets for addressing the COVID-19 pandemic and future outbreaks of both SARS-CoV-2 and novel coronaviruses. Inhibition of cysteine proteases by covalent modification of the active site cysteine, is the most common approach to drugging these enzyme targets and has been successful for 3Clpro inhibitors in clinical trials.42 This strategy is ineffective for PLpro owing to the featureless P1 and P2 sites (Gly-Gly recognition). 26 The known non-covalent SARS-CoV PLpro inhibitor, GRL0617, lacks sufficent potency for development as an antiviral agent; however, the co-crystal of GRL0617 with SARS-CoV Plpro, resolved by Ratia et al., provided an excellent platform for structure-based design.22 The benzamide of GRL0617 was essential for engaging the hydrogen bonding network with residues of the BL2 loop; thus, providing our starting scaffold.
Design and optimization of improved PLpro inhibitors was founded upon engagement of additional binding sites beyond those utilized by GRL0617. We anticipated that engaging additional binding interactions would lead to: i) increased inhibitor potency; ii) positive binding cooperativity; and ii) slow off-rates caused by the induced fit binding and the structural reorganization of the BL2 loop required for ligand dissociation. Five putative binding sites were identified primarily by studying interactions that contribute to binding of the PLpro substrates Ub and UbL; in addition to an enitirely novel “BL2 groove“ that is not involved in recognition of any known substrate or inhibitor and sits 15 Å from the active site. Our hypothesis was borne out by a structure-based drug design campaign, synthesizing almost 100 compounds, which yielded a series of novel 2-phenylthiophene Plpro inhibitors with potency and affinity improved tenfold over GRL0617 and with significantly slower rates of dissociation. Binding affinity measurements by SPR indicated positive cooperativity resulting from engagement of the BL2 groove and other binding sites. The BL2 groove is a new binding site identified in the search of novel binding interactions. New co-crystal structures with SARS-CoV-2 PLpro validated our design hypotheses.
SARS-CoV-2 infection is characterized by a strong dysregulation of the innate immune and the type I interferon (IFN-I) responses.43 The viral protein, PLpro, represents an excellent therapeutic target, because it acts as a Swiss Army Knife not only essential for replication as a viral peptidase, but also acting as a DUB/deISGylase that plays important roles: i) in mediating viral replication via processing of the viral polyprotein; and ii) in reversing host-mediated post-translational modifications in response to viral infection via its actions as a DUB. The novel 2-phenylthiophene PLpro inhibitors ablated DUB/deISGylase activity without inhibition of human DUBs. The PLpro inhibitors reported herein may be used as chemical probes to understand the PLpro-mediated disruption of both host immune response and autophagy that may contribute to infection, progression, “long-COVID”, and potential genetic bias.44, 45
Although, these inhibitors were not optimized for in vivo activity, measured plasma levels were above 10 μM, almost tenfold above the observed and antiviral potency in human lung epithelial cells (1.2–1.4 μM). In these cells, the response to XR8-24 (73) (20 μM) was not significantly different from the FDA-approved RdRp inhibitor remdesivir (20 μM). Combination therapy with remdesivir, or 3CLPro inhibitors such as PF-00835231 (in Phase I/II clinical trials) is an attractive strategy. Targeting multiple viral proteins in the replication process is a proven antiviral strategy in the clinic to treat viral infections, while reducing the chance of resistance. Genotyping of SARS-CoV-2 virus variants circulating worldwide has identified multiple recurrent non-synonymous mutations in the receptor-binding domain (RBD) of the spike protein; but, no variants of interest have been identified with mutations in PLpro.
In summary, the absence of druggable binding pockets in the Plpro active site, the induced fit mechanism of BL2 loop closure, and the need to engage multiple weak interactions for potent inhibition accounts for the very low hit rate in screening for Plpro inhibitors. Recognizing these features of PLpro, we designed novel, non-covalent PLpro inhibitors that in biochemical assays exhibited superior nanomolar potency and inhibited PLpro DUB activity. The design strategy exploiting binding cooperativity of multiple shallow binding sites was validated by new PLpro co-crystal structures. Novel, potent PLpro inhibitors such as XR8-23 (72) and XR8-24 (73) are development leads and to our knowledge are the most potent PLpro inhibitors reported, with demonstrated efficacy in blocking infection of human cells by SARS-CoV-2 PLpro. Further optimization and testing against the highly transmissible Delta variant are eagerly anticipated.
EXPERIMENTAL SECTION
Chemical synthesis.
Detailed methods are provided in Supporting Information, including characterization and purity. Unless otherwise specified, reactions were performed under an inert atmosphere of argon and monitored by thin-layer chromatography (TLC) and/or LCMS. All reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, Fisher Scientific, Ambeed, Combi-Blocks, Enamine) and used as provided. Synthetic intermediates were purified using a CombiFlash chromatography system on 230–400 mesh silica gel or Shimadzu prep-HPLC system. 1H and 13C NMR spectra were obtained using Bruker DPX-400 or AVANCE-400 spectrometer at 400 and 100 MHz, respectively. NMR chemical shifts were described in δ (ppm) using residual solvent peaks as standard. High resolution mass spectral data were measured in-house using a Shimadzu IT-TOF LC/MS for all final compounds. Optical rotations were measured with a Perkin-Elmer 241 polarimeter operating on the mercury lamp line (546 nm), using a 100 mm pathlength cell. All compounds submitted for biochemical and biological testing were confirmed to be ≥ 95% pure by analytical HPLC.
SARS-CoV-2 PLpro expression and purification:
pET11a vector containing SARS-CoV-2 PLpro protein (pp1ab aa 1564–1878) with N-terminal, TEV-cleavable His-tag was transformed into BL21(DE3) cells and maintained in media containing 100 ug/mL carbenicillin. Protein expression was induced using an auto-induction protocol modified from Studier et al.46 Briefly, 1 mL day cultures were used to inoculate a 2L flask of 500 mL of Super LB containing 100 ug/mL carbenicillin. Cells were grown for 24h at 25°C and then harvested by centrifugation. All steps of SARS-CoV2 PLpro purification were performed at 4°C. Protein yield at each step was monitored by Bradford assay using BSA as a standard. Frozen cells pellets were lysed by sonication in Buffer A (50 mM HEPES, pH 8, 0.5 M NaCl) containing 10 ug/mL lysozyme. The lysate was clarified by centrifugation and loaded onto a 2-mL HiTrap Talon crude column equilibrated with Buffer A. Bound His6-PLpro was eluted with a linear gradient of 0–150 mM imidazole in Buffer A, and fractions containing His6-PLpro were pooled and exchanged into cleavage buffer (20 mM Tris-HCl pH 8.5, 5 mM DTT, 0.5 mM EDTA, 5% glycerol). A 1:100 molar ratio of TEV protease to PLpro was incubated at 4°C overnight to cleave the His6-tag. To remove the tag and TEV protease, the reaction was loaded onto a UNO-Q column equilibrated with 20 mM Tris HCl, pH 8.5, 3 mM DTT. Cleaved PLpro eluted first in a gradient from 0–150 mM NaCl over 20 column volumes. Fractions containing cleaved PLpro were pooled and concentrated to 12 mg/mL, frozen in liquid nitrogen, and stored at −80 °C.
PLpro primary assay:
The PLpro primary assay, which measures protease activity with the short peptide substrate Z-RLRGG-AMC (Bachem), was performed in black, flat-bottom 384-well plates containing a final reaction volume of 50 μL. The assays were assembled at room temperature as follows: 40 μL of 50 nM PLpro in Buffer B (50 mM HEPES, pH 7.5, 0.1 mg/mL BSA, 0.01% Triton-X 100, and 5 mM DTT) was dispensed into wells containing 0.1–1 μL of inhibitor in DMSO or appropriate controls. The enzyme was incubated with inhibitor for 10 min prior to substrate addition. Reactions were initiated with 10 μL of 62.5 μM RLRGG-AMC in Buffer B. Plates were shaken vigorously for 30 s, and fluorescence from the release of AMC from peptide was monitored continuously for 15 min on a Tecan Infinite M200 Pro plate reader (λexcitation=360 nm; λemission=460 nm). Slopes from the linear portions of each progress curve were recorded and normalized to plate-based controls. Positive control wells, representing 100% inhibition, included 10 μM GRL0617; negative control wells, representing 0% inhibition, included vehicle.
The selectivity of the most potent inhibitors was tested against the human deubiquitinating enzymes USP7 and USP14 (Boston Biochem). Assay conditions were similar to the PLpro primary assay, with the following substitutions: USP7 assays contained 4 nM USP7 and 0.5 uM Ub-AMC (Boston Biochem); USP14 assays contained 1.7 uM USP14, 4 uM Ub-AMC, and the addition of 5% glycerol to Buffer B. PLpro activity with ISG15-AMC and Ub-AMC were assayed in a manner similar to the PLpro primary assay. PLpro and substrate concentrations were modified as follows: 80 nM PLpro was assayed with 0.5 uM Ub-AMC, and 4 nM PLpro was assayed with 0.5 uM ISG15-AMC.
Crystallization:
Crystals of SARS-CoV-2 PLpro complexed with compounds were grown by hanging drop vapor diffusion at 16°C. Prior to crystallization, 12 mg/mL PLpro protein was incubated with 2 mM 73 (or 86, 89, 92) for 30 min on ice. Crystals of the complexes were grown by mixing 1–2 uL of PLpro:inhibitor complex with 2 uL of reservoir solution containing 0.1 M MIB buffer, pH 7.2, 0.2 M (NH4)2SO4, and 24–28% PEG 4000 or 0.1 MIB buffer, pH 6.0–6.8, 0.2 M (NH4)2SO4, 13–16% PEG 3350, and 20% glycerol. Crystals grew overnight from the PEG 4000 conditions and were used to streak seed drops of PLpro:inhibitor equilibrating against the PEG 3350 conditions.
Data collection and structure refinement.
The glycerol present in the crystallization solution was sufficient to cryo-protect crystals, which were flash-cooled in liquid nitrogen. Data were collected at the Life Sciences Collaborative Access Team beamlines 21-ID-D, 21-ID-G, and 21-ID-F at the Advanced Photon Source, Argonne National Laboratory. Data indexing and integration were performed using XDS.47 Because the complex with 73 was strongly anisotropic, with diffraction limits beyond 2.8 Å along the b* and c* directions, but 3.5 Å along the a* direction, ellipsoidal truncation and anisotropic scaling were performed by the UCLA-DOE lab’s Diffraction Anisotropy Server for the 73 complex.48. The server truncated data that fell outside an ellipse centered at the reciprocal lattice origin and having vertices at 1/3.5, 1/2.8, and 1/2.8 Å along a*, b*, and c*, respectively. The data were then anisotropically scaled by the server. These corrections significantly improved electron density maps and refinement statistics. Complexes with 86, 89, and 92 only displayed mild anisotropy and were not corrected in the same manner.
For all complexes, phases were determined by molecular replacement using Molrep49 and a SARS-CoV-2 PLpro: GRL0617 complex (PDB entry: 7JRN) as search model. Rigid body refinement followed by iterative rounds of restrained refinement and model building were performed with CCP4i modules Refmac550 and Coot.51 The coordinates and structure factors have been deposited and released with PDB accession codes 7LBS (73 complex), 7LOS (86 complex), 7LLZ (89 complex), and 7LLF (92 complex).
Secondary binding analysis by Surface Plasmon Resonance (SPR).
The His-tagged SARS-CoV-2 PLpro enzyme was initially prepared in phosphate buffer and diluted to 50 μg/mL with 10 mM sodium acetate (pH 5.5) and immobilized on a CM5 sensor chip by standard amine-coupling with running buffer PBSP (10 mM phosphate, pH 7.4, 2.7 mM KCl, 137 mM NaCl, 0.05 % Tween-20). The CM5 sensor chip surface was first activated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC)/N-hydroxy succinimide (NHS) mixture using a Biacore 8K instrument (Cytiva). SARS-CoV-2 PLpro enzyme was immobilized to flow channels 1 through 4 followed by ethanolamine blocking on the unoccupied surface area, and immobilization levels for all four channels were similar at ~12,000 RU. Each flow channel has its own reference channel, and blank immobilization using EDC/NHS and ethanolamine was done for all reference channels. Compound solutions with a series of increasing concentrations (0.049 – 30 μM at 2.5-fold dilution) were applied to all active and reference channels in SPR binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, and 0.05% Tween-20, 0.5 mM TCEP, and 2% DMSO) at a 30 μL/min flow rate at 25 °C. The data were double referenced with a reference channel and zero concentration (2% DMSO) responses, and reference subtracted sensorgrams were fitted with 1 to 1 Langmuir kinetic model using a Biacore Insight evaluation software, producing two rate constants ( and ) (Figure S1). The equilibrium dissociation constants were determined from two rate constants . For steady-state affinity fittings, response units at each concentration were measured during the equilibration phase, and the values were determined by fitting the data to a single rectangular hyperbolic curve equation, where is the response, is the maximum response and is the compound concentration.
Cell Culture and cytotoxicity:
Human alveolar epithelial cell line (A549) that stably express hACE2 are from BEI Resources (NR-53821). They were grown DMEM supplemented with 10% fetal bovine serum (Gibco), 100 units of penicillin and 100 μg/mL streptomycin (Invitrogen), 1% nonessential amino acids (NEAA) with 100 μg/mL Blasticidin S. HCl for selection. All cells were grown at 37 °C and 5% CO2. Low passage A549 cells (5000 cells/well) were seeded in 96-well plates and incubated at 37 °C and 5% CO2 for 24 hours prior to a 48h treatment. All compounds were dissolved in DMSO and final DMSO concentrations never exceeded 1%. The cytotoxicity of compounds (100 μM to 1 μM, 3-fold dilution) was examined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Cell cytotoxicity data was normalized to DMSO control as 0% cell death.
Pharmacokinetics Studies.
The Animal Care and Use Committee of the University of Illinois at Chicago approved all the procedures involving animals. PK profiling was conducted by Pharmaron Inc. with details provided in Figure S5.
Antiviral activity assay:
A549-hACE2 cells were seeded 1.5×105 cells/well in DMEM complete into 24-well plates (0.5 mL/well) then incubated for 16 hours at 37 °C and 5% CO2. Cells were pretreated with compound for 1-hour prior to infection performed using a clinical isolate of SARS-CoV-2 (SARS-CoV-2, Isolate USA-WA1/2020) from BEI Resources. 2-fold serial dilutions of compound (0.15–20 uM; remdesivir: 10 uM) added to the same volume of SARS-CoV-2 (final MOI = 0.01), the mixture was added to the monolayer cells and incubated for 1 hour at 37 °C and 5% CO2. After, the mixture was removed and replaced with 0.5 mL of infection media and incubated at 37 °C and 5% CO2. After 48 hours, supernatants were harvested and processed for RT- qPCR.
RNA Extraction and RT-qPCR:
250 μL of culture fluids were mixed with 750 μL of TRIzol™ LS Reagent (Thermo Fisher Scientific). RNA was purified following phase separation by chloroform as recommended by the manufacturer. RNA in the aqueous phase was collected and further purified using PureLink RNA Mini Kits (Invitrogen) according to manufacturer’s protocol. Viral RNA was quantified by reverse-transcription quantitative PCR (RT-qPCR) using a 7500 Real-Time PCR System (Applied Biosystems) using TaqMan Fast Virus 1-Step Master Mix chemistry (Applied Bio- systems). SARS-CoV-2 N1 gene RNA was amplified using forward (5’- GACCCCAAAATCAGCGAAAT) and reverse (5’- TCTGGTTACTGCCAGTTGAATCTG) primers and probe (5’- FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1) designed by the United States Centers for Disease Control and Prevention (oligonucleotides produced by IDT, cat# 10006713). RNA copy numbers were determined from a standard curve produced with serial 10-fold dilutions of RNA standard material of the amplicon region from BEI Resources (NR-52358). All data was normalized to virus alone. All error bars represent S.D. from three replicates.
Statistical analysis.
GraphPad Prism 8 software package (GraphPad Software, USA) was used to perform statistical analysis. All data were presented as the mean ± SD unless otherwise noted. One-way analysis of variance (ANOVA) with appropriate post-hoc tests (3+ groups) and Student’s t-test (2 groups) were used to calculate statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Human microsome stability study.
Two separated experiments were performed as follows. a) With NADPH: 10 μL of 20 mg/mL liver microsomes and 40 μL of 10 mM NADPH were added to the incubations. The final concentrations of microsomes and NADPH were 0.5 mg/mL and 1 mM, respectively. b) Without NADPH: 10 μL of 20 mg/mL liver microsomes and 40 μL of ultra-pure H2O were added to the incubations. The final concentration of microsomes was 0.5 mg/mL. The reaction was started with the addition of 4 μL of 200 μM test compound solution or control compound solution at the final concentration of 2 μM and carried out at 37 °C. 4. Aliquots of 50 μL were taken from the reaction solution at 0, 15, 30, 45 and 60 min. The reaction was stopped by the addition of 4 volumes of cold acetonitrile with IS (100 nM alprazolam, 200 nM labetalol, 200 nM caffeine and 2 μM ketoprofen). Samples were centrifuged at 3, 220 g for 40 minutes. Aliquot of 100 μL of the supernatant was mixed with 100 μL of ultra-pure H2O and then used for LC-MS/MS analysis. Peak areas were determined from extracted ion chromatograms. The slope value, k, was determined by linear regression of the natural logarithm of the remaining percentage of the parent drug vs. incubation time curve. The in vitro half-life (in vitro t1/2) was determined from the slope value:
Conversion of the in vitro (min) into the in vitro intrinsic clearance (in vitro , in μL/min/mg protein) was done using the following equation (mean of duplicate determinations):
Conversion of the in vitro (min) into the scale-up unbound intrinsic clearance (Scale-up , in mL/min/kg) was done using the following equation (mean of duplicate determinations, for human the scaling factor is 1254.2):
Supplementary Material
Acknowledgements
This study is supported by NIH grant, UL1TR002003, via UICentre. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank Dr. Andrew Riley and Dr. Michael Flavin for their edits.
Abbreviations
- 3CLpro
3C-like protease
- ACE2
angiotensin-converting enzyme 2
- Arg
arginine
- Asp
aspartic acid
- Boc
tert-butyloxycarbonyl
- BSA
bovine serum albumin
- COVID-19
coronavirus disease 2019
- DTT
dithiothreitol
- DUB
deubiquitinase
- Gln
glutamine
- Glu
Glutamic acid
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HTS
high-throughput screening
- IFN-I
type I interferon
- ISG15
Interferon-stimulated gene 15
- MERS-CoV
middle East respiratory syndrome coronavirus
- Mpro
SARS coronavirus main proteinase
- NADPH
nicotinamide adenine dinucleotide phosphate hydrogen
- nsps
non-structural proteins
- PDB
protein data bank
- PLpro
papain-like protease
- Pro248
proline 248
- RBD
receptor-binding domain
- RT-qPCR
reverse transcription quantitative polymerase chain reaction
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SPR
surface plasmon resonance
- TEV
tobacco etch virus
- TLC
thin-layer chromatography
- LCMS
Liquid chromatography–mass spectrometry
- TMPRSS2
transmembrane protease serine 2
- Tyr
tyrosine
- Ub
ubiquitin
- UbL
ubiquitin-like proteins
- USP7
ubiquitin-specific-processing protease
Footnotes
Conflict of Interest
G.T. is an inventor on patents assigned to the University of Illinois. R.X., G.T., K.R., S.Z., L.R. and L.C. are inventors on the patent application related to PLpro inhibitors.
Supporting Information
Synthesis, characterization, and SAR for all compounds; and further experimental details of biophysical assays, cytotoxicity assays, and pharmacokinetic properties; HPLC of representative compounds.
The X-ray coordinates have been deposited with the Protein Data Bank.
Molecular formula strings.
Reference
- 1.Zhou P; Yang XL; Wang XG; Hu B; Zhang L; Zhang W; Si HR; Zhu Y; Li B; Huang CL; Chen HD; Chen J; Luo Y; Guo H; Jiang RD; Liu MQ; Chen Y; Shen XR; Wang X; Zheng XS; Zhao K; Chen QJ; Deng F; Liu LL; Yan B; Zhan FX; Wang YY; Xiao GF; Shi ZL A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabino EC; Buss LF; Carvalho MPS; Prete CA Jr.; Crispim MAE; Fraiji NA; Pereira RHM; Parag KV; da Silva Peixoto P; Kraemer MUG; Oikawa MK; Salomon T; Cucunuba ZM; Castro MC; de Souza Santos AA; Nascimento VH; Pereira HS; Ferguson NM; Pybus OG; Kucharski A; Busch MP; Dye C; Faria NR Resurgence of covid-19 in manaus, brazil, despite high seroprevalence. Lancet 2021, 397, 452–455, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C; Ginn HM; Dejnirattisai W; Supasa P; Wang B; Tuekprakhon A; Nutalai R; Zhou D; Mentzer AJ; Zhao Y; Duyvesteyn HME; Lopez-Camacho C; Slon-Campos J; Walter TS; Skelly D; Johnson SA; Ritter TG; Mason C; Costa Clemens SA; Gomes Naveca F; Nascimento V; Nascimento F; Fernandes da Costa C; Resende PC; Pauvolid-Correa A; Siqueira MM; Dold C; Temperton N; Dong T; Pollard AJ; Knight JC; Crook D; Lambe T; Clutterbuck E; Bibi S; Flaxman A; Bittaye M; Belij-Rammerstorfer S; Gilbert SC; Malik T; Carroll MW; Klenerman P; Barnes E; Dunachie SJ; Baillie V; Serafin N; Ditse Z; Da Silva K; Paterson NG; Williams MA; Hall DR; Madhi S; Nunes MC; Goulder P; Fry EE; Mongkolsapaya J; Ren J; Stuart DI; Screaton GR Reduced neutralization of sars-cov-2 b.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236 e4213, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh CL; Abiona O; Graham BS; McLellan JS Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science 2020, 367, 1260–1263, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.V’Kovski P; Kratzel A; Steiner S; Stalder H; Thiel V Coronavirus biology and replication: Implications for sars-cov-2. Nat Rev Microbiol 2021, 19, 155–170, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruse RL Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in wuhan, china. F1000Res 2020, 9, 72, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiel V; Ivanov KA; Putics Á; Hertzig T; Schelle B; Bayer S; Weißbrich B; Snijder EJ; Rabenau H; Doerr HW; Gorbalenya AE; Ziebuhr J Mechanisms and enzymes involved in sars coronavirus genome expression. J Gen Virol 2003, 84, 2305–2315, [DOI] [PubMed] [Google Scholar]
- 8.Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh M.-d; Ruiz-Palacios GM; Benfield T; Fätkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC. Remdesivir for the treatment of covid-19 — final report. N Engl J Med 2020, 383, 1813–1826, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lei J; Kusov Y; Hilgenfeld R Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res 2018, 149, 58–74, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ratia K; Saikatendu KS; Santarsiero BD; Barretto N; Baker SC; Stevens RC; Mesecar AD Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A 2006, 103, 5717–5722, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barretto N; Jukneliene D; Ratia K; Chen Z; Mesecar AD; Baker SC The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 2005, 79, 15189–15198, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X; Yang X; Zheng Y; Yang Y; Xing Y; Chen Z Sars coronavirus papain-like protease inhibits the type i interferon signaling pathway through interaction with the sting-traf3-tbk1 complex. Protein Cell 2014, 5, 369–381, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frieman M; Ratia K; Johnston RE; Mesecar AD; Baric RS Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of irf3 and nf-κb signaling. J Virol 2009, 83, 6689–6705, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratia K; Kilianski A; Baez-Santos YM; Baker SC; Mesecar A Structural basis for the ubiquitin-linkage specificity and deisgylating activity of sars-cov papain-like protease. PLoS Pathog 2014, 10, e1004113, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin D; Mukherjee R; Grewe D; Bojkova D; Baek K; Bhattacharya A; Schulz L; Widera M; Mehdipour AR; Tascher G; Geurink PP; Wilhelm A; van der Heden van Noort GJ; Ovaa H; Müller S; Knobeloch K-P; Rajalingam K; Schulman BA; Cinatl J; Hummer G; Ciesek S; Dikic I. Papain-like protease regulates sars-cov-2 viral spread and innate immunity. Nature 2020, 587, 657–662, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freitas BT; Durie IA; Murray J; Longo JE; Miller HC; Crich D; Hogan RJ; Tripp RA; Pegan SD Characterization and noncovalent inhibition of the deubiquitinase and deisgylase activity of sars-cov-2 papain-like protease. ACS Infect Dis 2020, 6, 2099–2109, [DOI] [PubMed] [Google Scholar]
- 17.Lindner HA; Fotouhi-Ardakani N; Lytvyn V; Lachance P; Sulea T; Ménard R The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 2005, 79, 15199–15208, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Valle DM; Kim-Schulze S; Huang H-H; Beckmann ND; Nirenberg S; Wang B; Lavin Y; Swartz TH; Madduri D; Stock A; Marron TU; Xie H; Patel M; Tuballes K; Van Oekelen O; Rahman A; Kovatch P; Aberg JA; Schadt E; Jagannath S; Mazumdar M; Charney AW; Firpo-Betancourt A; Mendu DR; Jhang J; Reich D; Sigel K; Cordon-Cardo C; Feldmann M; Parekh S; Merad M; Gnjatic S An inflammatory cytokine signature predicts covid-19 severity and survival. Nat Med 2020, 26, 1636–1643, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siklos M; BenAissa M; Thatcher GR Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B 2015, 5, 506–519, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, gc-376, and calpain inhibitors ii, xii inhibit sars-cov-2 viral replication by targeting the viral main protease. Cell Res 2020, 30, 678–692, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ Jr.; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ 3rd; Solowiej JE; Steppan CM; Taggart B Discovery of ketone-based covalent inhibitors of coronavirus 3cl proteases for the potential therapeutic treatment of covid-19. J Med Chem 2020, 63, 12725–12747, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratia K; Pegan S; Takayama J; Sleeman K; Coughlin M; Baliji S; Chaudhuri R; Fu W; Prabhakar BS; Johnson ME; Baker SC; Ghosh AK; Mesecar AD A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks sars virus replication. Proc Natl Acad Sci U S A 2008, 105, 16119–16124, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK; Takayama J; Aubin Y; Ratia K; Chaudhuri R; Baez Y; Sleeman K; Coughlin M; Nichols DB; Mulhearn DC; Prabhakar BS; Baker SC; Johnson ME; Mesecar AD Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome–coronavirus papain-like protease. J Med Chem 2009, 52, 5228–5240, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK; Takayama J; Rao KV; Ratia K; Chaudhuri R; Mulhearn DC; Lee H; Nichols DB; Baliji S; Baker SC; Johnson ME; Mesecar AD Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: Design, synthesis, protein–ligand x-ray structure and biological evaluation. J Med Chem 2010, 53, 4968–4979, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Báez-Santos YM; Barraza SJ; Wilson MW; Agius MP; Mielech AM; Davis NM; Baker SC; Larsen SD; Mesecar AD X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J Med Chem 2014, 57, 2393–2412, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug development and medicinal chemistry efforts toward sars-coronavirus and covid-19 therapeutics. ChemMedChem 2020, 15, 907–932, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baez-Santos YM; St John SE; Mesecar AD The sars-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res 2015, 115, 21–38, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klemm T; Ebert G; Calleja DJ; Allison CC; Richardson LW; Bernardini JP; Lu BG; Kuchel NW; Grohmann C; Shibata Y; Gan ZY; Cooney JP; Doerflinger M; Au AE; Blackmore TR; van der Heden van Noort GJ; Geurink PP; Ovaa H; Newman J; Riboldi-Tunnicliffe A; Czabotar PE; Mitchell JP; Feltham R; Lechtenberg BC; Lowes KN; Dewson G; Pellegrini M; Lessene G; Komander D. Mechanism and inhibition of the papain-like protease, plpro, of sars-cov-2. EMBO J 2020, 39, e106275, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rut W; Lv Z; Zmudzinski M; Patchett S; Nayak D; Snipas SJ; El Oualid F; Huang TT; Bekes M; Drag M; Olsen SK Activity profiling and crystal structures of inhibitor-bound sars-cov-2 papain-like protease: A framework for anti-covid-19 drug design. Sci Adv 2020, 6, 10.1126/sciadv.abd4596, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muley L; Baum B; Smolinski M; Freindorf M; Heine A; Klebe G; Hangauer DG Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: Synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin inhibitors. J Med Chem 2010, 53, 2126–2135, [DOI] [PubMed] [Google Scholar]
- 31.Lafleur K; Huang D; Zhou T; Caflisch A; Nevado C Structure-based optimization of potent and selective inhibitors of the tyrosine kinase erythropoietin producing human hepatocellular carcinoma receptor b4 (ephb4). J Med Chem 2009, 52, 6433–6446, [DOI] [PubMed] [Google Scholar]
- 32.Smith E; Davis-Gardner ME; Garcia-Ordonez RD; Nguyen T-T; Hull M; Chen E; Baillargeon P; Scampavia L; Strutzenberg T; Griffin PR; Farzan M; Spicer TP High-throughput screening for drugs that inhibit papain-like protease in sars-cov-2. SLAS Discov 2020, 25, 1152–1161, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ratia K; Pegan S; Takayama J; Sleeman K; Coughlin M; Baliji S; Chaudhuri R; Fu W; Prabhakar BS; Johnson ME; Baker SC; Ghosh AK; Mesecar AD A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks sars virus replication. Proc Natl Acad Sci U S A 2008, 105, 16119–16124, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai C-T; Li H-J; Yu W; Shah S; Bommineni GR; Perrone V; Garcia-Diaz M; Tonge PJ; Simmerling C Rational modulation of the induced-fit conformational change for slow-onset inhibition in mycobacterium tuberculosis inha. Biochemistry 2015, 54, 4683–4691, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tonge PJ Drug–target kinetics in drug discovery. ACS Chem Neurosci 2018, 9, 29–39, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barretto N; Jukneliene D; Ratia K; Chen Z; Mesecar AD; Baker SC The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 2005, 79, 15189–15198, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X; Yang X; Zheng Y; Yang Y; Xing Y; Chen Z Sars coronavirus papain-like protease inhibits the type i interferon signaling pathway through interaction with the sting-traf3-tbk1 complex. Protein Cell 2014, 5, 369–381, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frieman M; Ratia K; Johnston RE; Mesecar AD; Baric RS Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of irf3 and nf-kappab signaling. J Virol 2009, 83, 6689–6705, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratia K; Kilianski A; Baez-Santos YM; Baker SC; Mesecar A Structural basis for the ubiquitin-linkage specificity and deisgylating activity of sars-cov papain-like protease. PLOS Pathogens 2014, 10, e1004113, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freitas BT; Durie IA; Murray J; Longo JE; Miller HC; Crich D; Hogan RJ; Tripp RA; Pegan SD Characterization and noncovalent inhibition of the deubiquitinase and deisgylase activity of sars-cov-2 papain-like protease. ACS Infect Dis 2020, 6, 2099–2109, [DOI] [PubMed] [Google Scholar]
- 41.Shen Z; Ratia K; Cooper L; Kong D; Lee H; Kwon Y; Li Y; Alqarni S; Huang F; Dubrovskyi O; Rong L; Thatcher GR; Xiong R Potent, novel sars-cov-2 plpro inhibitors block viral replication in monkey and human cell cultures. bioRxiv 2021, DOI 10.1101/2021.1102.1113.431008, [DOI] [Google Scholar]
- 42.Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ Jr.; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ 3rd; Solowiej JE; Steppan CM; Taggart B Discovery of ketone-based covalent inhibitors of coronavirus 3cl proteases for the potential therapeutic treatment of covid-19. J Med Chem 2020, 63, 12725–12747, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vabret N; Britton GJ; Gruber C; Hegde S; Kim J; Kuksin M; Levantovsky R; Malle L; Moreira A; Park MD; Pia L; Risson E; Saffern M; Salomé B; Esai Selvan M; Spindler MP; Tan J; van der Heide V; Gregory JK; Alexandropoulos K; Bhardwaj N; Brown BD; Greenbaum B; Gümüş ZH; Homann D; Horowitz A; Kamphorst AO; Curotto de Lafaille MA; Mehandru S; Merad M; Samstein RM; Agrawal M; Aleynick M; Belabed M; Brown M; Casanova-Acebes M; Catalan J; Centa M; Charap A; Chan A; Chen ST; Chung J; Bozkus CC; Cody E; Cossarini F; Dalla E; Fernandez N; Grout J; Ruan DF; Hamon P; Humblin E; Jha D; Kodysh J; Leader A; Lin M; Lindblad K; Lozano-Ojalvo D; Lubitz G; Magen A; Mahmood Z; Martinez-Delgado G; Mateus-Tique J; Meritt E; Moon C; Noel J; O’Donnell T; Ota M; Plitt T; Pothula V; Redes J; Reyes Torres I; Roberto M; Sanchez-Paulete AR; Shang J; Schanoski AS; Suprun M; Tran M; Vaninov N; Wilk CM; Aguirre-Ghiso J; Bogunovic D; Cho J; Faith J; Grasset E; Heeger P; Kenigsberg E; Krammer F; Laserson U Immunology of covid-19: Current state of the science. Immunity 2020, 52, 910–941, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Del Ser T; Fernandez-Blazquez MA; Valenti M; Zea-Sevilla MA; Frades B; Alfayate E; Saiz L; Calero O; Garcia-Lopez FJ; Rabano A; Medina M; Calero M Residence, clinical features, and genetic risk factors associated with symptoms of covid-19 in a cohort of older people in madrid. Gerontology 2021, 1–9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang C; Zhang M; Garcia G Jr.; Tian E; Cui Q; Chen X; Sun G; Wang J; Arumugaswami V; Shi Y Apoe-isoform-dependent sars-cov-2 neurotropism and cellular response. Cell Stem Cell 2021, 28, 331–342 e335, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Studier FW Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005, 41, 207–234, [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D 2010, 66, 133–144, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strong M; Sawaya MR; Wang S; Phillips M; Cascio D; Eisenberg D Toward the structural genomics of complexes: Crystal structure of a pe/ppe protein complex from mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2006, 103, 8060–8065, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vagin A; Teplyakov A Molecular replacement with molrep. Acta Crystallogr D Biol Crystallogr 2010, 66, 22–25, [DOI] [PubMed] [Google Scholar]
- 50.Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 1997, 53, 240–255, [DOI] [PubMed] [Google Scholar]
- 51.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of coot. Acta Crystallogr D 2010, 66, 486–501, [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.