
Figure 1. Structure-guided design of SARS-CoV-2 PLpro inhibitors to explore druggable binding sites.
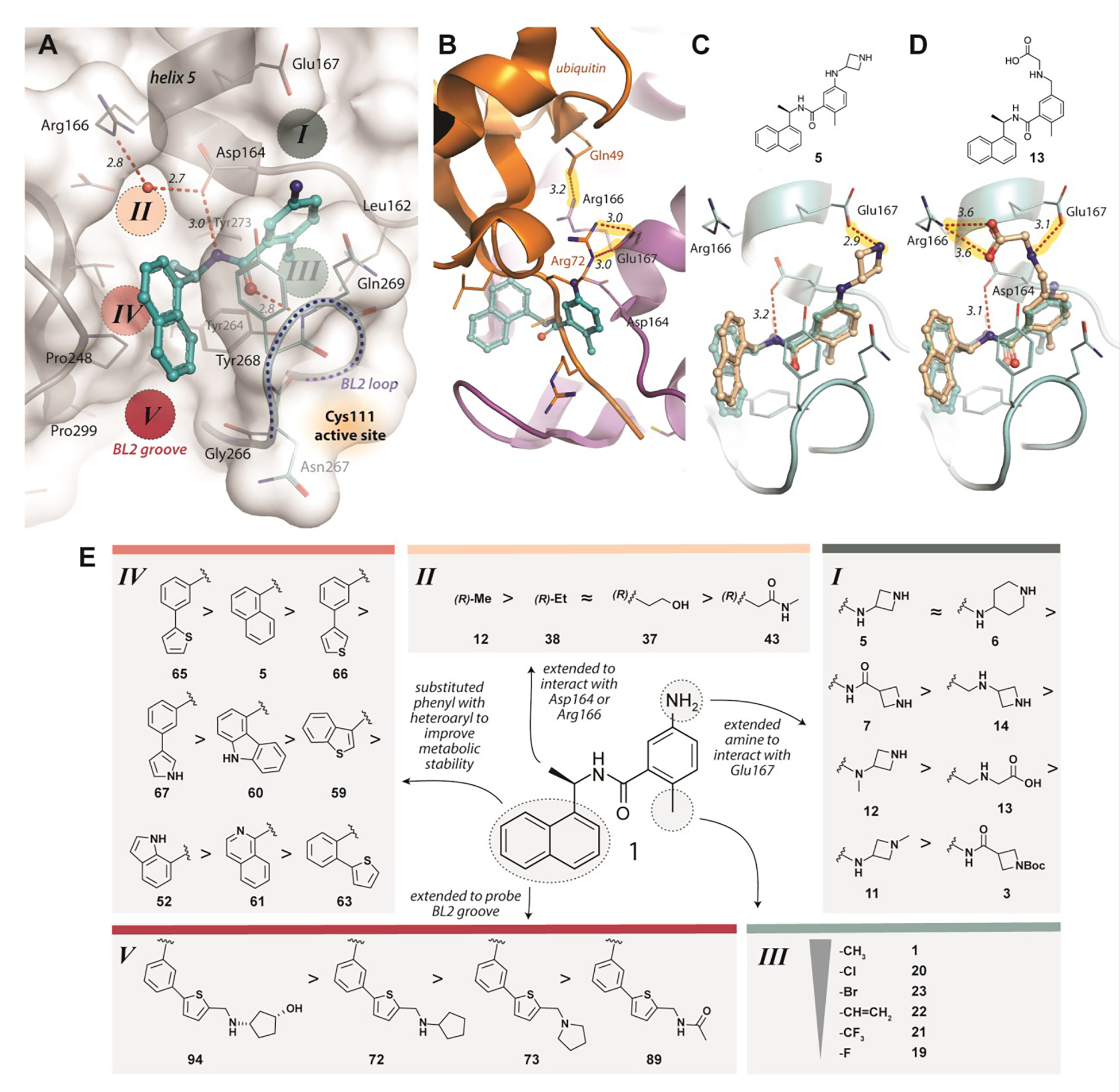
A) Identification of potential ligand binding Sites I-V (PDB: 3E9S). Key hydrogen bonds are shown as red, dashed lines, with distances (Å) labeled in italics. B) A superposition of GRL0617 (cyan; PDB 3E9S) onto the PLpro-ubiquitin structure (orange/magenta; PDB 4MM3) shows that Glu167 of PLpro (magenta) interacts with Arg72 of ubiquitin (orange) in Site I and Arg166 interacts with Gln49 of ubiquitin in Site II. New compounds were designed to mimic these two key interactions to improve binding affinity and to engage Sites I and II. C) Modeling of ZN-2-184 (5) (wheat) bound to PLpro, superimposed with PLpro-GRL0617 (cyan, PDB 3E9S), with the azetidine ring capturing the electrostatic interaction with Glu167 in Site I; D) Modeling of ZN-3-56 (13) (wheat) bound to PLpro, superimposed with PLpro-GRL0617 (cyan, PDB 3E9S; showing the glycine sidechain of ZN-3-56 (13) forming electrostatic interactions with Glu167 and Arg166. E) Summary of structure activity relationships of selected compounds designed to engage with Sites I-V of PLpro (Table 1 details potency and affinity for the selected compounds and full SAR is provided in Tables S1–S5).