

Abstract
Context:
In addition to genetic alterations, epigenetic alterations play a crucial role during prostate cancer progression. A better understanding of the epigenetic factors that promote prostate cancer progression may lead to the design of rational therapeutic strategies to target prostate cancer more effectively.
Objective:
To systematically review recent literature on the role of epigenetic factors in prostate cancer and highlight key preclinical and translational data with epigenetic therapies.
Evidence acquisition:
We performed a systemic literature search in PubMed. At the request of the editors, we limited our search to articles published between January 2015 and August 2020 in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines. Clinical trials targeting epigenetic factors were retrieved from clinicaltrials.gov.
Evidence synthesis:
We retrieved 1451 articles, and 62 were finally selected for review. Twelve additional foundational studies outside this time frame were also included. Findings from both preclinical and clinical studies were reviewed and summarized. We also discuss 12 ongoing clinical studies with epigenetic targeted therapies.
Conclusions:
Epigenetic mechanisms impact prostate cancer progression. Understanding the role of specific epigenetic factors is critical to determine how we may improve prostate cancer treatment and modulate resistance to standard therapies. Recent preclinical studies and ongoing or completed clinical studies with epigenetic therapies provide a useful roadmap for how to best deploy epigenetic therapies clinically to target prostate cancer.
Patient summary:
Epigenetics is a process by which gene expression is regulated without changes in the DNA sequence itself. Oftentimes, epigenetic changes influence cellular behavior and contributes to cancer development or progression. Understanding how epigenetic changes occur in prostate cancer is the first step toward therapeutic targeting in patients. Importantly, laboratory-based studies, and recently completed and ongoing clinical trials suggest that drugs targeting epigenetic factors are promising. More work is necessary to determine whether this class of drugs will add to our existing treatment arsenal in prostate cancer.
Keywords: Epigenetics, DNA methylation, DNA methyltransferase, Ten-Eleven Translocation, Histone acetyltransferase, Histone deacetylase, Histone methyltransferase, Histone demethylase, BET bromodomain, Chromatin, Prostate cancer
thm_compuscript
Recent preclinical studies with prostate cancer models and ongoing or completed clinical studies suggest that drugs targeting epigenetic factors are promising therapeutic agents. More work is necessary to determine how to best deploy epigenetic therapies clinically to target prostate cancer.
1. Introduction
Prostate cancer is the second most common cancer among men globally [1]. The androgen receptor (AR) is the central signaling molecule promoting both prostate cancer differentiation and proliferation. Owing to the role of the AR in prostate cancer cell survival, the principal systemic treatment for prostate cancer is androgen deprivation therapy (ADT) through chemical or surgical castration to block AR signaling. Although metastatic prostate cancers initially respond to ADT, the disease invariably progresses to lethal castration-resistant prostate cancer (CRPC) due to AR signaling reactivation [2]. Both genetic and epigenetic factors contribute to tumorigenesis and progression to CRPC. Genetic factors include, but are not limited to, the ETS family members SPOP, FOXA1, IDH1, TP53, RB1, and PTEN [3,4].
Epigenetics is the heritable control of gene expression in the absence of changes in the DNA coding sequence [5]. Epigenetic regulation is a complicated process in which many different factors, including those that add post-translational modifications to DNA or histone proteins, dictate whether a given gene is turned on or off. These epigenetic changes can occur at promoter regions that surround the transcriptional start site in addition to more distant regulatory regions called enhancers. Historically, epigenetic regulation through DNA methylation was the best studied mechanism. Methylation of cytosine residues is catalyzed by a family of enzymes called DNA methyltransferases (DNMTs) and removed by DNA demethylases, including the Ten-Eleven Translocation (TET) family of enzymes [6].
More recent work has determined that epigenetic regulation can also occur through chemical modifications of histone proteins that, along with DNA, make up chromatin [5]. Although there are many different histone modifications, this review focuses on methylation and acetylation of lysine on histone tails. Histone lysine methylation is mediated by a class of proteins called histone methyltransferases, while demethylation of lysines on histones is mediated by histone demethylases. Histone lysine acetylation is mediated by histone acetyltransferases, while deacetylation is mediated by histone deacetylases (HDACs). In addition to the histone writers (histone methyltransferases and histone acetyltransferases) and histone erasers (histone demethylases and HDACs), another class of proteins called chromatin readers are thought to interpret these histone marks and bind to chromatin. The best studied example of chromatin readers in prostate cancer is the bromodomain and extraterminal (BET) bromodomain family of proteins that detect acetylated lysines on histone tails and cooperate in multiprotein complexes to regulate transcription [7]. Major epigenetic factors implicated in prostate cancer are summarized in Figure 1.
Fig. 1 –
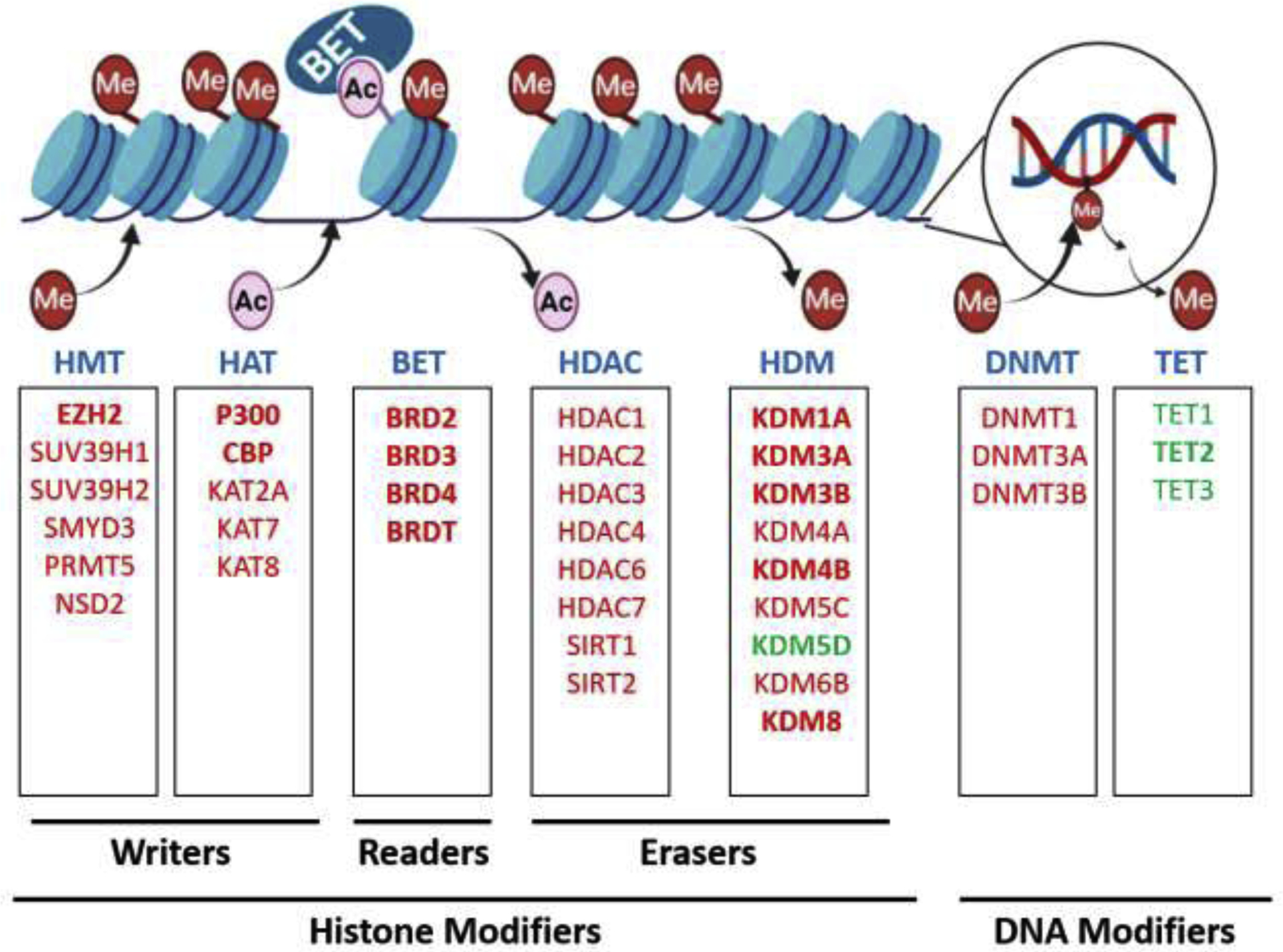
Overview of histone or DNA modifiers implicated in prostate cancer. Key factors that modulate or recognize epigenetic marks in prostate cancer are shown. Although this figure is not exhaustive, the best studied factors previously implicated in prostate cancer are depicted. Those that are reviewed in this manuscript are indicated in bold. Factors that are upregulated in prostate cancer are shown in red, and downregulated factors are shown in green. These factors can be classified broadly into histone modifiers (histone methyltransferases [HMTs], histone acetyltransferases [HATs], bromodomain and extraterminal [BET] chromatin readers, histone deacetylases [HDACs], and histone demethylases [HDMs]) or DNA modifiers (DNA methyltransferases [DNMTs] and Ten-Eleven Translocation [TETs]) based on their substrate. Further, the factors are classified as writers, readers, or erasers based on their biochemical activity. The epigenetic marks modulated by these factors are being explored as potential biomarkers. Finally, drugs targeting many of these factors have been developed or are in development.
Here, we systematically review the role of key epigenetic factors that belong to the classes described above in prostate cancer. We focus on both preclinical and clinical studies, and highlight current and future directions to develop epigenetic therapies in prostate cancer.
2. Evidence acquisition
2.1. Study design
A systematic review of recent literature on the role of epigenetic factors in prostate cancer was performed based on the Preferred Reporting Items for Systematic Review and Meta-analyses (PRISMA) guidelines.
2.2. Literature search
A systematic literature search was conducted using the PubMed database for articles published between January 2015 and August 2020. The search strategy contained two components linked by the AND operator. The first component was (Prostate cancer). The second component was one of the following terms: (Epigenetics), (DNA methylation), (DNMT), (DNA methyltransferase), (HDAC), (Histone deacetylase), (Bromodomain), (Chromodomain), (Histone methylation), (Histone acetylation), (HAT), (Histone acetyltransferase), (Histone demethylase), or (KDM). A systematic search for clinical trials was conducted in the clinicaltrials.gov database. The search strategy contained two components linked by the AND operator. The first component was (Prostate cancer). The second component was one of the following terms: (Epigenetics), (Metastatic), (Castration-resistant), (HDAC), (Histone), (EZH2), (p300), (CBP), or (bromodomain). Results from all the searches were aggregated.
2.3. Literature selection
The systematic search with the parameters detailed above yielded 1451 articles. From these, we refined the final list of articles based on selection criteria, including original articles—rather than a review, editorial, or commentary—discussing epigenetics in prostate cancer models or prostate cancer patient tumors and clinical relevance. After filtering based on these selection criteria, 62 articles were selected for inclusion in this review (Fig. 2). The search results for clinical trials from clinicaltrials.gov were filtered for trials targeting epigenetic factors, which yielded 31 studies (Tables 1–3).
Fig. 2 –
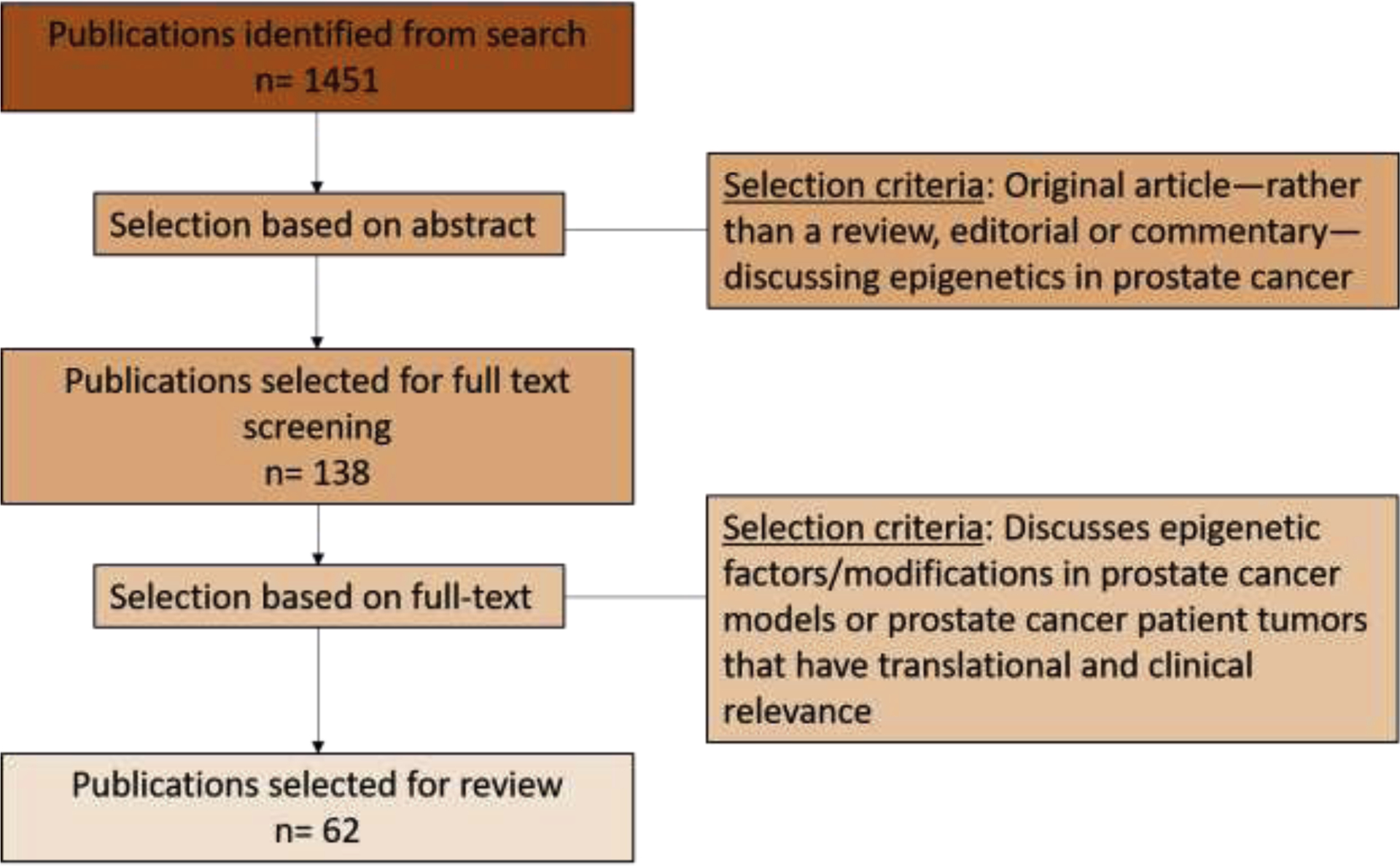
Consort diagram depicting the literature selection and filtering process. The articles from the search results as described in the evidence acquisition section were aggregated and screened by a two-step screening process. A final list of 62 articles was selected for review. Twelve additional foundational articles were also included.
Table 1 –
Ongoing clinical trials in prostate cancer with drugs targeting epigenetic factors
| S. no. | Drug | Target | Combination | Phase | Indication | Trial Identifier | Status |
|---|---|---|---|---|---|---|---|
| 1 | Decitabine | DNMT | Enzalutamide | 1, 2 | Metastatic castration-resistant prostate cancer | NCT03709550 | Not yet recruiting |
| 2 | ZEN003694 | BET | Enzalutamide and pembrolizumab | 2 | Metastatic castration-resistant prostate cancer | NCT04471974 | Not yet recruiting |
| 3 | GSK525762 | BET | Abiraterone/prednisone or enzalutamide | 1 | Metastatic castration-resistant prostate cancer | NCT03150056 | Active, not recawing |
| 4 | PLX2853 | BET | Abiraterone/prednisone or olaparib | 1, 2 | Metastatic castration-resistant prostate cancer | NCT04556617 | Recruiting |
| 5 | CCS1477 | p300/CBP | Abiraterone/prednisone or enzalutamide | 1, 2 | Metastatic castration-resistant prostate cancer | NCT03568656 | Recruiting |
| 6 | FT-7051 | p300/CBP | 1 | Metastatic castration-resistant prostate cancer | NCT04575766 | Recruiting | |
| 7 | MS-275 | HDAC | Enzalutamide | 1 | Castration-resistant prostate cancer | NCT03829930 | Recruiting |
| 8 | CPI-1205 | EZH2 | Abiraterone/prednisone or enzalutamide | 1, 2 | Metastatic castration-resistant prostate cancer | NCT03480646 | Active, not recruiting |
| 9 | PF-06821497 | EZH2 | 1 | Metastatic castration-resistant prostate cancer | NCT03460977 | Recruiting | |
| 10 | EPZ-6438 | EZH2 | Abiraterone/prednisone or enzalutamide | 1 | Metastatic castration-resistant prostate cancer | NCT04179864 | Recruiting |
| 11 | SHR2554 | EZH2 | SHR3680 (Novel Anti-Androgen) | 1, 2 | Metastatic castration-resistant prostate cancer | NCT03741712 | Recruiting |
| 12 | DS3201 | EZH1/2 | Ipilimumab | 1 | Metastatic castration-resistant prostate cancer | NCT04388852 | Recruiting |
BET = bromodomain and extraterminal; DNMT = DNA methyltransferase; EZH = Enhancer of Zeste; HDAC = histone deacetylase.
Table 3 –
Completed but unpublished clinical trials in prostate cancer with drugs targeting epigenetic factors
| S. no. | Drug | Target | Combination | Phase | Indication | Trial identifier | Status |
|---|---|---|---|---|---|---|---|
| 1 | ZEN003694 | BET | 1 | Metastatic castration-resistant prostate cancer | NCT02705469 | Completed | |
| 2 | LBH589 | HDAC | Bicalutamide | 1, 2 | Metastatic castration-resistant prostate cancer | NCT00878436 | Completed |
| 3 | LBH589 | HDAC | External beam radiotherapy | 1 | Localized prostate cancer | NCT00670553 | Completed |
| 4 | LBH589 | HDAC | 2 | Metastatic castration-resistant prostate cancer | NCT00667862 | Completed | |
| 5 | LBH589 | HDAC | Docetaxel/prednisone | 1 | Castration-resistant prostate cancer | NCT00663832 | Completed |
| 6 | SAHA | HDAC | Bicalutamide, goserelin acetate, or leuprolide acetate | 2 | Localized prostate cancer | NCT00589472 | Completed |
| 7 | SAHA | HDAC | 2 | Mtastatic castration-resistant prostate cancer | NCT00330161 | Completed | |
| 8 | SAHA | HDAC | 1 | Metastatic castration-resistant prostate cancer | NCT00005634 | Completed | |
| 9 | MS-275 | HDAC | 1 | Metastatic castration-resistant prostate cancer | NCT00020579 | Completed | |
| 10 | Azacitidine | DNMT | 2 | Metastatic or nonmetastatic castration-resistant prostate cancer | NCT00384839 | Completed | |
| 11 | Azacitidine | DNMT | Sodium phenylbutyrate | 2 | Metastatic castration-resistant prostate cancer | NCT00006019 | Completed |
BET = bromodomain and extraterminal; DNMT = DNA methyltransferase; HDAC = histone deacetylase.
3. Evidence synthesis
3.1. DNA methylation
3.1.1. DNA methylation markers
DNA methylation is among the best studied epigenetic changes in cancer [5]. DNA methylation occurs at the 5′ carbon of cytosine nucleotides flanked by a guanine, the so-called CpG sites. The DNA methylation pattern is often altered in cancer versus normal tissues, leading to aberrant gene expression. DNA methylation, particularly in promoter regions, often leads to gene silencing. Numerous studies have investigated genome-wide DNA methylation patterns and identified specific DNA methylation events in localized prostate cancer and metastatic CRPC, and within subtypes such as neuroendocrine prostate cancer (NEPC) versus adenocarcinoma. The goals of these efforts include identification of DNA methylation changes that (1) may drive tumor development and progression, or (2) may be reliable diagnostic, prognostic, or predictive biomarkers.
Localized prostate cancers exhibit widespread changes in hyper- or hypomethylation of specific regions that contribute to gene expression. In a study using formalin-fixed, paraffin-embedded tumor tissue cores and adjacent benign tissue from radical prostatectomies, Geybels et al [8] identified specific differentially methylated CpG sites. They confirmed that DNA methylation was associated with reduced gene expression and confirmed their findings using prostate cancer samples from The Cancer Genome Atlas (TCGA). A more recent study took a similar approach to identify specific DNA methylation changes that could be used to distinguish cancer from benign tissues [9]. By integrating their DNA methylation data with the Encyclopedia of DNA Elements (ENCODE) transcription factor binding database, the authors determined that regions with higher DNA methylation in cancer were associated with increased occupancy of the histone methyltransferase Enhancer of Zeste 2 (EZH2), thus linking DNA hypermethylation changes to a key protein that regulates histone methylation [9].
An even larger genome-wide study investigating the epigenetic landscape of 589 localized prostate tumors identified 1178 differentially methylated loci in intermediate-risk prostate cancer compared with benign tissue [10]. Many of the methylation differences between tumors were associated with differences in germline polymorphisms, demonstrating the potential influence of germline alterations on DNA methylation [10]. TCGA investigated the molecular taxonomy of 333 primary prostate cancer tumors and reported the DNA methylation patterns across seven genomically defined subtypes [3]. These authors also identified four clusters based on DNA methylation data, including a distinct TMPRSS2-ERG fusion-positive cluster with DNA hypermethylation [3]. This suggests a molecular interplay between TMPRSS2-ERG fusions and DNA methylation in specific prostate tumors. In early-onset prostate cancer, defined as prostate cancer diagnosed at age 55 yr or earlier, integrated DNA methylation and whole genome, and transcriptome analysis identified four subgroups that stratify patients into specific risk categories [11]. Importantly, one particularly aggressive subgroup was associated with genomic alterations and increased expression of ESPR1 that has been linked to epithelial-mesenchymal transition (EMT) and metastasis [11]. Finally, Patel et al [12] analyzed 15 frequently methylated loci in over 1300 primary prostate cancer and benign tissues from prostatectomy samples, and identified three genes, GAS6, GSTP1, and HAPLN3, whose DNA methylation status was sensitive and specific for distinguishing between prostate cancer and benign tissues.
DNA methylation changes may play an even more important role in metastatic prostate cancer. Zhao et al [13] recently reported the first whole-genome bisulfite sequencing analysis of metastatic CRPC, using 100 metastatic tumor biopsies. Using unsupervised, hierarchical clustering, they identified specific clusters of tumor samples. Importantly, 22% of samples comprised a previously unrecognized CpG methylator phenotype (CMP) with particularly high levels of DNA methylation [13]. That subtype was enriched for mutations in IDH1, DNMT3B, BRAF, and TET2, suggesting an interplay between DNA methylation and these genetic events and suggesting that these tumors may be particularly susceptible to DNMT inhibition [13]. The authors also report a cluster associated with treatment-emergent neuroendocrine prostate cancer (t-NEPC), demonstrating that unique DNA methylation patterns may contribute to t-NEPC development, which corroborates prior results from the literature [13,14]. The study by Zhao et al [13] also identified multiple hypomethylated regions near the AR gene, including a previously identified AR enhancer near the AR promoter, and other regions upstream and downstream of the AR [15]. These regions and AR mRNA expression had a strong correlation with hypomethylation, strongly suggesting that these hypomethylated regions are AR regulatory regions [13].
A more recent study examined DNA methylation in plasma samples from patients with NEPC [16]. This report demonstrated high concordance between DNA methylation results in cell-free DNA (cfDNA) and matched metastatic tumor biopsies. It also identified specific genes methylation of which could help distinguish NEPC from adenocarcinoma tumors—hypermethylation of ASXL3 and SPDEF and hypomethylation of INSM1 and CDH2 [16]. Finally, Wu et al [17] profiled DNA methylation in cfDNA from plasma and demonstrated frequent hypomethylation of regions containing androgen response element (ARE) sequences. Hypomethylation of these regions was associated with AR copy number gains and was linked to more aggressive clinical behavior, possibly due to heightened AR activity.
Multiple studies have used global or targeted DNA methylation measurements to identify DNA methylation biomarkers that may assist in the risk stratification of patients with prostate cancer. A quantitative assay evaluating the methylation status of promoter regions of GSTP1, RASSF1, and APC using prostate biopsy samples without cancer had a negative predictive value of 88% for subsequent detection of prostate cancer on repeat biopsy. This assay is currently used clinically to determine whether repeat prostate biopsy is warranted [18]. An integrated genomic and epigenomic analysis identified a multiomic biomarker, including ACTL6B hypermethylation and TCERGL1 hypomethylation, which was independently predictive of disease relapse in men with localized prostate cancer [19]. Wu et al [17] recently reported results of an AR methylation signature (AR-MethSig) based on DNA methylation analysis of cfDNA from patients with metastatic CRPC. This score distinguished less aggressive CRPC tumors from more aggressive CRPC tumors that harbored AR copy number gain [17]. Finally, Gordevicius et al [20] profiled cfDNA from 33 patients treated with abiraterone acetate and found specific CpG residues that were differentially methylated between responders and nonresponders, demonstrating the potential utility of noninvasive DNA methylation measurements for treatment response prediction. More studies are necessary to determine whether cfDNA methylation measurements will impact clinical practice. If successful, these studies may lead to new biomarkers that impact the care of men with advanced prostate cancer.
3.1.2. DNMT and TET enzymes
DNMT enzymes catalyze the addition of methyl groups to cytosine residues in DNA to form 5-methyl cytosine (5mC). These marks can be erased by the TET family of DNA demethylases [6]. TET catalyzes the demethylation of 5mC through sequential oxidation to 5-hydroxymethyl cytosine (5hmC), 5-formyl cytosine (5fC), and then to 5-carboxyl cytosine (5caC) [6]. Eventually, 5fC and 5caC are replaced by unmethylated cytosine by base excision repair [6]. Storebjerg et al [21] demonstrated that higher 5hmC levels in TMPRSS2-ERG fusion-positive prostate cancers, specifically, were associated with shorter time to biochemical recurrence, while high 5caC levels were associated with favorable prognosis in patients with TMPRSS2-ERG fusion-positive tumors.
Recent work sheds light on the molecular mechanisms of TET function and suggests that TET2 and the AR oppose each other’s function. Nickerson et al [22] determined that TET2 functions as a tumor suppressor that may act, in part, by suppressing AR activation. Further, the AR appears to repress TET2 mRNA expression by binding to a distal TET2 enhancer as well as by activating expression of microRNA family miR-29 that leads to reduced TET2 mRNA [23].
TET2 may also be a useful biomarker. Indeed, reduced TET2 mRNA levels correlated with tumor progression [22]. As mentioned above, mutations in TET2 help define the CMP subtype of CRPC identified by Zhao et al [13]. More studies on the function of TETs in prostate cancer will be necessary to understand the role of TET proteins and whether restoration of TET2 expression or function is a promising therapeutic direction.
Drug development efforts to target aberrant DNA hypermethylation led to the development of the DNMT inhibitor azacitidine, which has been tested clinically in numerous cancer types, including prostate cancer (NCT00503984) [24]. Based upon the idea that methylation-induced gene silencing promotes docetaxel resistance, a phase I/II study of azacitidine in combination with docetaxel and prednisone in patients with metastatic CRPC reported clinical benefit with no dose-limiting toxicity; a prostate-specific antigen (PSA) response was observed in ten of the 19 patients evaluated [24]. Three of these patients demonstrated objective responses, and decreased methylation of GADD45A in white blood cells was seen with azacitidine treatment [24]. The median progression-free survival (PFS) was 4.9 mo. However, the lack of a docetaxel/prednisone-only control arm makes interpretation of the trial results difficult.
In sum, these DNA methylation studies reveal the importance of DNA methylation alterations in localized and metastatic prostate cancer. Measuring DNA methylation in tissue or cfDNA is a promising approach to classify prostate cancer patients and predict clinical outcomes more accurately than clinical factors alone. More work is clearly needed to determine the effectiveness of targeting DNA methylation in clinical trials and whether specific molecular subgroups of patients, including those with CMP subtype identified by Zhao et al [13], may be most likely to benefit from this treatment.
3.2. Histone modifications
3.2.1. Histone acetylation
Post-translational modifications of histones govern chromatin organization and gene expression [25]. Acetylation of lysine tails on histones is linked to open and active chromatin, and a number of histone acetyltransferase enzymes have been described [25,26]. Pomerantz et al [27] demonstrated that genome-wide AR binding during the transition from benign tissue to localized castration-naïve disease to metastatic CRPC coincides with changes in acetylation of the lysine 27 position on histone H3 (H3K27ac). Further, integration of histone modification chromatin immunoprecipitation sequencing (ChIP-seq), including H3K27Ac data, with AR ChIP-seq helped define distinct prostate cancer subtypes [28].
3.2.2. Histone acetyltransferases
The histone acetyltransferase family is made up of 30 different proteins that transfer acetyl groups from acetyl-CoA to lysine tails on histones [26]. In general, histone acetylation has been linked to gene activation. Among the best-studied histone acetyltransferases in prostate cancer are p300 and CBP, which are highly homologous. Recently, several groups have developed inhibitors to target one or both of these histone acetyltransferases because of their role in regulating key genes in prostate cancer, including AR target genes [29]. A-485 is a novel p300/CBP catalytic inhibitor with much greater specificity and potency than previously developed histone acetyltransferase inhibitors [29]. Lasko et al [29] demonstrated that A-485 treatment blocked AR signaling and growth of AR-dependent CRPC xenografts in vivo, demonstrating the potential impact of targeting histone acetylation. More recently, p300/CBP inhibition by A-485 was also shown to inhibit secretion of exosomal PD-L1 by tumor cells; combination therapy with A-485 plus anti-PD-L1 antibodies increased T-cell infiltration in tumors and augmented the tumor immune response in a syngeneic model of prostate cancer. Together, these data suggest that combining histone acetyltransferase inhibitors with immunotherapy may be a promising approach to augment the activity of immunotherapy in prostate cancer. Importantly, clinical trials on p300/CBP histone acetyltransferase inhibitors in prostate cancer have recently begun (NCT03568656 and NCT04575766; Table 1), and preliminary clinical results with the p300/CBP inhibitor CCS1477 demonstrate modulation of PSA blood levels and biomarker expression in CRPC biopsies [31].
3.2.3. Histone deacetylases
The other class of proteins that regulate histone acetylation comprises the HDACs. There are five major classes of HDAC proteins: class I, class IIA, class IIB, class III, and class IV [32]. Drugs targeting HDAC proteins are among the best studied in both preclinical studies and clinical trials in prostate cancer. The number of publications related to HDAC inhibition is beyond the scope of this review, and the antitumor activity of HDAC inhibition has been ascribed to a variety of mechanisms, including suppression of EMT that contributes to resistance to AR targeting [33]. Interestingly, the HDAC inhibitor LBH589, also known as panobinostat, resensitized CRPC models that had undergone EMT to ADT [33]. Despite these promising preclinical studies, single agent HDAC inhibitor clinical trials have not shown significant activity [34]. However, a more recent clinical trial with LBH589 plus the AR inhibitor bicalutamide suggested that the combination may be a strategy to resensitize tumors to AR inhibition [35]. Since novel AR inhibitors are now more commonly used, these results suggest that it is rational to test HDAC inhibitors with those newer and more potent AR inhibitors.
3.2.4. Histone methyltransferases
Unlike histone acetylation that is primarily linked to gene activation, histone methylation can either activate or repress transcription depending on the histone lysine tail that is involved. Histone methyltransferases are the proteins that catalyze histone methylation, and several histone methyltransferases are of interest as therapeutic targets in prostate cancer. EZH2 is a member of the polycomb repressive complex 2 (PRC2), which is the best studied histone methyltransferase in prostate cancer, and recently several companies have developed EZH2 inhibitors [36].
EZH2 is upregulated in metastatic prostate cancer and has been shown to play key roles during prostate cancer progression [36]. In prostate cancer cells, irradiation induces epigenetic reprogramming, leading to reversion to a stem-like state [37]. One of the key alterations observed upon irradiation was hypermethylation of histone H3, including at H3K27 [37]. Consistent with that notion that EZH2 might contribute to this stem-like state, EZH2 inhibition blocked radiation-induced stemness [37].
It is now appreciated that low AR activity in prostate cancer is associated with resistance to AR inhibition [38]. EZH2 has been shown to be important for promoting lineage plasticity, or differentiation change, and reduced reliance on the AR that is now recognized as an important determinant of ADT resistance [39]. NEPC is one example of lineage plasticity, and EZH2 has been shown to cooperate with the oncogene N-Myc to block AR signaling in NEPC models by mediating methylation of the lysine 27 residue on histone H3 (H3K27) at AR target promoters, which leads to suppression of AR signaling [40]. Moreover, EZH2 is upregulated in tumors with loss of the tumor suppressors RB1 and TP53—genes that may prevent NEPC lineage plasticity [41]. Using models of lineage plasticity with loss of PTEN/RB1 or PTEN/RB1/TP53, Ku et al [41] demonstrated that EZH2 inhibition reactivates AR signaling and resensitizes these tumors to AR inhibition. Additionally, EZH2 was recently shown to activate AR mRNA expression and promote AR signaling; EHZ2 inhibition was synergistic with AR inhibition, resulting in complete suppression of AR signaling [42]. Further, cotargeting of EZH2 with antisense oligonucleotides plus AR inhibitors led to synergy [43]. Taken together, these studies suggest the potential of combination therapy with EZH2 and AR inhibition. Clinical trials with EZH2 inhibitors alone (NCT03460977), in combination with AR targeting agents, (NCT04179864, NCT03480646, and NCT03741712), or in combination with immunotherapy (NCT04388852) in prostate cancer have recently begun (Table 1).
3.2.5. Histone demethylases
Multiple histone demethylases that oppose the function of specific histone methyltransferases have been reported to be upregulated in advanced prostate cancer [44]. Histone demethylases are divided into two groups based on their enzymatic mechanisms: the Jumonji C domain-containing (JMJD) family and the flavin adenine dinucleotide (FAD)-dependent family [45]. Several histone demethylases have been shown to activate AR signaling and other important pathways. Lysine-specific demethylase 3A (KDM3A/JMJD1A) was shown to interact with the AR and co-operate in the activation of c-Myc expression by demethylating the dimethyl lysine 9 residue (H3K9me2) at the c-Myc enhancer region [46]. Further, KDM3A interacts with the E3 ubiquitin ligase HUWE1 and inhibits ubiquitination of the c-Myc protein [46].
Lysine-specific demethylase 1 (LSD1/KDM1A) demethylates dimethyl lysine 4 on histone H3 (H3K4me2). LSD1 has also been shown to co-operate with the AR and activate a subset of cell cycle genes, including CENPE [47]. Moreover, LSD1 was recently shown to demethylate the FOXA1 pioneer transcription factor, leading to FOXA1 activation—an example of LSD1 demethylation of a nonhistone substrate [48]. Sehrawat et al [49] showed that LSD1 has important AR-independent functions in prostate cancer. In that report, LSD1 was found to promote AR-independent cell survival in CRPC by activating a lethal prostate cancer gene network independently of its catalytic function [49]. Interestingly, Coleman et al [50] identified a neuronal-specific isoform of LSD1, called LSD1+8a, in NEPC tumors, suggesting that LSD1+8a may be a marker of neuronal lineage switching or may contribute to neuronal lineage switching. LSD1+8a was found to regulate a unique set of genes distinct from canonical LSD1, suggesting that this splice variant plays a distinct role from canonical LSD1 [50].
Several other histone demethylases have been studied in prostate cancer. Lysine-specific demethylase 5D (KDM5D), an H3K4me2 demethylase, regulates AR signaling by interacting with the AR, and loss of KDM5D promotes docetaxel resistance [51]. Furthermore, measuring KDM5D expression in metastatic castration-naive prostate cancer may help predict response to docetaxel [51]. In CRPC, lysine-specific demethylase 4B (KDM4B/JMJD2B) interacts with the splicing factor SF3B3, promotes expression of AR-V7, and confers resistance to AR targeting agents [52]. KDM4B may also promote AR-independent survival by cooperating with BMyb to activate cell cycle genes, including PLK1, by demethylating trimethyl lysine 9 on histone H3 (H3K9me3) on target promoters [53]. Targeting KDM4B blocked the growth of AR-negative prostate cancer cells, demonstrating the potential of KDM4B targeting in AR-independent CRPC [53]. Similarly, lysine-specific demethylase 3B (KDM3B), an H3K9me2 demethylase, was shown to promote androgen-independent growth of CRPC by altering cellular metabolism in CRPC cells [54]. Furthermore, suppression of KDM3B expression or treatment with pan-KDM3 inhibitors had greater effects on reducing the survival of CRPC cells versus castration-sensitive cells [54]. Finally, lysine-specific demethylase 8 (KDM8/JMJD5) demethylates dimethyl lysine 36 on histone H3 (H3K36me2) and was shown to regulate the function of several important factors in CRPC, including the AR, HIF-1a, and EZH2, and promotes prostate cancer cell survival [55]. Importantly, KDM8 knockdown resensitized resistant cells to enzalutamide, suggesting the potential of this combination strategy [55].
These recent studies demonstrate the importance of measuring histone demethylases as biomarkers or targeting these factors in prostate cancer. Understanding of how specific histone demethylases function is critical, as there is a clear appreciation now that many histone demethylases, such as KDM1A, have important noncatalytic, scaffold functions [49]. In some cases, inhibition of critical protein-protein interactions of specific histone demethylases or other chromatin-modifying enzymes may be a more effective approach to block these factors. Once these studies are completed, future clinical studies may be warranted to test the effectiveness of blocking specific histone demethylases alone or in combination with standard treatments.
3.3. Chromatin remodelers and readers
Alterations in proteins that regulate chromatin conformation have been implicated in prostate cancer progression [56,57]. Indeed, deletion of the gene encoding the chromatin remodeler CHD1 is commonly observed in prostate cancer [56]. Augello et al [56] experimentally deleted CHD1 in normal prostate cells, and determined that CHD1 loss altered the chromatin landscape and redistributed the AR to regions commonly bound by the AR in tumors and away from regions linked to lineage commitment. More recent work demonstrates that CHD1 loss contributes to enzalutamide resistance through upregulation of transcription factors that promote AR-independent cell survival, including the glucocorticoid receptor (GR) that is targetable with drugs [58].
The SWI/SNF complex, also known as the BAF complex, regulates chromatin accessibility, and recent work demonstrates that subunits of this complex are deregulated in specific prostate cancer subsets [57]. Importantly, the SMARCA4 (BRG1) subunit is much more highly expressed in NEPC than in its paralogue SMARCA2 (BRM), and SMARCA4 upregulation in tumors is associated with shorter overall survival; furthermore, the SWI/SNF complex interacts with unique transcription factors in NEPC versus adenocarcinoma cells—factors that may promote the unique chromatin conformation seen in NEPC or that mediate expression of DNA repair genes [57]. These data suggest that there may be opportunities to target specific dependencies in tumors with CHD1 loss or alterations in SWI/SNF complex composition.
Histone marks, including acetylation, are recognized by a class of proteins called chromatin readers. One example of particular relevance to prostate cancer is the BET family of proteins that include BRD2, BRD3, BRD4, and BRDT [7]. BET proteins recognize and bind to acetylated lysine residues via their two tandem bromodomains, BD1 and BD2, to regulate transcription [7]. BET protein mRNA and protein are overexpressed in prostate cancer compared with those in benign tissue and further increased in CRPC [59,60]. Furthermore, an analysis of prostate cancer tumor biopsies demonstrated that high BRD4 expression at diagnosis is associated a poor clinical outcome [60].
BET bromodomain proteins have been linked to AR signaling activation by promoting AR-mediated chromatin accessibility [59,60]. BET bromodomain protein expression correlates with AR activity, and BET bromodomain proteins appear to regulate expression of the splice variant AR-V7 [60,61]. Evidence suggests that BET proteins interact directly with the AR, potentially through the BD2 domain [61,62]. BET bromodomain proteins are also overexpressed in SPOP mutant prostate cancer. This may be due to the fact that SPOP normally mediates the ubiquitination and subsequent degradation of BET bromodomain proteins [63,64]. Preclinical studies suggest that these SPOP-mutant tumors may be particularly resistant to BET bromodomain inhibition, but combined BETi + AKT inhibition may be one approach to overcome resistance [64].
BET bromodomain proteins have numerous AR-independent functions, including activation of c-Myc, a known oncogene in prostate cancer [61,65–67]. Moreover, BET bromodomain proteins mediate TMPRSS2-ERG rearrangements by facilitating DNA repair and nonhomologous end joining (NHEJ); further, BETi was synergistic with DNA damaging agents in prostate cancer models [68,69]. BET bromodomain proteins may promote resistance to AR inhibitors by binding to an enhancer element that regulates GR expression [70]. BETi resensitizes tumors to enzalutamide by impairing GR expression in GR-dependent models [70]. BET bromodomain proteins also play an important role in AR-independent CRPC by activating AR-independent and c-Myc–independent transcriptional regulators upregulated in CRPC patient tumors [65].
Numerous BETi have been developed over the past decade, and many have demonstrated antitumor effects in prostate cancer preclinical models. In addition to small-molecule BET inhibitors, pan-BET degraders, including proteolysis-targeting chimeras (PROTACs), which induce degradation of BET proteins through ubiquitination followed by proteolysis, have been developed [71]. The pan-BET bromodomain competitive inhibitors JQ1 and I-BET151, and pan-BET PROTAC degrader ARV-771 block AR signaling in full-length AR-driven as well as AR-V7–driven prostate cancer cell lines [59,61,67,71]. Moreover, BETi inhibits cell survival in a variety of AR-driven and AR-independent prostate cancer cell lines, and patient-derived organoids and xenografts (PDXs) [60–62,65–67,71]. BETi also blocks AR-independent BET targets such as c-Myc [61,65–67]. Since the two bromodomains in BET proteins have distinct functions, inhibitors selective for a single bromodomain have been developed in an effort to increase efficacy and reduce the toxicity [62]. ABBV-744 is a selective inhibitor of the BD2 domain of BET bromodomain proteins [62]. Treatment with ABBV-744 reduced survival of AR-driven prostate cancer cell lines and PDXs; ABBV-744 also blocked AR-mediated transcription while impacting global transcription to a lesser extent than the BETi ABBV-075, which blocks both the BD1 and the BD2 domain [62]. In sum, these reports suggest that BETi may be effective in treating both AR-dependent and AR-independent CRPC.
Phase I BETi clinical trials have recently been completed, and several of these trials included prostate cancer patients. BETi monotherapy with MK-8628 (birabresib, NCT02259114) or ABBV-075 (mivebresib, NCT02391480) was tested in patients with solid tumors, including those with CRPC. Each study reported similar toxicity profiles, including reversible thrombocytopenia that warranted intermittent scheduling to reduce toxicity [72,73]. However, neither MK-8628 nor ABBV-075 demonstrated significant antitumor activity in CRPC patients [72,73]. Another phase Ib/IIa study studied the combination of enzalutamide with the BETi ZEN003694 in patients with metastatic CRPC resistant to enzalutamide and/or abiraterone (NCT02711956; Table 2) [74]. The combination of enzalutamide and ZEN003694 was well tolerated and resulted in longer PFS in a subset of patients [74]. Interestingly, patients with low AR signaling measured by RNA-seq in pretreatment biopsies had longer radiographic PFS than patients with high AR signaling, suggesting that there are important AR-independent mechanisms that contribute to BETi response [74]. A phase I clinical trial investigating the BETi GSK525762 (molibresib) in combination with abiraterone or enzalutamide in CRPC is ongoing (NCT03150056; Table 1). Recently, a phase Ib/IIa clinical trial investigating the combination of the BETi PLX2853 with abiraterone or olaparib in metastatic CRPC has been launched (NCT04556617; Table 1). Finally, a new phase II trial combining enzalutamide, ZEN003694, and the immune checkpoint inhibitor pembrolizumab in men with CRPC (NCT04471974) has recently been launched (Table 1). Of note, this trial includes a cohort with NEPC—tumors with reduced reliance on the AR for survival. Taken together, these data suggest that BETi is a promising approach worthy of further study. Further, results from ongoing clinical studies will determine the potential of using BETi with immunotherapy or AR targeting therapies to treat advanced prostate cancer.
Table 2 –
Published clinical trials in prostate cancer with drugs targeting epigenetic factors
| S. no. | Drug | Target | Combination | Phase | Indication | Trial identifier | PubMed ID | Year published |
|---|---|---|---|---|---|---|---|---|
| 1 | LBH589 | HDAC | 2 | Metastatic castration-resistant prostate cancer | NCT00667862 | 23820963 | 2013 | |
| 2 | Valproic acid | HDAC | Bevacizumab | 1 | Metastatic castration-resistant prostate cancer | NCT00530907 | 24435060 | 2014 |
| 3 | Azacitidine | DNMT | Docetaxel/prednisone | 1, 2 | Metastatic castration-resistant prostate cancer | NCT00503984 | 25178642 | 2015 |
| 4 | SB939 | HDAC | 2 | Metastatic castration-resistant prostate cancer | NCT01075308 | 25983041 | 2015 | |
| 5 | MK-8628 | BET | 1b | Metastatic castration- resistait prostate cancer | NCT02259114 | 29733771 | 2018 | |
| 6 | LBH589 | HDAC | Bicalutamide | 2 | Metastatic castration-resistant prostate cancer | NCT00878436 | 30224345 | 2019 |
| 7 | ABBV-075 | BET | 1 | Metastatic castration-resistant prostate cancer | NCT02391480 | 31420359 | 2019 | |
| 8 | ZEN003694 | BET | Enzalutamide | 1, 2 | Metastatic castration-resistant prostate cancer | NCT02711956 | 32694156 | 2020 |
BET = bromodomain and extraterminal; HDAC = histone deacetylase.
4. Conclusions
Prostate cancer is a complex disease driven by both genetic and epigenetic factors. A greater understanding of the role of epigenetic factors in prostate cancer development or progression is predicted to lead to new approaches to treat or prevent this disease. In addition, it is clear that measurement of the expression of epigenetic factors or epigenetic marks may improve our ability to diagnose prostate cancer earlier, provide more accurate prognostic information, or treat patients more precisely. Further studies in larger datasets are warranted to establish more epigenetic biomarkers for clinical use.
Emerging data on epigenetic factors extend the spectrum of promising targets from DNMTs to other chromatin modifiers or readers. Recent preclinical studies and clinical trials highlight the potential of epigenetic therapies plus standard prostate cancer treatments such as AR targeting agents [74]. Integration of data from epigenomic and genomic analyses may shed further light on novel targetable vulnerabilities in prostate cancer. Importantly, clinical trials targeting epigenetic factors alone or in combination with AR targeting agents are a particularly active research area. Clinical trials with DNMT inhibition (NCT03709550), p300/CBP inhibition (NCT03568656 and NCT04575766), HDAC inhibition (NCT03829930), BET inhibition (NCT03150056, NCT04556617, and NCT04471974), and EZH2 inhibition (NCT04179864, NCT03480646, NCT03741712, NCT04388852, and NCT03460977) are ongoing (Table 1). Analysis of pretreatment tumor samples from these trials with genomic, proteomic, and epigenomic assays (eg, DNA methylation, histone modifications, and chromatin accessibility) may provide a greater understanding of specific subsets of patients who are most likely to respond, and analysis of on-treatment or progression samples may provide a clearer understanding of whether these drugs engage their targets and how resistance develops. The results of these trials and ongoing preclinical studies may provide a useful roadmap for how to test these agents in specific prostate cancer patient populations, so that we might one day make precision epigenetic therapy a part of our treatment paradigm in the clinic.
Funding/Support and role of the sponsor:
Felix Y. Feng: Benioff Initiative for Prostate Cancer Research. Joshi J. Alumkal: NCI R01 CA251245, the Pacific Northwest Prostate Cancer SPORE/NCI (P50 CA097186); the Michigan Prostate SPORE/NCI (P50 CA186786 and P50 CA186786-07S1); University of Michigan Rogel Innovation Award (P30 CA046592); University of Michigan Rogel Scholar Award; and Department of Defense (DOD) Idea Award (PC190147).
Financial disclosures:
Joshi J. Alumkal certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (e.g., employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Shuang G. Zhao: patent applications on molecular signatures in prostate cancer with GenomeDx Biosciences and Celgene, and in breast cancer with PFS Genomics, none directly relevant to this work; a family member has a leadership role at PFS Genomics. Felix Y. Feng: personal fees from Astellas, Bayer, Blue Earth Diagnostics, Celgene, Genentech, Janssen, Myovant, Roivant, and Sanofi; he also is a cofounder of PFS Genomics and serves on the Scientific Advisory Board for SerImmune; his institution University of California at San Francisco has received research funding from Zenith Epigenetics. Joshi J. Alumkal: personal fees from Dendreon, Merck Sharp & Dohme, and Astellas Pharma; his prior institution Oregon Health & Science University received research funding from Aragon Pharmaceuticals, Astellas Pharma, Zenith Epigenetics, and Gilead Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–53. [DOI] [PubMed] [Google Scholar]
- [2].Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cancer Genome Atlas Research N. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;128:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016;30:733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov 2018;8:24–36. [DOI] [PubMed] [Google Scholar]
- [8].Geybels MS, Zhao S, Wong CJ, et al. Epigenomic profiling of DNA methylation in paired prostate cancer versus adjacent benign tissue. Prostate 2015;75:1941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kirby MK, Ramaker RC, Roberts BS, et al. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer 2017;17:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Houlahan KE, Shiah YJ, Gusev A, et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med 2019;25:1615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gerhauser C, Favero F, Risch T, et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell 2018;34:996–1011.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Patel PG, Wessel T, Kawashima A, et al. A three-gene DNA methylation biomarker accurately classifies early stage prostate cancer. Prostate 2019;79:1705–14. [DOI] [PubMed] [Google Scholar]
- [13].Zhao SG, Chen WS, Li H, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet 2020;52:778–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;174:758–69 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Beltran H, Romanel A, Conteduca V, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest 2020;130:1653–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu A, Cremaschi P, Wetterskog D, et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J Clin Invest 2020;130:1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Partin AW, Van Neste L, Klein EA, et al. Clinical validation of an epigenetic assay to predict negative histopathological results in repeat prostate biopsies. J Urol 2014;192:1081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fraser M, Sabelnykova VY, Yamaguchi TN, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64. [DOI] [PubMed] [Google Scholar]
- [20].Gordevicius J, Krisciunas A, Groot DE, et al. Cell-free DNA modification dynamics in abiraterone acetate-treated prostate cancer patients. Clin Cancer Res 2018;24:3317–24. [DOI] [PubMed] [Google Scholar]
- [21].Storebjerg TM, Strand SH, Hoyer S, et al. Dysregulation and prognostic potential of 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) levels in prostate cancer. Clin Epigenetics 2018;10:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nickerson ML, Das S, Im KM, et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 2017;36:2172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Takayama K, Misawa A, Suzuki T, et al. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat Commun 2015;6:8219. [DOI] [PubMed] [Google Scholar]
- [24].Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer 2015;13:22–31. [DOI] [PubMed] [Google Scholar]
- [25].Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21:381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 2007;8:284–95. [DOI] [PubMed] [Google Scholar]
- [27].Pomerantz MM, Qiu X, Zhu Y, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet 2020;52:790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stelloo S, Nevedomskaya E, Kim Y, et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun 2018;9:4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu J, He D, Cheng L, et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 2020;39:3939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Welti J, Sharp A, Brooks N, et al. Targeting p300/CBP axis in lethal prostate cancer. Cancer Discov. In press. 10.1158/2159-8290.CD-20-0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Burger M, Chory J. Structural and chemical biology of deacetylases for carbohydrates, proteins, small molecules and histones. Commun Biol 2018;1:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruscetti M, Dadashian EL, Guo W, et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016;35:3781–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Eigl BJ, North S, Winquist E, et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest New Drugs 2015;33:969–76. [DOI] [PubMed] [Google Scholar]
- [35].Ferrari AC, Alumkal JJ, Stein MN, et al. Epigenetic therapy with panobinostat combined with bicalutamide rechallenge in castration-resistant prostate cancer. Clin Cancer Res 2019;25:52–63. [DOI] [PubMed] [Google Scholar]
- [36].Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- [37].Peitzsch C, Cojoc M, Hein L, et al. An epigenetic reprogramming strategy to resensitize radioresistant prostate cancer cells. Cancer Res 2016;76:2637–51. [DOI] [PubMed] [Google Scholar]
- [38].Alumkal JJ, Sun D, Lu E, et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc Natl Acad Sci U S A 2020;117:12315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beltran H, Hruszkewycz A, Scher HI, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res 2019;25:6916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dardenne E, Beltran H, Benelli M, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016;30:563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim J, Lee Y, Lu X, et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep 2018;25:2808–20 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xiao L, Tien JC, Vo J, et al. Epigenetic reprogramming with antisense oligonucleotides enhances the effectiveness of androgen receptor inhibition in castration-resistant prostate cancer. Cancer Res 2018;78:5731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Crea F, Sun L, Mai A, et al. The emerging role of histone lysine demethylases in prostate cancer. Mol Cancer 2012;11:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Verrier L, Vandromme M, Trouche D. Histone demethylases in chromatin cross-talks. Biol Cell 2011;103:381–401. [DOI] [PubMed] [Google Scholar]
- [46].Fan L, Peng G, Sahgal N, et al. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2016;35:2441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liang Y, Ahmed M, Guo H, et al. LSD1-mediated epigenetic reprogramming drives CENPE expression and prostate cancer progression. Cancer Res 2017;77:5479–90. [DOI] [PubMed] [Google Scholar]
- [48].Gao S, Chen S, Han D, et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat Genet 2020;52:1011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sehrawat A, Gao L, Wang Y, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A 2018;115:E4179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Coleman DJ, Sampson DA, Sehrawat A, et al. Alternative splicing of LSD1+8a in neuroendocrine prostate cancer is mediated by SRRM4. Neoplasia 2020;22:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Komura K, Jeong SH, Hinohara K, et al. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc Natl Acad Sci U S A 2016;113:6259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Duan L, Chen Z, Lu J, et al. Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res 2019;47:11623–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Duan L, Rai G, Roggero C, et al. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol 2015;22:1185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sarac H, Morova T, Pires E, et al. Systematic characterization of chromatin modifying enzymes identifies KDM3B as a critical regulator in castration resistant prostate cancer. Oncogene 2020;39:2187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang HJ, Pochampalli M, Wang LY, et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019;38:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Augello MA, Liu D, Deonarine LD, et al. CHD1 loss alters AR binding at lineage-specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell 2019;35:603–17 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cyrta J, Augspach A, De Filippo MR, et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat Commun 2020;11:5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang Z, Zhou C, Li X, et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell 2020;37:584–98 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Urbanucci A, Barfeld SJ, Kytola V, et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep 2017;19:2045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Welti J, Sharp A, Yuan W, et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin Cancer Res 2018;24:3149–62. [DOI] [PubMed] [Google Scholar]
- [61].Asangani IA, Dommeti VL, Wang X, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Faivre EJ, McDaniel KF, Albert DH, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306–10. [DOI] [PubMed] [Google Scholar]
- [63].Dai X, Gan W, Li X, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 2017;23:1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang P, Wang D, Zhao Y, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med 2017;23:1055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Coleman DJ, Gao L, King CJ, et al. BET bromodomain inhibition blocks the function of a critical AR-independent master regulator network in lethal prostate cancer. Oncogene 2019;38:5658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Coleman DJ, Gao L, Schwartzman J, et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci Rep 2019;9:3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Asangani IA, Wilder-Romans K, Dommeti VL, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res 2016;14:324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li X, Baek G, Ramanand SG, et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep 2018;22:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vazquez R, Civenni G, Kokanovic A, et al. Efficacy of novel bromodomain and extraterminal inhibitors in combination with chemotherapy for castration-resistant prostate cancer. Eur Urol Oncol. In press. 10.1016/j.euo.2019.07.013 [DOI] [PubMed] [Google Scholar]
- [70].Shah N, Wang P, Wongvipat J, et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife 2017;6:e27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Raina K, Lu J, Qian Y, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016;113:7124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lewin J, Soria JC, Stathis A, et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol 2018;36:3007–14. [DOI] [PubMed] [Google Scholar]
- [73].Piha-Paul SA, Sachdev JC, Barve M, et al. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin Cancer Res 2019;25:6309–19. [DOI] [PubMed] [Google Scholar]
- [74].Aggarwal RR, Schweizer MT, Nanus DM, et al. A phase Ib/IIa study of the Pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res 2020;26:5338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]