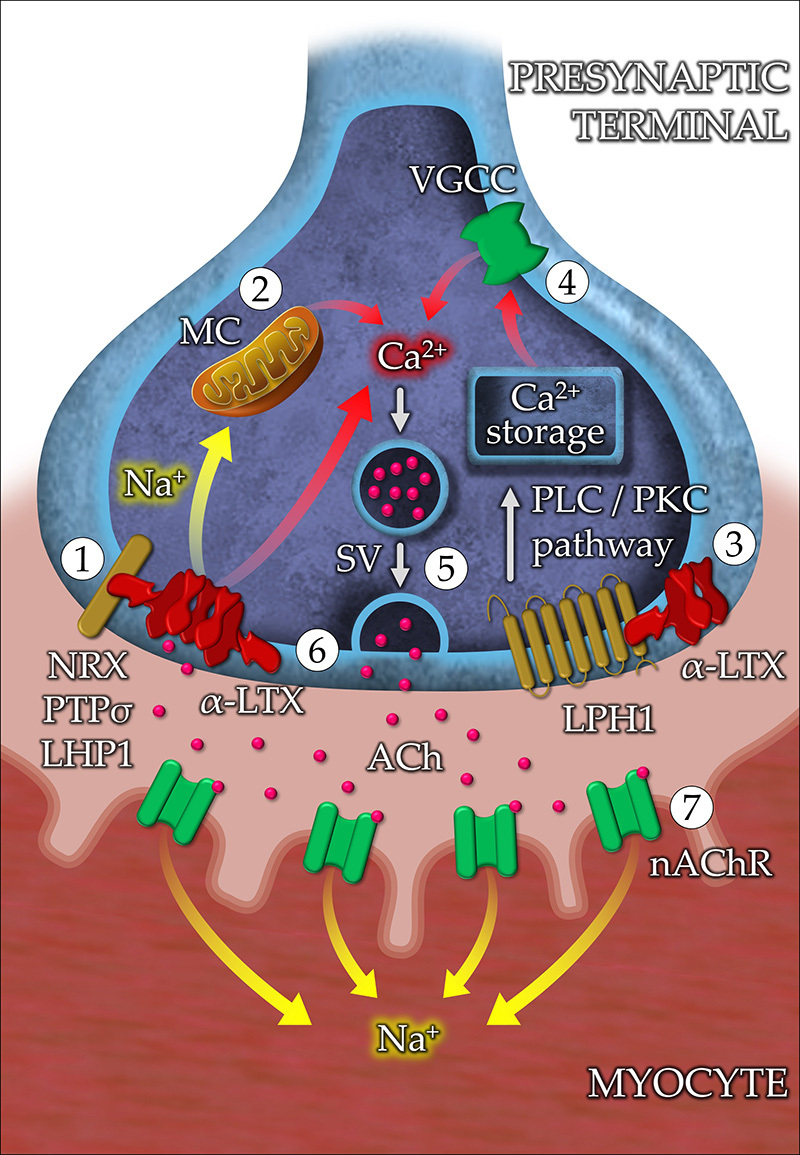
Figure 3. Proposed mechanisms of neurotoxicity of α-LTX in the neuromuscular junction. (1) In the presynaptic membrane, the wing region (N-terminal domain) of α-latrotoxin (α-LTX) interacts with neurexin-Iα (NXR-Iα), protein tyrosine phosphatase σ (PTPσ), and latrophilin 1 (LPH1). This interaction facilitates the insertion of the tetramer into the lipid bilayer, allowing the influx of monovalent (Li+, Cs+, Na+, K+) and divalent (Ca2+, Ba2+, Mg2+) alkali cations. (2) The excess of Na+ induces the release of more Ca2+ from the mitochondria into the cytoplasm. (3) α-LTX triggers LPH1 signaling, resulting in the activation of the phospholipase C (PLC)/protein kinase C (PKC) pathway. PLC increases the levels of inositol trisphosphate, promoting the release of intracellular Ca2+ stored in the endoplasmic reticulum. (4) The depolarization of the presynaptic terminal activates voltage-gated calcium channels (VGCC), allowing the influx of more Ca2+. (5) The massive increase in intracellular Ca2+ promotes the mobilization of synaptic vessels (SV) containing the neurotransmitter acetylcholine (Ach). (6) The release of Ach into the synaptic cleft can be either due to the fusion of SVs with the plasma membrane or due to the leakage of the neurotransmitter through the α-LTX pore. (7) The interaction of Ach with the nicotinic acetylcholine receptor (nAChR) present in the membrane of the myocyte causes depolarization and muscle contraction.