
Figure 1.
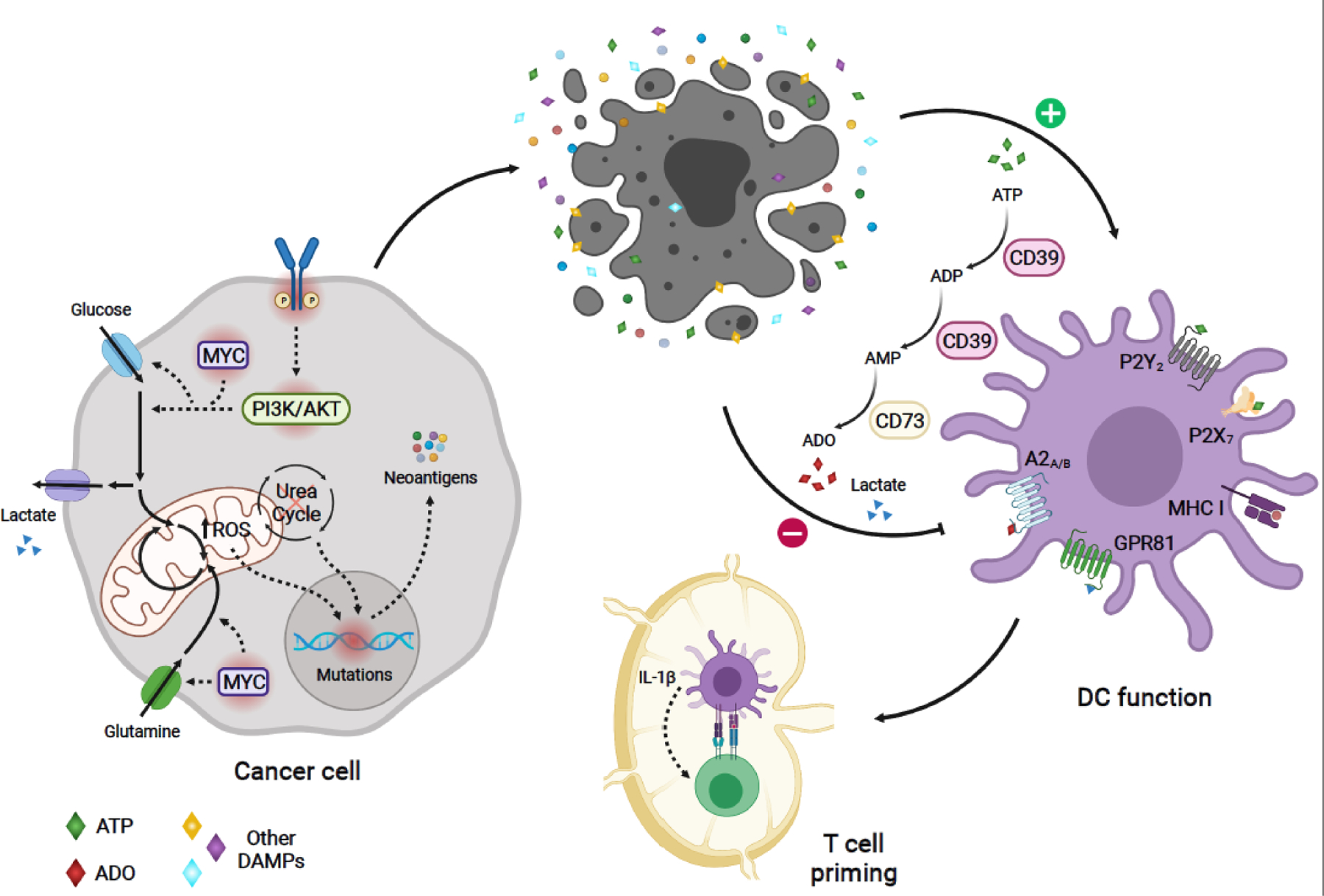
Model of metabolic regulation of tumor immunogenicity: Oncogenic-driven metabolic rewiring required for cancer cell growth can promote tumor immunogenicity. Oncogenic signaling (e.g. PI3K and MYC pathways), can boost nutrient uptake, leading to increased production of mitochondrial ROS and ROS-mediated mutagenesis [12]. Altered expression of urea cycle enzymes and transporters can result in urea cycle dysregulation, promoting diversion of nitrogen to pyrimidine synthesis, leading to increased purine to pyrimidine transversion mutations [21]. These metabolic-driven mutations generating tumor antigens can activate antitumor immune responses. Aberrant tumor growth leads to cell death and release of tumor antigens. In addition, dying cells in the TME release DAMPs, which enhance DC-mediated capture and presentation of cancer antigens to tumor antigen-specific CD8+ T-cells, as well as, priming and activation of these T-cells. One of such DAMPs, ATP, stimulates antitumor function of DCs in humans and mice [57,58]. However, sequential catabolism of ATP -- mediated by the ectonucleotidases CD39 and CD73 -- may lead to the production of ADO, which can suppress DC function through A2A and A2B receptor-mediated signaling in humans and mice [57,59–61]. Metabolic-derived lactate secreted by certain cancer cells can also hinder DC function through activation of the GPR81 receptor, as evidenced in breast cancer mouse models [73]. Solid arrows depict metabolic reactions, metabolite uptake and secretion, and modulation of cellular phenotypes. Dashed arrows indicate signaling or mutation-associated events. Oncogenic mutated molecules are surrounded by a red shadow. A2A, Adenosine A2a receptor; A2B, Adenosine A2b receptor; ADO, adenosine; DAMPs, damage-associated molecular patterns;; DC, dendritic cell; GPR81, G protein-coupled receptor 81;, bHLH transcription factor; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; ROS, reactive oxygen species; TME, tumor microenvironment. Figure created with BioRender.com