

Abstract
Rett syndrome is a neurodevelopmental disorder caused predominantly by loss‐of‐function mutations in MECP2, encoding transcriptional modulator methyl‐CpG‐binding protein 2 (MeCP2). Although no disease‐modifying therapies exist at this time, some proposed therapeutic strategies aim to supplement the mutant allele with a wild‐type allele producing typical levels of functional MeCP2, such as gene therapy. Because MECP2 is a dosage‐sensitive gene, with both loss and gain of function causing disease, these approaches must achieve a narrow therapeutic window to be both safe and effective. While MeCP2 supplementation rescues RTT‐like phenotypes in mouse models, the tolerable threshold of MeCP2 is not clear, particularly for partial loss‐of‐function mutations. We assessed the safety of genetically supplementing full‐length human MeCP2 in the context of the R294X allele, a common partial loss‐of‐function mutation retaining DNA‐binding capacity. We assessed the potential for adverse effects from MeCP2 supplementation of a partial loss‐of‐function mutant and the potential for dominant negative interactions between mutant and full‐length MeCP2. In male hemizygous R294X mice, MeCP2 supplementation rescued RTT‐like behavioral phenotypes and did not elicit behavioral evidence of excess MeCP2. In female heterozygous R294X mice, RTT‐specific phenotypes were similarly rescued. However, MeCP2 supplementation led to evidence of excess MeCP2 activity in a motor coordination assay, suggesting that the underlying motor circuitry is particularly sensitive to MeCP2 dosage in females. These results show that genetic supplementation of full‐length MeCP2 is safe in males and largely so females. However, careful consideration of risk for adverse motor effects may be warranted for girls and women with RTT.
Keywords: gene therapy, MECP2 duplication syndrome, methyl‐CpG‐binding protein 2, mouse model, Rett syndrome
RTT‐ and MECP2 duplication syndrome‐like behavioral phenotypes are rescued in female mice with the R294X Mecp2 mutation and a transgene overexpressing MeCP2 protein.
1. INTRODUCTION
Rett syndrome (RTT) is an X‐linked neurodevelopmental disorder caused predominantly by de novo mutations in the MECP2 gene encoding transcriptional modulator methyl‐CpG‐binding protein 2 (MeCP2). 1 , 2 Because MECP2 mutations are largely paternally inherited, RTT occurs mostly in females. 3 However, males with MECP2 mutations manifest a variety of clinical phenotypes as well. 4 Individuals with RTT experience typical development until approximately 18–30 months, at which age affected individuals experience developmental regression with loss of previously acquired skills such as expressive language and purposeful hand use, as well as onset of hand stereotypies and gait abnormalities. 5 , 6 RTT can also cause a variety of associated clinical features including seizures, breathing dysfunction, and autonomic abnormalities. 7 , 8 , 9 Promisingly, restoration of MeCP2 pre‐ or postsymptomatically has been shown to rescue premature death and gross RTT‐like phenotypes in mice, providing hope that disease‐modifying therapies could be developed for RTT. 10 , 11 , 12 , 13 While early clinical trials have shown some therapeutic promise, no disease‐modifying therapies are currently available. 14 , 15
One proposed therapeutic approach for RTT is supplementing the MECP2 mutant allele with a wild‐type (WT) MECP2 allele to produce typical levels of functioning protein, including specific strategies such as gene therapy and reactivation of MECP2 on the inactive X‐chromosome. Gene therapy achieved via viral delivery of MECP2, for example, has shown rescue of survival, gross phenotypic development and some behavioral phenotypes in mouse models of RTT. 12 , 13 , 16 , 17 , 18 One primary challenge to the realization of these therapies is that MECP2 is a dosage‐sensitive gene, and protein levels are tightly regulated bidirectionally in vivo. While loss of MeCP2 function causes RTT in humans, duplication and occasionally triplication of MECP2 with resultant gain of protein function causes a related neurodevelopmental disorder called MECP2 duplication syndrome (MDS). 19 , 20 RTT and MDS share some clinical features such as motor dysfunction, stereotypic movements, gait abnormalities and seizures. However, there are differences in onset and severity of these shared features. 21 MDS can also be differentiated from RTT by its predominance in males and associated immunodeficiency, which causes recurrent respiratory infections in early life. Given this narrow therapeutic window of MECP2 dosage, interventions for RTT that supplement full‐length MeCP2 protein must do so without eliciting excess MeCP2 levels and MDS‐like adverse effects.
The risk of MDS‐like adverse effects is predicted to depend on baseline level of functioning MeCP2, with lower and higher levels decreasing and increasing risk, respectively. A main factor influencing baseline level of functioning MeCP2 is mutation type. Specific MECP2 mutation type impacts clinical severity in humans, likely because of different mutations' impacts on protein function. 22 Mutations that cause significant disruption of DNA binding capacity (e.g., R106W) or do not produce any protein product (e.g., R255X) cause disease with higher clinical severity in people. Mutations that are predicted to retain partial DNA binding (e.g., R133C) or co‐repressor interaction capacity (e.g., R294X) cause disease with lower clinical severity in people. These genotype–phenotype correlations have been recapitulated in mouse models and suggest that partial loss‐of‐function mutations would confer higher risk for MDS‐like adverse effects with MeCP2 supplementation. 23 , 24 , 25 , 26 Previous work has evaluated MeCP2 supplementation in the context of loss‐of‐function mutations T158M, R255X and R306C. 16 , 23 , 27 , 28 However, these studies focused largely on mutations associated with higher clinical severity and on rescue of RTT‐like phenotypes in male mice. Further preclinical evaluation of the MeCP2 dosage therapeutic window in the context of partial loss‐of‐function mutations, particularly in female mice, is needed to determine the safety of therapeutic MeCP2 supplementation approaches.
To address this, we used a mouse model of the R294X MECP2 mutation. 25 R294X is a common mutation that truncates the protein within the NCoR interaction domain (NID) and eliminates the entire C‐terminus. 29 In humans and in mice, the R294X mutation is associated with milder disease compared with other MECP2 mutations, likely attributable to the presence of a stable truncated protein product. 22 , 25 , 30 Because this truncation product lacks the domain required for co‐repressor interaction, the R294X mutation is predicted to cause RTT via partial loss‐of‐function. Moreover, the R294X truncation product binds chromatin more tightly than WT MeCP2, creating the potential for dominant negative interactions with supplementation of full‐length MeCP2. 25 For these two reasons, the R294X mouse model is advantageous to study the feasibility of therapeutic approaches aimed at restoring levels of functional MeCP2.
A fundamental question for therapeutic development is whether approaches that restore MeCP2 function, such as gene therapy, are safe and efficacious in partial loss‐of‐function mutations that cause RTT, especially those that produce a truncated protein with both the ability and an increased avidity to bind DNA, such as R294X. To answer this question, we crossed mice harboring the R294X allele with mice carrying a human MECP2 transgene on an autosome, as a model of idealized gene therapy. We found that addition of MECP2 transgene normalized MeCP2 expression level in both male and female mice, and behavioral phenotypes because of MeCP2 loss‐of‐function were rescued in both sexes. We did not observe adverse MDS‐like behavioral phenotypes in male R294X animals. However, we observed MDS‐like motor coordination and gait phenotypes in female R294X mice expressing the MECP2 transgene. Collectively, these findings suggest that genetic supplementation of MeCP2 in the context of the partial loss‐of‐function R294X mutation is safe and effective in males, and largely so in females. However, careful consideration of baseline motor function and risk for potential adverse motor effects with MeCP2 supplementation therapy may be warranted for girls and women with RTT.
2. MATERIALS AND METHODS
2.1. Mouse care and breeding
All methods and animal care procedures were approved by the Vanderbilt Animal Care and Use Committee. Mice were housed in AAALAC‐approved facilities at Vanderbilt University. All mice were bred on a C57BL/6J background. Male WT C57BL/6J mice were mated to female Mecp2 R294X/+;MECP2 Tg mice to generate male and female experimental mice, including Mecp2 +/Y and Mecp2 +/+ (WT), Mecp2 R294X/Y and Mecp2 R294X/+ (R294X), MECP2 Tg (TG), and Mecp2 R294X/Y;MECP2 Tg and Mecp2 R294X/+;MECP2 Tg (R294X TG) mice. 23 , 25 , 31 Mice were genotyped as previously described using allele‐specific PCR. 25 All experiments were performed using WT littermates as controls.
2.2. Mouse behavior
All behavioral experiments were performed in the Vanderbilt University Neurobehavioral Core Facility. Behavioral tests were performed using male and female mice. Mice were group housed (3–5 mice/cage) on a 12‐h light/dark cycle with food and water available ad libitum. Mice were transferred to test rooms and acclimated to the environment for 30 min prior to testing. Unless otherwise stated, all equipment was cleaned with 70% ethanol between trials to provide a standardized testing environment. All behavioral experiments were performed during the light phase.
Behavioral batteries were conducted across three independent cohorts for both males and females. Male mice underwent a behavioral battery at 20–22 weeks of age, including the following tests in order: elevated zero maze, open field, whole‐body plethysmography, rotarod, Crawley 3‐chamber and fear conditioning. Female mice underwent a behavioral battery at 30–31 weeks of age, including the following tests in order: open field, whole‐body plethysmography, rotarod, fear conditioning and forced gait.
2.2.1. Physical characterization
Mouse weights were measured prior to general phenotyping (described below). Mouse lengths were measured as distance from nose to base of the tail.
2.2.2. General phenotyping
Mice were scored for development of Rett‐like phenotypes based on Bird scoring. 10 Phenotypes including activity level, hindlimb clasping, tremor, breathing irregularity, coat condition, hunch and gait were scored in a clean cage containing only bedding. Scores were recorded as 0, 1 or 2 for absent, present or severe phenotype according to the following table:
| Phenotype | Score of 0 (absent) | Score of 1 (present) | Score of 2 (severe) |
|---|---|---|---|
| Activity | Typically responsive | Minimally responsive | Nonresponsive |
| Hindlimb clasping | Legs typically spread | One leg held against body | Both legs held against body |
| Tremor | No tremor | Intermittent tremor | Continuous tremor |
| Breathing | Typical breathing | Hyperventilation or pauses in breathing | Gasps or panting |
| Coat condition | Typically groomed fur | Fur poorly groomed on close inspection (during handling) | Fur obviously poorly groomed (noticed immediately when cage is opened) |
| Hunch | No hunch | Hunch present on palpation | Hunch visualized without palpation |
| Gait | Typical gait | Wide stance or waddling gait | Uncoordinated or “hopping” gait |
2.2.3. Elevated zero maze
Anxiety‐like behavior was assessed using a white elevated zero maze (Stoelting: 50 cm inner diameter, 5 cm lane width, 15 cm closed arm wall height and 50 cm apparatus leg height). Illuminance was approximately 300 lux in open arms and approximately 70 lux in closed arms. Animals were selected in random order to minimize handling prior to testing. Animals were placed in the center of one open arm of the maze at the beginning of each trial. At the end of each trial, test mice were placed in a new cage to leave untested cage mates undisturbed. Each trial was recorded by a video camera mounted to the ceiling, and data were analyzed by ANY‐maze software (Stoelting).
2.2.4. Open field
Spontaneous locomotor activity was assessed in MedAssociates 27 × 27 × 20.5 cm activity chambers placed within sound‐attenuating 64 × 27 × 42 cm boxes that were light‐ and air‐controlled. Locomotion was detected by the disruption of infrared beams with mouse body movement. Horizontal activity was measured by 32 photocells located 1 cm above the chamber floor (16 each in the X‐ and Y‐directions). Vertical activity was measured by 16 photocells located 4 cm above the chamber floor (in the Z‐direction). Mice were placed in the center of the activity chambers at the beginning of each trial, and their locomotion was tracked with the Activity Monitor software for 30 min.
2.2.5. Unrestrained whole‐body plethysmography
Respiratory function was assessed using unrestrained whole‐body plethysmography. Mice were placed into plethysmography chambers (DSI: Buxco, ~500 ml in volume) with a continuous air flow rate of 1 L/min. Breathing was recorded for 60 min and raw breathing data were collected through Ponemah software. Concurrent time‐locked chamber video data were collected to track mouse movement throughout the experiment. Between experiments, chambers were cleaned with water. A custom Python script was used to quantify movement and filter out segments of the experiment in which the mouse was active. The remaining “calm” segments were subjected to breathing waveform analysis. Derived parameters included breathing rate, breathing irregularity and apnea count. Breathing irregularity was calculated as the instantaneous change in breath duration between adjacent breaths. Apneas were called as pauses that were greater than 0.5 s in duration and also greater than twice the local and overall average breath duration. Apnea count was normalized to count per 1000 breaths. A minimum of 200 total called breaths was required for inclusion in analyses. One male mouse (R294X) and three female mice (2 WT, 1 R294X TG) met exclusion criteria.
2.2.6. Rotarod
Motor coordination was assessed with a five‐lane MedAssociates rotarod machine (ENV‐574M). The rubber rotarod drum was 3.18 cm in diameter and textured to prevent slippage. The drum was located 30.68 cm above the apparatus floor. Mice were loaded onto the rotarod at a rotation speed of 4 rpm. Once all animals were loaded, the rotarod was set to initiate preprogrammed acceleration from 4 to 40 rpm over a period of 5 min. The time to fall was manually recorded for a maximum time of 5 min. To account for passivity on the rotarod, if an animal completed two passive rotations (i.e., underwent two full rotations without making a step forward) it was considered to have fallen. Animals completed three trials a day for three consecutive days, with at least 30 min rest between trials. Time to fall was averaged across trials for each mouse for each day.
2.2.7. Crawley 3‐chamber
Social behavior was assessed using the Crawley 3‐chamber assay. 32 The 3‐chamber apparatus consists of a clear 60 × 42 × 22 cm box divided into three adjacent and equally sized compartments. Walls separating the compartments contained openings for mice to travel freely between compartments. Empty inverted wire pencil cups were placed in same‐sided corners of each the left and right compartments. The test was conducted in three consecutive stages on a single day. In Stage 1 (Habituation), mice were allowed to freely explore the apparatus for 5 min. In Stage 2 (Sociability), an unfamiliar sex‐, age‐ and weight‐matched WT mouse (Stranger 1) was placed under one of the pencil cups. Test mice were allowed to explore the apparatus for 7.5 min. The side of Stranger 1 was alternated between successive test mice and balanced across genotypes to account for potential side bias. In Stage 3 (Social Novelty Preference), another unfamiliar sex‐, age‐ and weight‐matched WT mouse (Stranger 2) was placed under the remaining pencil cup. Test mice were again allowed to explore the apparatus for 7.5 min. Stranger 1 and Stranger 2 were always derived from different home cages and had no previous contact with any experimental mice. Additionally, all stranger mice were habituated to the wire pencil cups by placing them underneath for 30 min/day for 2 days prior to testing. After Stage 3, test mice were placed in a new cage, which prevented contact with untested cage mates. A camera mounted to the ceiling captured video data for all stages. Video data were manually scored for location of the mouse (left, center, or right chamber) for Stages 1, 2 and 3. Video data were also manually scored for interaction with pencil cups (left or right pencil cup) for Stages 2 and 3. Interaction was defined as sniffing, pawing or rearing onto the cup. Scoring was performed with the free and open‐source Behavioral Observation Research Interactive Software (BORIS v7.9.7). Discrimination indices were calculated for Stage 2 (Sociability) and Stage 3 (Social Novelty Preference) according to the following calculations:
Sociability Discrimination Index:
Social Novelty Preference Discrimination Index:
2.2.8. Fear conditioning
Learning and memory was assessed using the fear conditioning assay. The assay consisted of one training day and one test day. On training day, mice were acclimated for 30 min in a dedicated acclimation room adjacent to the test room. When ready for testing, mice were placed into 29.53 × 23.5 × 20.96 cm MedAssociates chambers equipped with stainless steel grid floors capable of delivering an electric shock. Mice were allowed to freely explore for 2 min and then a 30‐s 90 dB tone stimulus was administered, with the final 2 s accompanied by a 0.5 mA foot shock. After an additional 2‐min period, this 30‐s tone‐shock pairing was repeated. Mice remained in the chamber for a final 1‐min period and were then removed. Mice were placed into a fresh cage to avoid contact with untested cage mates, within a recovery room adjacent to the test room. Twenty‐four hours later, mice were tested for context‐dependent fear memory by re‐introducing them to the training chamber for 4 min. Two hours later, mice were tested for cue‐dependent fear memory by placing them into an altered environment for 4 min, the final 2 min of which were accompanied by the 90 dB tone stimulus. This altered environment included red lighting in the acclimation/test/recovery rooms, lack of white light in the chamber, a flat baseboard, rounded walls, and vanilla scent. In both tests, a video camera mounted to the front door of the chamber recorded mouse movement. Percentage of time spent freezing was recorded across the full 4‐min context test and the final 2 min of the cue test (when the tone stimulus was played). Freezing was defined as behavior below a motion threshold of 18 arbitrary units for 30 frames (1 s) minimum freeze duration and % time freezing was calculated using the default linear method.
2.2.9. Forced gait analysis
Mouse gait was assessed using the CleverSys TreadScan for forced gait analysis. The TreadScan apparatus consists of a treadmill chamber placed above a transparent belt treadmill illuminated by external light sources. A high‐speed digital camera is placed underneath the treadmill chamber to capture ventral videos of mice on the treadmill. Mice were placed into the treadmill chamber and allowed to freely explore for 1 min. The treadmill was turned on and off at a speed of 20 cm/s until the test mouse was continuously running. A 20‐s video was then captured for footprint placement and movement analysis. Video files were automatically segmented using TreadScan software to include only periods of continual running. Video files were additionally manually clipped to eliminate periods when the mouse slowed running before a stop or exhibited lateral movement. This list of segments was subjected to footprint analysis with paw identification training modules built in accordance with the manufacturer's instructions. All analyzed gait metrics were weighted by number of counted steps per foot.
2.3. MeCP2 Western blotting
Mice were humanely euthanized and perfused with ice‐cold PBS prior to isolation of brain tissue and dissection into hemibrains. Hemibrains were homogenized in buffer containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.05% NP‐40, 0.5 mM DTT and 1× Mammalian Protease Inhibitor Cocktail (Sigma P8340), pH 7.9. The cytosolic fraction was removed following centrifugation and nuclear pellets were resuspended in buffer containing 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.5 mM DTT and 1× Mammalian Protease Inhibitor Cocktail, pH 7.9. Suspended nuclei were treated with benzonase for 1 h on ice (Sigma E1014). Following nuclease treatment, NaCl concentration was increased to 600 mM and nuclei were incubated on ice for 1 h. Cellular debris was removed by high‐speed centrifugation and nuclear protein concentration was determined with the 660 nm Protein Assay Reagent (Pierce, Waltham, MA). 5ug of nuclear protein was loaded into individual wells of 10% house‐made acrylamide SDS‐PAGE gels and subjected to standard Western blotting procedures. An Odyssey CLx (LI‐COR, Lincoln, NE) imaging system was used for blot detection and quantification was performed in Image Studio Lite (LI‐COR). Full‐length MeCP2 was detected with an antibody specific to the carboxy terminus of MeCP2 (Cell Signaling, Danvers, MA; D4F3, monoclonal rabbit anti‐C'MeCP2, 1:5000 dilution). R294X MeCP2 was detected with an antibody specific to the amino terminus of MeCP2 (Sigma; M7443, monoclonal mouse anti‐N' MeCP2, 1:1000 dilution). TATA‐binding protein (TBP) was detected as a loading control (Full‐length MeCP2 Westerns: abcam ab51841, monoclonal mouse anti‐TBP, 1:2000 dilution) (R294X MeCP2 Westerns: Cell Signaling 8515S, polyclonal rabbit anti‐TBP, 1:2000 dilution). Secondary antibodies used for detection were goat anti‐rabbit 800CW (LI‐COR 926‐32211, 1:10,000 dilution) and goat anti‐mouse 680RD (LI‐COR 926‐68070, 1:10,000 dilution).
2.4. Statistical analysis
Statistical analysis was performed within Prism 9 (GraphPad, San Diego, CA). Behavioral data were analyzed with one‐way ANOVA (factor = genotype) and Dunnett's post‐hoc comparison to WT. Rotarod and fear conditioning training data were analyzed with two‐way repeated measures ANOVA (factors = genotype, time) and Dunnett's post‐hoc comparison to WT. Quantitative Western blots were analyzed with one‐way ANOVA (factor = genotype) and Dunnett's post‐hoc comparison to WT (Figure S1(A), (B)) or unpaired t‐test (Figure S1(C), (D)). All plots display mean ± standard error of the mean. Statistical significance is represented in all plots as follows: *p <0.05, **p <0.01, ***p <0.001 and ****p <0.0001.
3. RESULTS
3.1. Gross RTT‐like phenotypes are rescued with MECP2 transgene in male and female R294X mice
We set out to determine whether genetically introducing full‐length MeCP2 in the context of the partial loss‐of‐function R294X allele could rescue RTT‐like phenotypes without eliciting adverse MDS‐like effects. To do so, we drew on an established mouse model of MDS that expresses an autosomal human MECP2 transgene under its native promoter. 31 These transgenic mice (MECP2 Tg) express full‐length MeCP2 at levels approximately twice that of endogenous mouse MeCP2 in males and females (Figure S1(A), (B)). 28 , 31 MECP2 Tg mice display some phenotypes, such as those relevant to motor behavior and cognition, that are opposite to those observed in mice with Mecp2 loss‐of‐function mutations, providing a behavioral readout of MeCP2 dosage. 23 , 31 , 33 , 34 We crossed mice harboring the Mecp2 R294X allele with MECP2 Tg mice to generate Mecp2 +/Y and Mecp2 +/+ (WT), Mecp2 R294X/Y and Mecp2 R294X/+ (R294X), MECP2 Tg (TG), and Mecp2 R294X/Y;MECP2 Tg and Mecp2 R294X/+;MECP2 Tg (R294X TG) mice. 25 R294X mice of both sexes express truncated MeCP2, and this is supplemented by transgenic full‐length MeCP2 in R294X TG animals (Figure S1(C), (D)). We assayed and analyzed male and female cohorts separately because MECP2 mutations cause RTT in females and variety of clinical phenotypes in males, including a subset with a more severe clinical course compared with females with RTT (RTT encephalopathy). 2 , 4 , 5
We first evaluated gross physical features and symptomatic progression in male and female mice. Consistent with previous characterization, we found that male R294X mice have low body weight compared with WT littermates as early as 4 weeks of age (Figure 1(A)). 25 Additionally, male R294X mice display progression of RTT‐like symptoms (Figure 1(B)). 10 Both of these phenotypes are rescued with addition of MECP2 transgene, as male R294X TG mice are indistinguishable from WT littermates. Unlike the males, we found that female R294X mice do not have a low body weight phenotype (Figure 1(C)). However, female R294X mice do display progression of RTT‐like symptoms, and these are also rescued to WT levels by addition of MECP2 transgene (Figure 1(D)).
FIGURE 1.
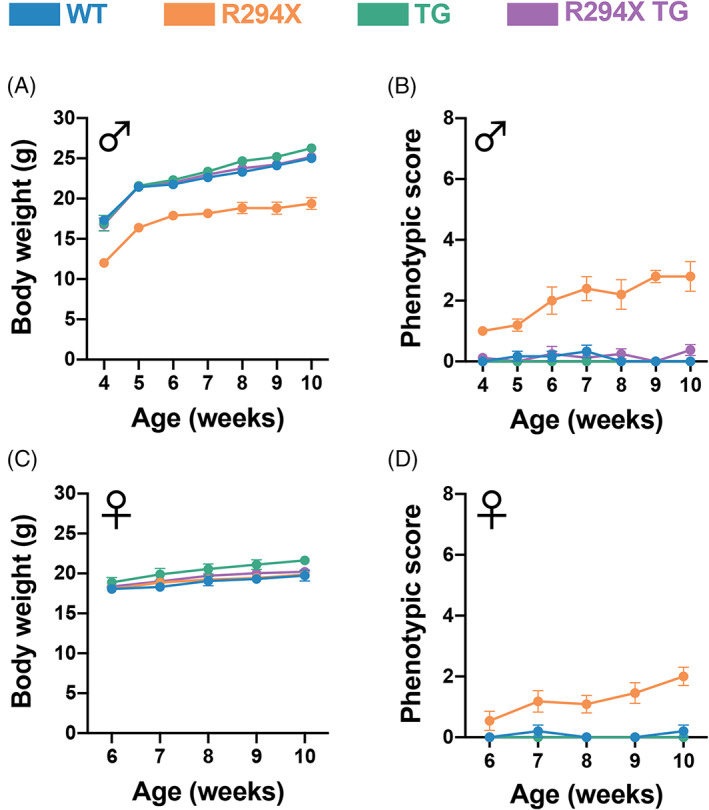
Gross RTT‐like phenotypes are rescued with MECP2 transgene in male and female R294X mice. (A) Body weights of male R294X mice are lower than that of WT littermates at 4 weeks of age and beyond. This phenotype is rescued by MECP2 transgene in male R294X TG mice, whose body weights are not different from WT. (WT n = 6, blue; R294X n = 5, orange; TG n = 9, green; R294X TG n = 8, purple) (B) Male R294X mice exhibit progression of RTT‐like phenotypes, as reflected by an increase in phenotypic score, which is rescued by MECP2 transgene. (WT n = 6; R294X n = 5; TG n = 9; R294X TG n = 8) (C) Female R294X mouse body weights are not different from that of WT littermates. (WT n = 5; R294X n = 11; TG n = 7; R294X TG n = 9) (D) Female R294X mice exhibit progression of RTT‐like phenotypes, as reflected by an increase in phenotypic score, which is rescued by MECP2 transgene. (WT n = 5; R294X n = 11; TG n = 7; R294X TG n = 9)
3.2. RTT‐specific breathing phenotypes are rescued with MECP2 transgene in male R294X mice
To test the hypothesis that MeCP2 supplementation provides safe and effective behavioral rescue in R294X mice, we conducted a behavioral battery including assays for RTT‐specific phenotypes and assays for bidirectionally dosage‐sensitive phenotypes. We evaluated these phenotypes at different ages for male (20–22 weeks old) and female mice (30–31 weeks old, with exception of gait performed at 34–40 weeks old), as male mice lacking MeCP2 function have an earlier onset of phenotypic abnormalities compared with female RTT mice. This is expected, as male Mecp2 mutant mice are hemizygous for the mutation. Here we present results from the male mice as in this condition all somatic cells should express both the Mecp2 R294X and MECP2 TG alleles, providing a genetically homogenous readout of MeCP2 supplementation. The batteries included tests for spontaneous locomotor activity, anxiety‐like behavior, breathing function, motor coordination, sociability and social novelty preference, learning and memory and gait (Figure S2).
Breathing disturbances are highly prevalent in RTT and, although complex and individually variable, often include bouts of hypoventilation alternating with irregular breathing or hyperventilation. 35 Mice with common Mecp2 mutations also reproducibly display breathing dysfunction. 23 , 25 , 27 , 36 To investigate breathing function in the context of the R294X allele, we conducted unrestrained whole‐body plethysmography experiments in male WT, R294X, TG and R294X TG mice. 37 , 38 , 39 We found that male R294X mice display abnormal breathing function, which is not observed in R249X TG littermates (Figure 2(A)). Quantitatively, this abnormal breathing function reflects an increased basal breathing rate (Figure 2(B): one‐way ANOVA F (3,55) = 19.39, p <0.0001; Dunnett's post‐hoc comparison to WT p <0.0001, p = 0.1359 and p = 0.6527 for R294X, TG and R294X TG, respectively), increased breathing irregularity (Figure 2(C): one‐way ANOVA F (3,55) = 29.35, p <0.0001; Dunnett's post‐hoc comparison to WT p <0.0001, p = 0.7114 and p = 0.7631 for R294X, TG and R294X TG, respectively), and increased incidence of apneic events (Figure 2(D): one‐way ANOVA F (3,55) = 41.64, p <0.0001; Dunnett's post‐hoc comparison to WT p <0.0001, p = 0.9971 and p = 0.9931 for R294X, TG and R294X TG, respectively) compared with WT littermates. Each of these RTT‐specific breathing phenotypes is rescued to WT levels with addition of MECP2 transgene.
FIGURE 2.
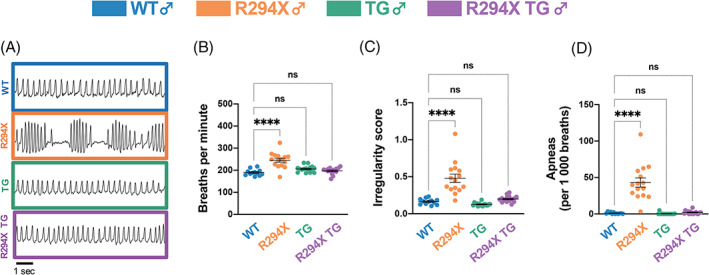
RTT‐specific breathing phenotypes are rescued with MECP2 transgene in male R294X mice. (A) Male R294X mice have an altered breathing pattern compared with WT littermates at 20 weeks of age, which is rescued by addition of MECP2 transgene in male R294X TG mice. (Colors as in Figure 1). (B) Basal breathing rate is elevated in male R294X mice compared with WT littermates, which is rescued by addition of MECP2 transgene. (Age: 20 weeks. WT n = 12; R294X n = 15; TG n = 16; R294X TG n = 16) (C) Breathing irregularity score, which reflects the instantaneous rate of change in total breath time, is elevated in male R294X mice compared with WT littermates. This phenotype is rescued by addition of MECP2 transgene in male R294X TG mice. (Age: 20 weeks. WT n = 12; R294X n = 15; TG n = 16; R294X TG n = 16) (D) Male R294X mice have increased apneic events compared with WT littermates, which is rescued by addition of MECP2 transgene. (Age: 20 weeks. WT n = 12; R294X n = 15; TG n = 16; R294X TG n = 16)
3.3. Dosage‐sensitive behavioral phenotypes are normalized with MECP2 transgene in male R294X mice
In addition to RTT‐specific breathing phenotypes, we also assayed behaviors that are bidirectionally dosage‐sensitive in male mice. One such behavior is spontaneous locomotor activity. Individuals with RTT have gross motor dysfunction and can experience motor deterioration in the late course of disease, with loss of ambulation and development of parkinsonian gait. Individuals with MDS display early onset of severe motor dysfunction, including infantile hypotonia and progressive spasticity. 5 , 19 , 40 , 41 , 42 Mice with Mecp2 mutations and transgenic MECP2 overexpressing mice have both been reported to display decreased spontaneous locomotor activity. 25 , 27 , 29 , 33 , 36 We observe a similar phenotype in male R294X and male TG mice, which display decreased total distance traveled in a 30‐min open field assay, compared with WT littermates (Figures 3(A), (B) one‐way ANOVA F (3,55) = 7.825, p = 0.0002; Dunnett's post‐hoc comparison to WT p = 0.0003, p = 0.0156 and p = 0.6554 for R294X, TG and R294X TG, respectively). The total distance that male R294X TG mice traveled is not different from that of WT littermates, indicating normalization of spontaneous locomotor activity.
FIGURE 3.
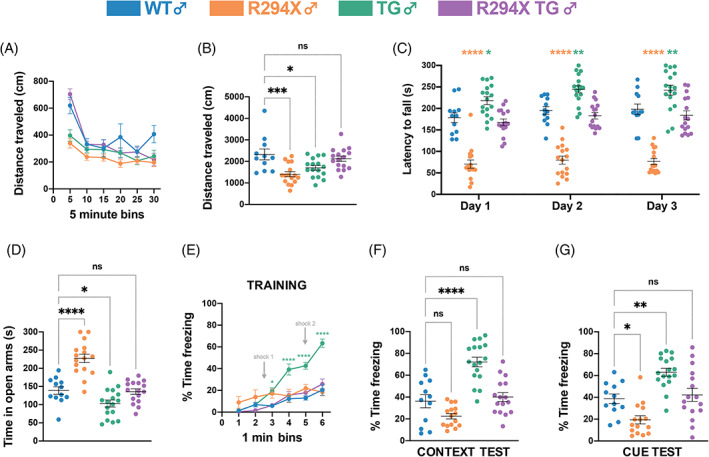
Bidirectionally dosage‐sensitive behavioral phenotypes are normalized with MECP2 transgene in male R294X mice. (A,B) Both male R294X and TG mice have decreased locomotor activity in the open field assay compared with WT littermates. Locomotor activity of male R294X TG mice is not different from WT littermates. (A) depicts distance traveled in centimeters across 5‐min bins. (B) quantifies total distance traveled across the 30‐min assay. (Colors as in Figure 1. Age: 20 weeks. WT n = 11; R294X n = 16; TG n = 16; R294X TG n = 16) (C) Male R294X mice have reduced motor coordination in the rotarod assay, while male TG mice have enhanced motor coordination, compared with WT littermates. Motor coordination in male R294X TG mice is not different from that of WT littermates, indicating normalization of phenotypes. (Age: 20 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16) (D) Male R294X mice have decreased anxiety‐like behavior in the elevated zero maze assay, while male TG mice display increased anxiety‐like behavior, compared with WT littermates. Addition of MECP2 transgene normalizes anxiety‐like behavior in male R294X TG mice. (Age: 20 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16) (E–G) During the training session of the fear conditioning assay (E), male TG mice display increased freezing after the first shock compared with WT mice, which is rescued in R294X TG mice. In the context (F) and cue (G) tests of the fear conditioning assay, male TG mice display increased freezing compared with WT littermates. Male R294X mice display decreased freezing relative to WT littermates in the cue test. Freezing levels of male R294X TG mice are not different from that of WT littermates. (Age: 22 weeks. WT n = 12; R294X n = 15; TG n = 17; R294X TG n = 16)
To evaluate motor coordination, we performed the rotarod assay. Literature suggests that mice harboring Mecp2 mutations generally underperform, while transgenic MECP2 overexpressing mice generally overperform, relative to WT controls. 23 , 25 , 26 , 27 , 29 , 31 , 33 , 36 We too observe this difference, as male R294X mice display decreased latency to fall and male TG mice display increased latency to fall compared with WT littermates across all 3 days of the assay (Figure 3(C): two‐way repeated measures ANOVA, Genotype F (3,57) = 73.58 and p <0.0001, Day F (2,114) = 10.38 and p <0.0001, Genotype × Day F (6,114) = 0.5766 and p = 0.7483). Addition of MECP2 transgene normalizes motor coordination in males, as the latency to fall of male R294X TG mice is not different from that of WT littermates.
We assayed anxiety‐like behavior using the elevated zero maze assay, as there is a high prevalence of comorbid anxiety in individuals with RTT and MDS. 42 , 43 , 44 Previous reports indicate that mice harboring common loss‐of‐function Mecp2 mutations generally display decreased anxiety‐like behavior, while transgenic MECP2 overexpressing mice generally display increased anxiety‐like behavior. 23 , 25 , 26 , 33 , 34 , 36 Consistently, we observe that male R294X mice spend more time in the open arms of the maze, while male TG mice spend less time in the open arms of the maze, compared with WT littermates, indicating decreased and increased anxiety‐like behavior, respectively (Figure 3(D): one‐way ANOVA F (3,57) = 30.49, p <0.0001; Dunnett's post‐hoc comparison to WT p <0.0001, p = 0.0464 and p = 0.9945 for R294X, TG and R294X TG, respectively). The time male R294X TG mice spend in the open arms of the maze is not different from that of WT littermates, demonstrating that addition of MECP2 transgene normalizes anxiety‐like behavior. As a complementary metric for anxiety‐like behavior, we analyzed the percent distance traveled in the center of the open field arena during the open field assay. Male R294X mice travel a higher percentage of distance in the center compared with WT littermates, consistently indicative of decreased anxiety‐like behavior (Figure S3(A): one‐way ANOVA F (3,55) = 6.108, p = 0.0012; Dunnett's post‐hoc comparison to WT p = 0.0002, p = 0.0747, p = 0.0504 for R294X, TG and R294X TG, respectively). Both TG and R294X TG male mice traveled a percentage of distance in the center similar to WT littermates.
As an evaluation of social behavior, we used the Crawley 3‐chamber assay for sociability and social novelty preference. Although social behavior phenotypes vary widely amongst mouse models of RTT depending on allele, mouse strain and testing/analysis methods, some work has found elevated sociability and social novelty preference in Mecp2 mutant mice and reduced sociability and social novelty preference in transgenic MECP2 overexpressing mice. 33 , 34 , 45 , 46 , 47 We used a single‐day protocol and evaluated the sociability of mice (preference to interact with an unfamiliar mouse over an empty cup) and social novelty preference (preference to interact with an unfamiliar mouse over a familiar mouse). We found that mice of all genotypes exhibited typical sociability, with positive mean discrimination indices (Figure S4(A): one‐way ANOVA F (3,57) = 1.642, p = 0.1898; Dunnett's post‐hoc comparison to WT p = 0.2486, p = 0.9966 and p >0.9999 for R294X, TG and R294X TG, respectively). Additionally, mice of all genotypes exhibited typical social novelty preference (Figure S4(B): one‐way ANOVA F (3,57) = 0.0431, p = 0.9880; Dunnett's post‐hoc comparison to WT p = 0.9931, p = 0.9821 and p = 0.9703 for R294X, TG and R294X TG, respectively). These data suggest that neither R294X nor TG male mice show social behavior phenotypes in this assay.
Finally, we employed contextual and cued fear conditioning to investigate learning and memory. Previous reports have showed that mice with Mecp2 mutations display decreased freezing, while transgenic MECP2 overexpressing mice display increased freezing, relative to WT controls across both tests. 23 , 25 , 31 , 45 We found that, during the training session, male TG mice displayed higher freezing levels than WT littermates, beginning and continuing after the first tone‐shock pairing (Figure 3(E): two‐way repeated measures ANOVA, Genotype F (3,55) = 13.09 and p <0.0001, Day F (5,275) = 59.19 and p <0.0001, Genotype × Day F (15,275) = 11.38 and p <0.0001). Neither R294X nor R294X TG male mice displayed freezing levels different from WT mice in the training session. Additionally, although not significant in the context recall test, male R294X mice displayed decreased freezing relative to WT mice during the cue recall test. Male TG mice displayed increased freezing relative to WT during both context and cue recall tests (Figure 3(F): one‐way ANOVA F (3,56) = 26.05, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.0889, p <0.0001 and p = 0.8655 for R294X, TG and R294X TG, respectively; Figure 3(G): one‐way ANOVA F (3,56) = 16.50, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.0164, p = 0.0016 and p = 0.9072 for R294X, TG and R294X TG, respectively). Male R294X TG mice displayed freezing levels that were not different from WT during context and cue recall tests, indicating that addition of MECP2 transgene normalizes fear memory.
3.4. RTT‐ and MDS‐like behavioral phenotypes are rescued in female R294X TG mice
No adverse effects of MeCP2 supplementation were observed in our behavioral battery with male mice. However, we hypothesized that the therapeutic window for MECP2 elevation might differ for female mice. Females with MECP2 mutations are mosaic because of inactivation of either the WT or mutant MECP2 allele in each somatic cell. 48 Most individuals with RTT have balanced XCI, but highly skewed XCI has been observed in some individuals with mild RTT and asymptomatic carriers of disease‐causing MECP2 mutations. 49 , 50 , 51 Importantly, while MECP2 gene therapy in 46,XY males should lead to co‐expression of endogenous mutant and exogenous WT alleles in all somatic cells, MECP2 gene therapy in 46,XX females should lead to two populations of somatic cells—(1) cells that co‐express endogenous mutant and exogenous WT alleles, as occurs in males and (2) cells that express both endogenous and exogenous WT alleles. We hypothesized that modeling gene therapy in females would specifically address potential MDS‐like adverse effects arising from this second population of somatic cells that are not present in males.
We began the female behavioral battery at 30 weeks with the open field assay to evaluate spontaneous locomotor activity. Similar to male mice, female R294X mice traveled decreased total distance compared with WT littermates over a 30‐min period (Figure 4(A), (B): one‐way ANOVA F (3,55) = 5.186, p = 0.0032; Dunnett's post‐hoc comparison to WT p = 0.0028, p = 0.2303 and p = 0.9705 for R294X, TG and R294X TG, respectively). Unlike males, female MDS model mice did not display a phenotype in this assay, as female TG mice traveled a total distance that was not different from WT littermates. In female R294X TG mice, spontaneous locomotor activity was rescued to WT levels with addition of MECP2 transgene. We next analyzed breathing function through unrestrained whole‐body plethysmography. Female R294X mice displayed an abnormal breathing pattern (Figure 4(C)), with an increased basal breathing rate (Figure 4(D): one‐way ANOVA F (3,55) = 6.516, p = 0.0008; Dunnett's post‐hoc comparison to WT p = 0.0081, p = 0.9586 and p = 0.9267 for R294X, TG and R294X TG, respectively), increased breathing irregularity (Figure 4(E): one‐way ANOVA F (3,55) = 17.41, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.0014, p = 0.0089 and p = 0.9298 for R294X, TG and R294X TG, respectively), and increased incidence of apneic events (Figure 4(F): one‐way ANOVA F (3,55) = 15.27, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.0020, p = 0.0510 and p = 0.3989 for R294X, TG and R294X TG, respectively) compared with WT littermates. Unexpectedly, female TG mice also showed relatively decreased breathing irregularity (increased regularity) (Figure 4(E)). These RTT‐specific breathing phenotypes were each rescued by addition of MECP2 transgene in R294X TG littermates. Finally, we assayed learning and memory with fear conditioning, an assay in which Mecp2 −/+ mice have been shown to exhibit deficits. 52 , 53 , 54 In the training session, female TG mice displayed increased freezing beginning and continuing after the first tone‐shock pairing, similar to male TG mice (Figure 4(G): two‐way repeated measures ANOVA, Genotype F (3,58) = 16.13 and p <0.0001, Minute F (5,290) = 221.4 and p <0.0001, Genotype × Minute F (15,290) = 4.905 and p <0.0001). This phenotype is not observed in female R294X TG mice. In the contextual and cue recall tests, female TG mice again displayed increased freezing compared with WT littermates (Figure 4(H): one‐way ANOVA F (3,58) = 5.050, p = 0.0035; Dunnett's post‐hoc comparison to WT p = 0.8863, p = 0.0114 and p = 0.6172 for R294X, TG and R294X TG, respectively; Figure 4(I): one‐way ANOVA F (3,58) = 2.320, p = 0.0847; Dunnett's post‐hoc comparison to WT p = 0.2994, p = 0.0292 and p = 0.3981 for R294X, TG and R294X TG, respectively). However, the percentage of time that female R294X mice spent freezing was not different from WT mice. In female R294X TG mice, the MDS‐like phenotypes were not apparent, demonstrating that transgenic MECP2 supplementation does not elicit adverse hippocampal‐dependent learning phenotypes in the context of the R294X allele.
FIGURE 4.
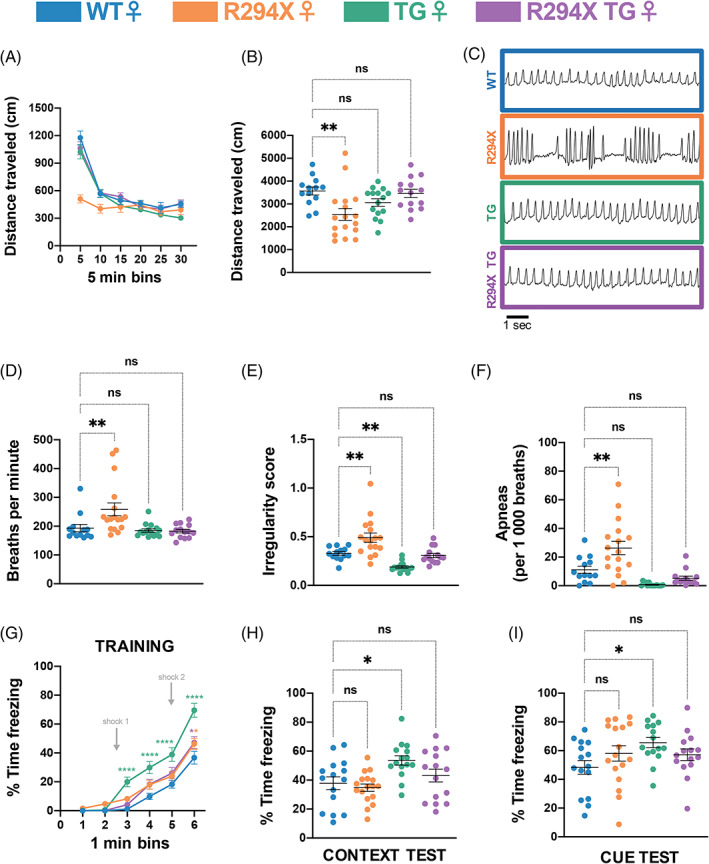
RTT‐ and MDS‐like behavioral phenotypes are rescued in female R294X TG mice. (A,B) Female R294X mice have decreased locomotor activity in the open field assay compared with WT littermates, which is rescued by addition of MECP2 transgene. (A) depicts distance traveled in centimeters across 5‐min bins. (B) quantifies total distance traveled across the 30‐min assay. (Colors as in Figure 1. Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14) (C) Mecp2 R294X/+ mice have an altered breathing pattern compared with WT littermates at 30 weeks of age, which is rescued by addition of MECP2 transgene in female R294X TG mice. (D) Basal breathing rate is elevated in female R294X mice compared with WT littermates, which is rescued by addition of MECP2 transgene. (Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14) (E) Breathing irregularity score, which reflects the instantaneous rate of change in total breath time, is elevated in female R294X mice and reduced in female TG mice compared with WT littermates. These phenotypes are rescued by addition of MECP2 transgene. (Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14) (F) Female R294X mice have increased apneic events compared with WT littermates, which is rescued by addition of MECP2 transgene. (Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14) (G–I) During the training session of the fear conditioning assay (G), female TG mice display increased freezing after the first shock compared with WT littermates, which is rescued in female R294X TG mice. In the context (H) and cue (I) tests of the fear conditioning assay, female TG mice display increased freezing compared with WT mice. Freezing levels of female R294X TG mice are not different from that of WT mice. (Age: 31 weeks. WT n = 15; R294X n = 17; TG n = 15; R294X TG n = 15)
3.5. Transgenic MECP2 supplementation leads to an MDS‐like motor coordination phenotype in female R294X mice
Our behavioral battery also evaluated a motor behavior that is bidirectionally dosage‐sensitive in females – motor coordination—through the rotarod assay. Similar to findings in the male mice, female R294X mice displayed a decreased latency to fall while female TG mice displayed an increased latency to fall relative to WT littermates (Figure 5(A): two‐way repeated measures ANOVA, Genotype F (3,59) = 60.68 and p <0.0001, Day F (2,118) = 13.74 and p <0.0001, Genotype × Day F (6,118) = 0.5522 and p = 0.7674). However, female R294X TG mice overperformed compared with WT mice, with an increased latency to fall mice that reached statistical significance on the second day of the assay. We wondered whether the enhanced motor coordination in female R294X TG mice stemmed from differences in gait function. To address this, we performed forced gait analysis in female mice aged 34–40 weeks. We found that front paw stride length was increased in both TG mice and R294X TG mice compared with WT littermates (Figure 5(B): one‐way ANOVA F (3,58) = 9.460, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.1535, p = 0.0354 and p = 0.0490 for R294X, TG and R294X TG, respectively). Rear paw stride length was only increased in R294X TG mice compared with WT littermates (Figure 5(C): one‐way ANOVA F (3,58) = 8.361, p = 0.0001; Dunnett's post‐hoc comparison to WT p = 0.2234, p = 0.0522 and p = 0.0489 for R294X, TG and R294X TG, respectively). Because differences in gross physical features can impact forced gait performance, we measured body weight and length. While there were no differences in body weight across genotype, female R294X mice had decreased body length and female TG mice had increased body length compared with WT mice (Figure 5(D): one‐way ANOVA F (3,58) = 2.030, p = 0.1197; Dunnett's post‐hoc comparison to WT p = 0.0787, p = 0.9506 and p = 0.9791 for R294X, TG and R294X TG, respectively). Body length of female R294X TG mice was not different from that of WT mice (Figure 5(E): one‐way ANOVA F (3,58) = 13.46, p <0.0001; Dunnett's post‐hoc comparison to WT p = 0.0092, p = 0.0092 and p = 0.6014 for R294X, TG and R294X TG, respectively).
FIGURE 5.
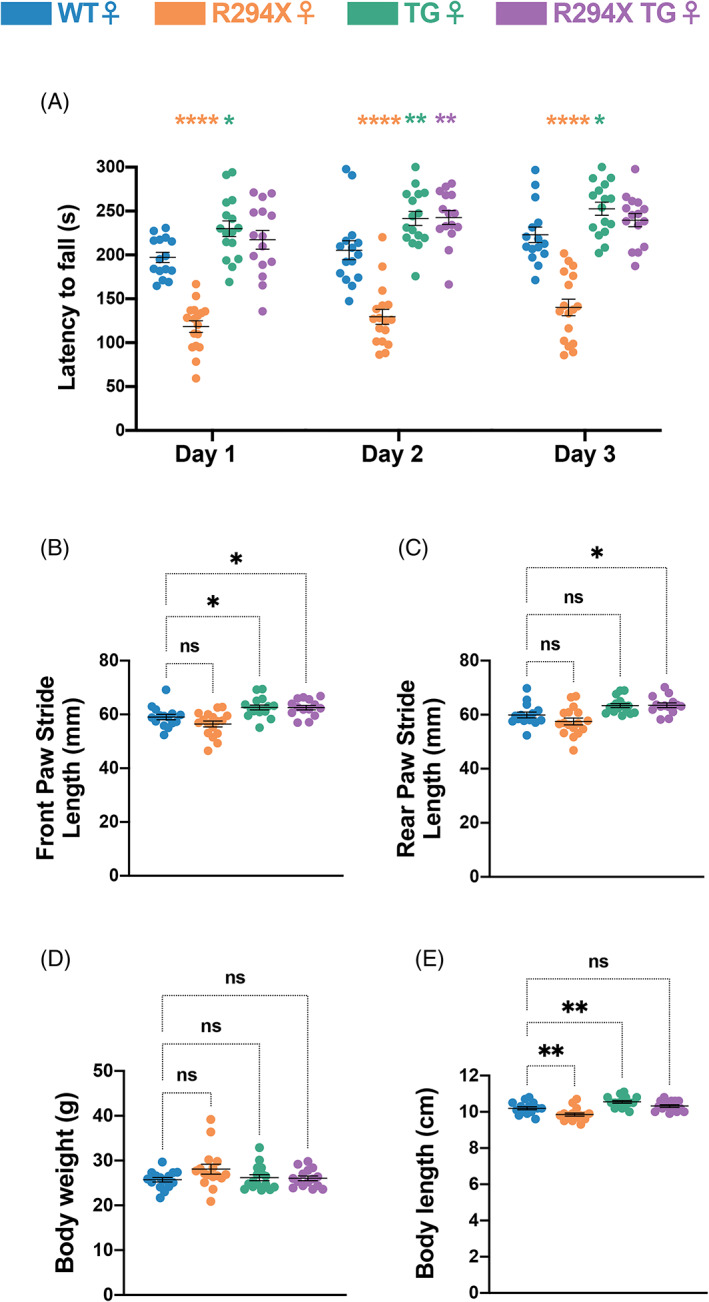
Transgenic MECP2 supplementation leads to an MDS‐like motor coordination phenotype in female R294X mice. (A) Female R294X mice have reduced motor coordination in the rotarod assay, while female TG mice have enhanced motor coordination, compared with WT littermates. Motor coordination of female R294X TG mice is elevated compared with WT. (Colors as in Figure 1. Age: 31 weeks. WT n = 15; R294X n = 17; TG n = 16; R294X TG n = 15) (B,C) In the forced gait assay, front paw stride length in millimeters (B) is increased in both female TG and R294X TG mice compared with WT littermates. Rear paw stride length (C) is increased in female R294X TG mice compared with WT littermates. (Age: 34–40 weeks. WT n = 15; R294X n = 17; TG n = 16; R294X TG n = 14) (D) There are no differences in body weights of female R294X,TG or R294X TG mice compared with WT littermates. (Age: 34–40 weeks. WT n = 15; R294X n = 16; TG n = 16; R294X TG n = 15) (E) The body length of female R294X mice is decreased relative to WT littermates, while body length of female TG mice is relatively increased. Body length of female R294X TG mice is not different from that of WT. (Age: 34–40 weeks. WT n = 15; R294X n = 16; TG n = 16; R294X TG n = 15)
4. DISCUSSION
Within the past decade, preclinical evidence has emerged supporting MeCP2 supplementation as a viable therapeutic strategy for RTT. Promising amongst such approaches is gene therapy, with viral delivery of full‐length MECP2 to the central nervous system (CNS). Numerous studies have evaluated the safety and efficacy of gene therapy in hemizygous Mecp2 −/Y mice and have showed rescue of survival, body weight and select behavioral phenotypes. 12 , 13 , 16 , 17 , 18 , 55 However, only one study thus far has evaluated the safety and efficacy of gene therapy in heterozygous Mecp2 −/+ mice, where authors showed that systemic viral delivery in adulthood rescued gross phenotypic progression as well as motor and cognitive behavioral phenotypes. 13 Collectively these studies have also identified potential barriers to the use of gene therapy in people, most prominently hepatotoxicity occurring with systemic delivery. 16 , 18 However, MDS phenotypes, and subsequently the potential for MDS‐like adverse effects because of excess MeCP2 in the CNS, have not been thoroughly evaluated in these experiments. Additional preclinical research into the therapeutic window of MECP2 gene therapy in mice harboring partial loss‐of‐function Mecp2 mutations, particularly in female mice, is needed to determine the safety of approaches involving MeCP2 supplementation.
We addressed both of these by genetically modeling MeCP2 supplementation in the context of the partial loss‐of‐function R294X allele and screening for behavioral evidence of excess MeCP2 in males and females separately. We found that male R294X TG mice express full‐length MeCP2 at levels comparable to male WT littermates, indicating normalization of protein level. In comparison, female R294X TG mice express full‐length MeCP2 at levels approximately 1.5× that of female WT littermates, consistent with random X‐chromosome inactivation of endogenous WT and R294X alleles. We carried out a broad behavioral battery designed to evaluate rescue of RTT‐like phenotypes observed in male and female R294X mice, as well as potential MDS‐like adverse effects. We found that male R294X and female R294X mice share some RTT‐like phenotypes, including decreased spontaneous locomotor activity, breathing dysfunction and reduced motor coordination. Male and female TG mice displayed opposing phenotypes, notably enhanced motor coordination and enhanced fear memory. RTT‐specific phenotypes were rescued and phenotypes that are bidirectionally MeCP2 dosage‐sensitive were normalized in male R294X TG mice. RTT‐specific phenotypes were also rescued by addition of MECP2 transgene in females. However, female R294X TG mice performed similarly to MDS model mice in assays for motor coordination and gait, suggesting that the underlying motor circuitry is particularly sensitive to MeCP2 dosage in females.
We chose to examine the partial loss‐of‐function R294X mutation, which we previously engineered into mice using CRISPR/Cas9 technology. 25 R294X is a common mutation in MECP2 that truncates the coding sequence within the NID and eliminates the C‐terminal portion of the protein. In mice, the R294X allele produces a truncated protein product, rendering it unique amongst the common RTT‐causing MECP2 truncation mutations. This truncation product retains DNA binding capacity with resultant partial functioning, likely the reason why R294X is associated with milder clinical disease in humans. 22 We replicated behavioral results in male R294X mice, finding that they display decreased spontaneous locomotor activity, elevated anxiety‐like behavior, breathing dysfunction, reduced motor coordination and impaired memory formation. 25 Our results in the fear conditioning assay differ slightly from previous findings in this model. 25 While we report significant differences between male WT and R294X mice in the cue but not context recall test at 20 weeks, Merritt et al. reported significant differences in both at 16 weeks. This difference might be because of experimental conditions and/or mouse age causing generally lower freezing in our WT mice. Additionally, we found that R294X mice do not display deficits in sociability or social novelty preference via the Crawley 3‐chamber assay. Of note, social behavior phenotypes vary widely amongst mouse models of RTT depending on allele, mouse strain and testing/analysis methods. 56 We evaluated a partial loss‐of‐function allele on a pure C57BL6/J background, using a single‐day protocol and blinded manual scoring of interaction time from a video of the assay—any/several of which could account for differences between our results and previous reports.
We extended this work with behavioral evaluation of female R294X mice. Gross physical characterization showed that, unlike many male models, female R294X mice do not have a low body weight phenotype. This is consistent with a report from Mecp2 R168X/+ mice, in which there was no statistically significant difference in body weight from WT until 10 weeks of age. 36 We also did not observe a high body weight phenotype, as was notably reported in Mecp2 R255X/+ mice beginning at 10 weeks of age. At 34–40 weeks, however, several individual female R294X mice displayed very high body weights, although the R294X genotype average did not reach statistically significant difference from WT. Genetic background strongly influences body weight phenotypes in RTT model mice and it is possible that genetic variation mediates expression of this phenotype in individual mice. 57 As evidence of this, one study documented differences in body weight phenotypes amongst Mecp2 +/− mice, noting elevated body weight in F1 FVB/N × 129S6/SvEv mice and reduced body weight in F1 FVB/N × C57BL/6 mice. 54 Differences in strain may account for variation between female R294X weight and that of previous reports such as R255X females, given that the R294X data are derived from >10 generations of a backcross to C57BL/6J.
Behaviorally, we found that similar to males, the females showed RTT‐like phenotypes in assays for spontaneous locomotor activity, breathing function and motor coordination. However, unlike the males, female R294X mice did not display deficits in fear learning. Contextual memory deficits have been reported in female Mecp2 −/+ mice on FVB/N × 129S6/SvEv F1, 129S6/SvEv × C57BL/6F1, and C57BL/6 backgrounds but not, to our knowledge, in a model of a human point mutation. 52 , 53 , 54 We hypothesize that the lack of memory deficits in female R294X mice reflects the milder phenotype observed in people with the R294X mutation and can be attributed to partial functioning of the R294X truncation product. However, our experiments do not address the alternate possibility that memory deficits have a delayed manifestation in female R294X mice, past 30 weeks of age. Together, our behavioral evaluation of male and female R294X mice contributes insights into the behavioral consequences of partial loss‐of‐function MECP2 mutations like R133C, R294X and R306C. 26 , 28 Additionally, our work reinforces the utility of the R294X model, with relatively prolonged survival and delayed onset of RTT‐like phenotypes, for preclinical work. 25
An important consideration for any RTT therapeutic in preclinical development is its effect on MeCP2 dosage. Several neuronal ASD‐associated genes have been determined to be bidirectionally dosage‐sensitive (RAI1, SHANK3 and UBE3A) in addition to MECP2, meaning that both decreases and increases in gene function cause neurological disease in people. 58 Because of this, therapeutic approaches that elevate functional MeCP2 beyond typical levels, such as gene therapy and reactivation of MECP2 on the inactive X‐chromosome, have the potential for excess dosage‐related adverse effects. We considered the potential for these dosage‐related adverse effects by genetically modeling idealized gene therapy capable of fully restoring MeCP2, via supplementation of transgenic human MeCP2 in the context of the R294X allele. While this genetic approach permits precise control of MeCP2 dosage, one limitation is that it models MeCP2 supplementation presymptomatically, whereas putative gene therapy would be administered postsymptomatically. Postsymptomatic genetic and viral delivery of MECP2 have both shown symptomatic benefit in mice, providing hope that this is achievable clinically. 10 , 17 , 55 However, the experimental paradigm used here does not address postsymptomatic MeCP2 supplementation.
We first assessed male mice because they develop RTT‐like phenotypes with earlier onset compared with females and model genetically homogenous MeCP2 supplementation throughout the CNS. When assessed at 20 weeks, we found that addition of MECP2 transgene in male R294X mice was sufficient to rescue RTT‐specific breathing phenotypes. We additionally evaluated behaviors that are bidirectionally dosage‐sensitive in males. At this age, male TG mice showed phenotypes in spontaneous locomotor activity, anxiety‐like behavior, motor coordination and fear memory assays, consistent with previous reports. 23 , 33 , 34 , 45 Unexpectedly, we also found that male TG mice had elevated freezing levels in the training session of the fear conditioning assay after the first tone‐shock pairing. Yu et al. reported a similar phenotype in MECP2 transgenic mice, as well as elevated freezing in a new context after training, suggesting elevated startle and generalization of learned contextual fear. 45 Male R294X TG mice do not display any of these MDS‐like phenotypes, demonstrating that MeCP2 supplementation is safe in the male R294X model. Only one other study by Heckman et al. has evaluated the behavioral consequences of transgenic MeCP2 supplementation in the context of a partial loss‐of‐function MECP2 mutation. 28 In this work, authors evaluated the common R306C mutation in male mice aged 5‐ and 11‐weeks expressing mutant MeCP2 with a C‐terminal GFP tag, finding that motor and cognitive behaviors were fully rescued with MECP2 transgene. Our work complements these findings, supporting the notion that introduction of full‐length MeCP2 to males is safe and effective. Specifically, we note that MeCP2 supplementation is effective even in the presence of a truncation product with DNA‐binding capacity, alleviating potential concerns for dominant negative interactions.
We next assessed female mice, which undergo XCI and heterogeneously express WT or mutant MeCP2 throughout the CNS. XCI generates two considerations for MeCP2 supplementation that are specific to females. First, unlike the males, females with transgenic MECP2 expression will have a population of neurons that co‐express the full‐length MECP2 transgene and the endogenous WT allele. Second, XCI skewing can alter the predominance of this population of neurons. Although most individuals with RTT display balanced XCI, skewed XCI that occurs in a minority of individuals could decrease or increase severity depending on whether the WT or mutant allele predominates. 49 , 51 Together, mutation type and XCI status contribute to individual variation in functioning MeCP2 that likely impacts clinical severity, and both factors should be considered with therapeutic approaches involving MeCP2 supplementation. Although our experimental design could not evaluate different degrees of XCI skew, we were able to assess safety in the context of a partial loss‐of‐function allele. We found that, similar to males, RTT‐specific phenotypes such as decreased spontaneous locomotor activity and breathing dysfunction were rescued by addition of MECP2 transgene.
Females with MECP2 duplications on the X‐chromosome often display neuropsychiatric disease but not the MDS clinical phenotype seen in males. This is because of nearly 100% XCI skewing in favor of the WT allele, as measured in peripheral blood samples. 42 However, if the MECP2 duplication is on an autosome or if there is unskewed XCI, females can manifest an abnormal phenotype to varying degrees, making this model relevant to females as well. 59 MDS‐like phenotypes were milder in females as compared with males, as female TG mice displayed differences from WT only in assays for motor coordination and contextual learning. While enhanced freezing in fear conditioning was rescued by transgenic MeCP2 supplementation of the R294X allele in females, female R294X TG mice overperformed on the rotarod assay for motor coordination, similar to female TG mice. Furthermore, both TG and R294X TG female mice displayed increases in stride length compared with WT littermates as determined by forced gait analysis. These changes in gait did not correlate with gross physical parameters across both genotypes, as there were no differences in weight and female TG mice, but not female R294X TG mice, had increased body length relative to WT controls. These results point to a potential sex‐specific motor coordination side effect of MeCP2 supplementation in the female brain. MeCP2 supplementation of the R255X allele in female mice leads to similarly subtle overperformance, with significantly elevated latency to fall reported on the first of two test days compared with WT controls. 23 Because this has now been observed across both complete (R255X) and partial (R294X) loss‐of‐function mutations, we predict that MeCP2 supplementation of other RTT‐causing mutations would elicit a similar motor coordination phenotype. One possible explanation is that this subtle MDS‐like motor coordination phenotype is driven by the population of cells expressing both endogenous Mecp2 as well as transgenic human MECP2. This population of cells is exposed to excess levels of full‐length MeCP2 and may uniquely impact behavioral phenotypes in females, regardless of mutation type.
The circuitry underlying the enhanced motor coordination phenotype has been dissected by Rothwell et al. using a mouse model of autism‐associated neurologin‐3 (NL3) mutations. 60 The authors found that these mutations specifically decreased synaptic inhibition onto D1‐expressing medium spiny neurons in the nucleus accumbens in ventral striatum, causing acquired repetitive motor behavior. Consistently, selective loss of MeCP2 from forebrain GABAergic neurons causes impaired motor coordination in male mice, and selective preservation of MeCP2 in forebrain GABAergic neurons in floxed‐STOP‐Mecp2 female mice rescues this impaired motor coordination phenotype. 61 , 62 Interestingly, the latter study also found that female floxed‐STOP‐Mecp2 mice with MeCP2 levels restored in the striatum overperformed on the rotarod when retested 1 month after the original test compared with WT controls. 62 Together with our findings, these data suggest that the striatal circuitry responsible for motor coordination is particularly sensitive to MeCP2 dosage in females, reinforcing the notion that MeCP2 dosage can have sex‐specific effects and highlighting the importance of using female mice in preclinical studies for RTT. 60
CONFLICT OF INTEREST
Jeffrey L. Neul receives personal consultancy from Acadia Pharmaceuticals and GW Pharmaceuticals, and is on the advisory board of Alcyone Therapeutics and Taysha.
Supporting information
FIGURE S1 Full‐length and truncated R294X MeCP2 are expressed in male and female mice. (A) Male TG mice express full‐length MeCP2 at ~2× of WT levels. Male R294X TG mice express full‐length MeCP2 at ~1× of WT levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Colors as in Figure 1. Age: 24–25 weeks. WT n = 4; TG n = 4; R294X TG n = 4) (B) Female R294X mice express full‐length MeCP2 at ~0.5× of WT levels. Female TG mice express full‐length MeCP2 at ~2× of WT levels. Female R294X TG mice express full‐length MeCP2 at ~1.5× of WT levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 36–39 weeks. WT n = 3; R294X n = 3; TG n = 3; R294X TG n = 3) (C) Male R294X and R294X TG mice express R294X truncation product at similar levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 24–25 weeks. R294X n = 3, R294X TG n = 3) (D) Female R294X mice express R294X truncation product ~2× of R294X TG levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 38–39 weeks. R294X n = 3, R294X TG n = 3)
FIGURE S2 Timeline of mouse behavioral experiments. (Top) Three cohorts of male mice were weighed and phenotypically scored between weeks 4–14, and the behavioral battery run between weeks 20–22. The behavioral battery included the following tests in order: elevated zero maze, open field, plethysmography, rotarod, Crawley 3‐chamber and fear conditioning. (Bottom) Three cohorts of female mice were weighed and phenotypically scored between weeks 6–14, and the behavioral battery run between weeks 30–31. The behavioral battery included the following tests in order: open field, plethysmography, rotarod, and fear conditioning.
FIGURE S3 Male R294X exhibit a decreased anxiety‐like behavior phenotype in the open field assay. (A) Male R294X mice have a higher percentage of distance traveled in the center of the open field arena, indicating decreased anxiety‐like behavior. Addition of MECP2 transgene rescues anxiety‐like behavior in male R294X TG mice. (Colors as in Figure 1. Age: 20 weeks. WT n = 11; R294X n = 16; TG n = 16; R294X TG n = 16) (B) Female R294X, TG and R294X TG mice have a percentage of distance traveled in the center of the open field arena that is not different from WT littermates. (Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14)
FIGURE S4 Neither R294X nor TG male mice exhibit deficits in social behavior. (A) Sociability in the Crawley 3‐chamber assay (Stage 2) in male R294X, TG and R294X TG mice is not different from WT. (Colors as in Figure 1. Age: 21 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16) (B) Social novelty preference in the Crawley 3‐chamber assay (Stage 3) in male R294X, TG and R294X TG mice is not different from WT. (Age: 21 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16)
ACKNOWLEDGMENTS
We are grateful to Dr. John Allison for his expertise and gracious assistance with mouse behavioral experiments performed through the Mouse Neurobehavioral Core Facility at Vanderbilt University. This work was supported by the National Institutes of Health grants U54HD083211 (JLN) Neuroscience Core Subproject 5907, 1P50HD103537 (JLN) Behavioral Phenotyping Core Subproject 7968, R01HD083181 (JLN), F30MH122064 (BEC), and T32GM007347 (BEC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Eunice Kennedy Shriver Child Health and Human Development Institute (NICHD).
Collins BE, Merritt JK, Erickson KR, Neul JL. Safety and efficacy of genetic MECP2 supplementation in the R294X mouse model of Rett syndrome. Genes, Brain and Behavior. 2022;21(1):e12739. 10.1111/gbb.12739
Funding information Eunice Kennedy Shriver National Institute of Child Health and Human Development, Grant/Award Numbers: P50HD103537, R01HD083181, U54HD083211; National Institute of General Medical Sciences, Grant/Award Number: T32GM007347; National Institute of Mental Health, Grant/Award Number: F30MH122064; National Institutes of Health, Grant/Award Numbers: T32GM007347, F30MH122064, R01HD083181, 1P50HD103537, U54HD083211; Vanderbilt University
REFERENCES
- 1. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet. 1999;23(2):185‐188. [DOI] [PubMed] [Google Scholar]
- 2. Rett A. On a remarkable syndrome of cerebral atrophy associated with hyperammonaemia in childhood. Wiener Medizinische Wochenschrift. 1966;116:723‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trappe R, Laccone F, Cobilanschi J, et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet. 2001;68(5):1093‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neul JL, Benke TA, Marsh ED, et al. The array of clinical phenotypes of males with mutations in methyl‐CpG binding protein 2. Am J Med Genet Part B Neuropsychiatr Genet. 2019;180(1):55‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471‐479. [DOI] [PubMed] [Google Scholar]
- 6. Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guideri F, Acampa M, DiPerri T, Zappella M, Hayek Y. Progressive cardiac Dysautonomia observed in patients affected by classic Rett syndrome and not in the preserved speech variant. J Child Neurol. 2001;16(5):370‐373. [DOI] [PubMed] [Google Scholar]
- 8. Tarquinio DC, Hou W, Neul JL, et al. The course of awake breathing disturbances across the lifespan in Rett syndrome. Brain Dev. 2018;40(7):515‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74(11):909‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. PNAS. 2007;104(6):1931‐1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gadalla KK, Bailey ME, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21(1):18‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33(34):13612‐13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glaze DG, Neul JL, Percy A, et al. A double‐blind, randomized, placebo‐controlled clinical study of trofinetide in the treatment of Rett syndrome. Pediatr Neurol. 2017;76:37‐46. [DOI] [PubMed] [Google Scholar]
- 15. Glaze DG, Neul JL, Kaufmann WE, et al. Double‐blind, randomized, placebo‐controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019;92(16):e1912‐e1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gadalla KKE, Vudhironarit T, Hector RD, et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther‐ Methods Clin Dev. 2017;5:180‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matagne V, Ehinger Y, Saidi L, et al. A codon‐optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol Dis. 2017;99:1‐11. [DOI] [PubMed] [Google Scholar]
- 18. Matagne V, Borloz E, Ehinger Y, Saidi L, Villard L, Roux J‐C. Severe offtarget effects following intravenous delivery of AAV9‐MECP2 in a female mouse model of Rett syndrome. Neurobiol Dis. 2021;149:105235. [DOI] [PubMed] [Google Scholar]
- 19. Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77(3):442‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. del Gaudio D, Fang P, Scaglia F, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8(12):784‐792. [DOI] [PubMed] [Google Scholar]
- 21. Sandweiss AJ, Brandt VL, Zoghbi HY. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: prospects for future therapies. Lancet Neurol. 2020;19(8):689‐698. [DOI] [PubMed] [Google Scholar]
- 22. Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl‐CpG‐binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70(16):1313‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pitcher MR, Herrera JA, Buffington SA, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. 2015;24(9):2662‐2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson BS, Zhao YT, Fasolino M, et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med. 2017;23(10):1203‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merritt JK, Collins BE, Erickson KR, Dong H, Neul JL. Pharmacological read‐through of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum Mol Genet. 2020;29(15):2461‐2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown K, Selfridge J, Lagger S, et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet. 2016;25(3):558‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamonica JM, Kwon DY, Goffin D, et al. Elevating expression of MeCP2 T158M rescues DNA binding and Rett syndrome–like phenotypes. J Clin Invest. 2017;127(5):1889‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heckman LD, Chahrour MH, Zoghbi HY. Rett‐causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lyst MJ, Ekiert R, Ebert DH, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co‐repressor. Nat Neurosci. 2013;16(7):898‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl‐CpG‐binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51(3):152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679‐2689. [DOI] [PubMed] [Google Scholar]
- 32. Moy SS, Nadler JJ, Perez A, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic‐like behavior in mice. Genes Brain Behav. 2004;3(5):287‐302. [DOI] [PubMed] [Google Scholar]
- 33. Sztainberg Y, Chen H, Swann JW, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature. 2015;528(7580):123‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Samaco RC, Mandel‐Brehm C, McGraw CM, Shaw CA, McGill BE, Zoghbi HY. Crh and Oprm1 mediate anxiety‐related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet. 2012;44(2):206‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramirez JM, Ward CS, Neul JL. Breathing challenges in Rett syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 2013;189(2):280‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wegener E, Brendel C, Fischer A, Hülsmann S, Gärtner J, Huppke P. Characterization of the MeCP2R168X Knockin mouse model for Rett syndrome. PLoS One. 2014;9(12):e115444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 is critical within HoxB1‐derived tissues of mice for Normal lifespan. J Neurosci. 2011;31(28):10359‐10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang TW, Kochukov MY, Ward CS, et al. Progressive changes in a distributed neural circuit underlie breathing abnormalities in mice lacking MeCP2. J Neurosci. 2016;36(20):5572‐5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ward CS, Huang TW, Herrera JA, et al. Loss of MeCP2 function across several neuronal populations impairs breathing response to acute hypoxia. Front Neurol. 2020;11:593554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lombardi LM, Baker SA, Zoghbi HY. MECP2 disorders: from the clinic to mice and back. J Clin Invest. 2015;125(8):2914‐2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Friez MJ, Jones JR, Clarkson K, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118(6):e1687‐e1695. [DOI] [PubMed] [Google Scholar]
- 42. Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann Neurol. 2009;66(6):771‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sansom D, Krishnan VHR, Corbett J, Kerr A. Emotional and behavioural aspects of Rett syndrome. Dev Med Child Neurol. 1993;35(4):340‐345. [DOI] [PubMed] [Google Scholar]
- 44. Mount RH, Charman T, Hastings RP, Reilly S, Cass H. The Rett syndrome behaviour questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry. 2002;43(8):1099‐1110. [DOI] [PubMed] [Google Scholar]
- 45. Yu B, Yuan B, Dai J‐K, et al. Reversal of social recognition deficit in adult mice with MECP2 duplication via normalization of MeCP2 in the medial prefrontal cortex. Neurosci Bull. 2020;36(6):570‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kerr B, Alvarez‐Saavedra M, Sáez MA, Saona A, Young JI. Defective body‐weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum Mol Genet. 2008;17(12):1707‐1717. [DOI] [PubMed] [Google Scholar]
- 47. Schaevitz LR, Moriuchi JM, Nag N, Mellot TJ, Berger‐Sweeney J. Cognitive and social functions and growth factors in a mouse model of Rett syndrome. Physiol Behav. 2010;100(3):255‐263. [DOI] [PubMed] [Google Scholar]
- 48. Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X‐chromosome inactivation ratios affect wild‐type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13(12):1275‐1286. [DOI] [PubMed] [Google Scholar]
- 49. Shahbazian MD, Sun Y, Zoghbi HY. Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am J Med Genet. 2002;111(2):164‐168. [DOI] [PubMed] [Google Scholar]
- 50. Amir RE, Van Den Veyver IB, Schultz R, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol. 2000;47(5):670‐679. [PubMed] [Google Scholar]
- 51. Knudsen GPS, Neilson TCS, Pedersen J, et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006;14(11):1189‐1194. [DOI] [PubMed] [Google Scholar]
- 52. Gogliotti RG, Senter RK, Fisher NM, et al. mGlu7 potentiation rescues cognitive, social, and respiratory phenotypes in a mouse model of Rett syndrome. Sci Transl Med. 2017;9(403):eaai7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gogliotti RG, Fisher NM, Stansley BJ, et al. Total RNA sequencing of Rett syndrome autopsy samples identifies the M4 muscarinic receptor as a novel therapeutic target. J Pharmacol Exp Ther. 2018;365(2):291‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samaco RC, Mcgraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre‐clinical studies. Hum Mol Genet. 2013;22(1):96‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sinnett SE, Hector RD, Gadalla KKE, et al. Improved MECP2 gene therapy extends the survival of MeCP2‐null mice without apparent toxicity after intracisternal delivery. Mol Ther ‐ Methods Clin Dev. 2017;5:106‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Katz DM, Berger‐Sweeney JE, Eubanks JH, et al. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis Model Mech. 2012;5(6):733‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2‐null mutation causes neurological symptoms that mimic rett syndrome. Nat Genet. 2001;27(3):322‐326. [DOI] [PubMed] [Google Scholar]
- 58. Javed S, Selliah T, Lee Y‐J, Huang W‐H. Dosage‐sensitive genes in autism spectrum disorders: from neurobiology to therapy. Neurosci Biobehav Rev. 2020;118:538‐567. [DOI] [PubMed] [Google Scholar]
- 59. El Chehadeh S, Touraine R, Prieur F, et al. Xq28 duplication including MECP2 in six unreported affected females: what can we learn for diagnosis and genetic counselling? Clin Genet. 2017;91(4):576‐588. [DOI] [PubMed] [Google Scholar]
- 60. Rothwell PE, Fuccillo MV, Maxeiner S, et al. Autism‐associated Neuroligin‐3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158(1):198‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chao HT, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism‐like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Su SH, Kao FC, Huang YB, Liao W. MeCP2 in the rostral striatum maintains local dopamine content critical for psychomotor control. J Neurosci. 2015;35(15):6209‐6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Full‐length and truncated R294X MeCP2 are expressed in male and female mice. (A) Male TG mice express full‐length MeCP2 at ~2× of WT levels. Male R294X TG mice express full‐length MeCP2 at ~1× of WT levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Colors as in Figure 1. Age: 24–25 weeks. WT n = 4; TG n = 4; R294X TG n = 4) (B) Female R294X mice express full‐length MeCP2 at ~0.5× of WT levels. Female TG mice express full‐length MeCP2 at ~2× of WT levels. Female R294X TG mice express full‐length MeCP2 at ~1.5× of WT levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 36–39 weeks. WT n = 3; R294X n = 3; TG n = 3; R294X TG n = 3) (C) Male R294X and R294X TG mice express R294X truncation product at similar levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 24–25 weeks. R294X n = 3, R294X TG n = 3) (D) Female R294X mice express R294X truncation product ~2× of R294X TG levels. Representative Western blot of hemibrain nuclear lysates next to quantification. (Age: 38–39 weeks. R294X n = 3, R294X TG n = 3)
FIGURE S2 Timeline of mouse behavioral experiments. (Top) Three cohorts of male mice were weighed and phenotypically scored between weeks 4–14, and the behavioral battery run between weeks 20–22. The behavioral battery included the following tests in order: elevated zero maze, open field, plethysmography, rotarod, Crawley 3‐chamber and fear conditioning. (Bottom) Three cohorts of female mice were weighed and phenotypically scored between weeks 6–14, and the behavioral battery run between weeks 30–31. The behavioral battery included the following tests in order: open field, plethysmography, rotarod, and fear conditioning.
FIGURE S3 Male R294X exhibit a decreased anxiety‐like behavior phenotype in the open field assay. (A) Male R294X mice have a higher percentage of distance traveled in the center of the open field arena, indicating decreased anxiety‐like behavior. Addition of MECP2 transgene rescues anxiety‐like behavior in male R294X TG mice. (Colors as in Figure 1. Age: 20 weeks. WT n = 11; R294X n = 16; TG n = 16; R294X TG n = 16) (B) Female R294X, TG and R294X TG mice have a percentage of distance traveled in the center of the open field arena that is not different from WT littermates. (Age: 30 weeks. WT n = 13; R294X n = 17; TG n = 15; R294X TG n = 14)
FIGURE S4 Neither R294X nor TG male mice exhibit deficits in social behavior. (A) Sociability in the Crawley 3‐chamber assay (Stage 2) in male R294X, TG and R294X TG mice is not different from WT. (Colors as in Figure 1. Age: 21 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16) (B) Social novelty preference in the Crawley 3‐chamber assay (Stage 3) in male R294X, TG and R294X TG mice is not different from WT. (Age: 21 weeks. WT n = 12; R294X n = 16; TG n = 17; R294X TG n = 16)