
Figure 6.
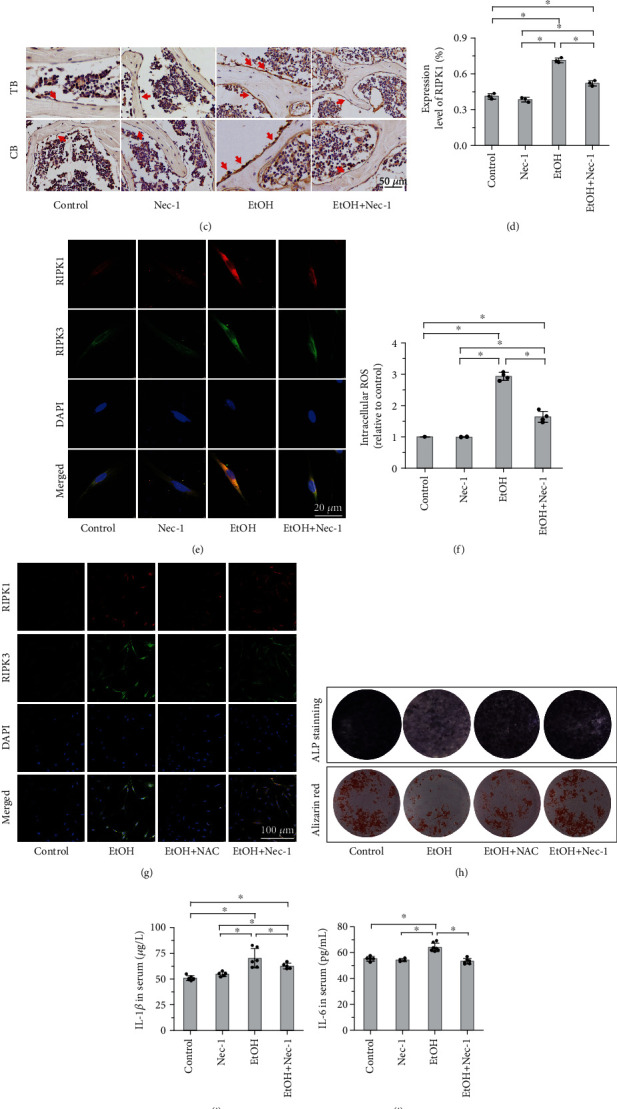
Necrostatin-1 and NAC treatment inhibited the RIPK1/RIPK3/MLKL signaling. (a, b) Western blot analysis showed that EtOH treatment activated the expression levels of the RIPK1 signaling pathway, including p-RIPK1, p-RIPK3, and p-MLKL compared to control mice and MC3T3-E1 cells, and necrostatin-1 treatment lowered the elevated levels both in vivo (a) and in vitro (b). (c, d) Immunohistochemistry staining showed that the expression of RIPK1 in bone tissue increased with EtOH treatment and inhibited by necrostatin-1. The red arrow indicates the positive expression in osteoblasts. Bar: 50 μm. Confocal laser scanning microscopy revealed that Nec-1 treatment alleviated the increase of RIPK1- and RIPK3-positive osteoblasts in vitro (e). Bar: 20 μm. (f-h) High dose of EtOH consumption increased the intracellular ROS levels compared with the control group, and Nec-1 directly inhibited the elevated ROS by flow cytometry. The addition of NAC effectively reduced EtOH treatment necroptosis of the osteoblastic cells as NAC attenuated the upregulation of RIPK1 and RIPK3 (g). NAC treatment significantly increased bone formation, as indicated by the increase in ALP staining and the formation of mineralized nodules (alizarin red staining) in osteoblasts (h). (i, j) The levels of serum proinflammatory cytokines (IL-1β and IL-6) were significantly downregulated by Nec-1 treatment compared to the EtOH-treated mice. The level of TNF-α elevated in mouse serum (k) and osteoblast culture medium (l) after EtOH treatment by ELISA assay, but had no significant difference after Nec-1 treatment. Error bars represent the SD from the mean values. ∗p < 0.05 compared with the control group. #p < 0.05 compared with EtOH treatment. TB: trabecular bone; CB: cortical bone; ROS: reactive oxygen species; NAC: N-acetylcysteine; TNF-α: tumor necrosis factor-α.