

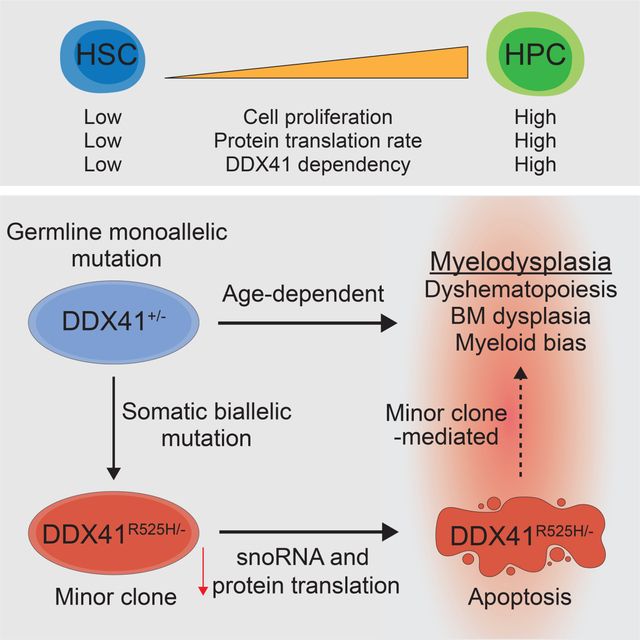
Summary
DDX41 mutations are the most common germline alteration in adult myelodysplastic syndromes (MDS). The majority of patients harbor germline monoallelic frameshift DDX41 mutations and subsequently acquire somatic mutations in their other DDX41 allele, typically missense R525H. Hematopoietic progenitor cells (HPCs) with biallelic frameshift and R525H mutations undergo cell cycle arrest and apoptosis, causing bone marrow failure in mice. Mechanistically, DDX41 is essential for snoRNA processing, ribosome assembly, and protein synthesis. Although monoallelic DDX41 mutations do not affect hematopoiesis in young mice, a subset of aged mice develops features of MDS. Biallelic mutations in DDX41 are observed at a low frequency in non-dominant hematopoietic stem cell clones in MDS patient bone marrow (BM). Mice chimeric for monoallelic DDX41-mutant BM cells and a minor population of biallelic mutant BM cells develop hematopoietic defects at a younger age, suggesting that biallelic DDX41-mutant cells are disease-modifying in the context of a monoallelic DDX41-mutant BM.
eTOC Blurb
Germline mutations in DDX41 cause susceptibility to myeloid neoplasms. Starczynowski and colleagues demonstrate that DDX41 is required for hematopoiesis by regulating snoRNA processing and ribosome biogenesis. Acquired biallelic mutations in DDX41 are tolerated in hematopoietic stem cells and contribute to disease as minor clones.
Graphical Abstract
Introduction
Myelodysplastic syndromes (MDS) are bone marrow (BM) failure disorders derived from abnormal hematopoietic stem cells (HSCs) that give rise to dysplastic myeloid cells and cause multilineage blood cytopenias (Gangat et al., 2016). MDS can transform to secondary acute myeloid leukemia (AML), which has a poor prognosis (Sperling et al., 2016). Germline heterozygous mutations in DDX41, a DEAD-box RNA helicase gene, cause inherited susceptibility to adult MDS and/or AML with a median age of disease onset of 69 years (Polprasert et al., 2015, Lewinsohn et al., 2016, Cardoso et al., 2016, Li et al., 2016, Quesada et al., 2019). DDX41 mutation carriers also have an increased incidence of idiopathic cytopenia of undetermined significance (ICUS), suggesting that germline DDX41 mutations predispose to BM defects prior to MDS/AML (Choi et al., 2021). MDS in patients with DDX41 mutations is unique in that the BM is often hypocellular, whereas adult MDS is typically hypercellular (Choi et al., 2021).
Many functions have been ascribed to DDX41 and thus the role of these mutations in MDS pathogenesis remains undefined. In dendritic cells and macrophages, DDX41 responds to cytosolic viral ligands, such as dsDNA, and induces innate immune signaling through STING (Lee et al., 2015, Zhang et al., 2011, Parvatiyar et al., 2012). DDX41 is also thought to function in splicing, RNA:DNA hybrid accumulation, and ribosomal RNA transcription (Polprasert et al., 2015, Kadono et al., 2016, Weinreb et al., 2021). The most common germline mutations in DDX41 MDS patients are frameshift mutations at aspartic acid (D) 52 or D140, which likely result in an inactive protein(Cheah et al., 2017). The majority of these patients also acquire a somatic mutation at arginine 525 to histidine (R525H) in their residual DDX41 allele (Polprasert et al., 2015, Sébert et al., 2019, Qu et al., 2020). The mutation at R525 diminishes the ATPase function of the helicase domain, indicating that both DDX41 alleles encode inactive or hypomorphic proteins in the somatically acquired biallelic DDX41-mutant cells (Kadono et al., 2016). Collectively, these observations suggest that decreased function of DDX41 promotes hematopoietic cell defects associated with the pathogenesis of MDS. Here, we set out to determine the effect of DDX41 mutations on hematopoietic stem and progenitor cells (HSPCs) to better understand their role in disease. We show that DDX41 is required for snoRNA processing and snoRNA-mediated ribosomal RNA pseudouridylation in HSPCs. We also report that biallelic DDX41-mutant HSPCs exhibit reduced protein synthesis and are a disease-modifying minor clone in the context of monoallelic DDX41-mutant BM. Our findings suggest that co-existence of monoallelic and biallelic DDX41-mutant cells contributes to hematopoietic defects in DDX41-mutated patients.
Results
DDX41 function is critical for hematopoiesis and HSPC proliferation and viability
To recapitulate the DDX41 frameshift mutation at D140 that is present in the germline of the majority of patients with inherited susceptibility to MDS, we generated a conditional knockout mouse wherein exons 7–9 of Ddx41 are floxed (Ddx41flox) resulting in a truncated non-expressed peptide (Figure 1A,B). Since DDX41 patients often acquire a second somatic mutation in DDX41 at R525H, we also generated a conditional Ddx41R525H knockin (KI) mouse by inserting a floxed wild-type cDNA cassette coding for exons 12–17 of Ddx41 into intron 11 of the endogenous gene and then creating a mutation in exon 15 to code for R525H (Figure 1B). After Cre expression, the wild-type Ddx41 cassette is excised, allowing for expression of Ddx41R525H from the endogenous allele (Figure 1B). To model DDX41 patient genetics, we crossed the Ddx41R525H mice with Ddx41flox and Rosa26-CreERT2 mice, resulting in mice with combined loss-of-function (flox) and R525H (KI) mutations upon tamoxifen-inducible Cre-activation (herein, KI/−). To confirm expression of the Ddx41 alleles, we harvested lineage negative (Lin−) BM cells from Ddx41KI/+ or Ddx41KI/flox;Rosa26-CreERT2 mice and treated the cells with 4-OH-tamoxifen in vitro. We confirmed the expected Ddx41 cDNA sequence in each genotype and protein expression in Lin− BM cells (Figure 1C,D; Supplemental Figure 1A). To determine the requirement of Ddx41 on hematopoietic function, we first assessed hematopoietic reconstitution in lethally-irradiated recipient mice. BM cells isolated from wild-type (+/+), Ddx41flox (+/− or −/−), and Ddx41KI (KI/+ or KI/−) mice administered with tamoxifen daily for three days were transplanted into lethally-irradiated recipient mice (Figure 1E). All mice reconstituted with BM cells from biallelic mutant Ddx41−/− or Ddx41KI/− mice died within 20 days from BM failure (BMF) (Figure 1F). In contrast, mice reconstituted with BM cells from monoallelic mutant Ddx41+/− or Ddx41KI/+ mice survived for the duration of the experiment and were indistinguishable from Ddx41+/+ mice. Mice reconstituted with BM cells from biallelic mutant Ddx41−/− or Ddx41KI/− mice exhibited a hypocellular BM, extramedullary hematopoiesis, and pancytopenia (Figure 1G,H). However, mice reconstituted with Ddx41+/+, Ddx41+/−, or Ddx41KI/+ BM cells had comparable BM and spleen pathology, and did not exhibit significant changes in PB counts up to 12 weeks (Supplemental Figure 1B). These findings indicate that Ddx41 is required for regeneration of hematopoiesis, while a single copy of Ddx41 is sufficient for maintaining hematopoiesis without evidence of hematologic disease for up to 12 weeks. We excluded defective HSPC homing to the BM as a reason for the hematopoietic failure in mice transplanted with DDX41-deficient BM cells (Supplemental Figure 1C). To determine the requirement of Ddx41 on hematopoiesis after engraftment of BM cells has been established, we transplanted recipients with BM cells from wild-type and the monoallelic and biallelic Ddx41-mutant mice, allowed the BM to engraft for 4 weeks and then treated mice with tamoxifen (Figure 1I). At 10–18 weeks post-tamoxifen treatment, a subset of the Ddx41−/− or Ddx41KI/− mice became moribund due to pancytopenia (Figure 1J,K). These mice also exhibited a hypocellular BM and extramedullary hematopoiesis in the spleen, suggestive of BMF (Supplemental Figure 1D). The surviving Ddx41−/− or Ddx41KI/− mice had one unexcised Ddx41 allele, indicating a strong selection for cells escaping excision of both Ddx41 alleles (Supplemental Figure 1E).
Figure 1. DDX41 function is required for hematopoiesis.
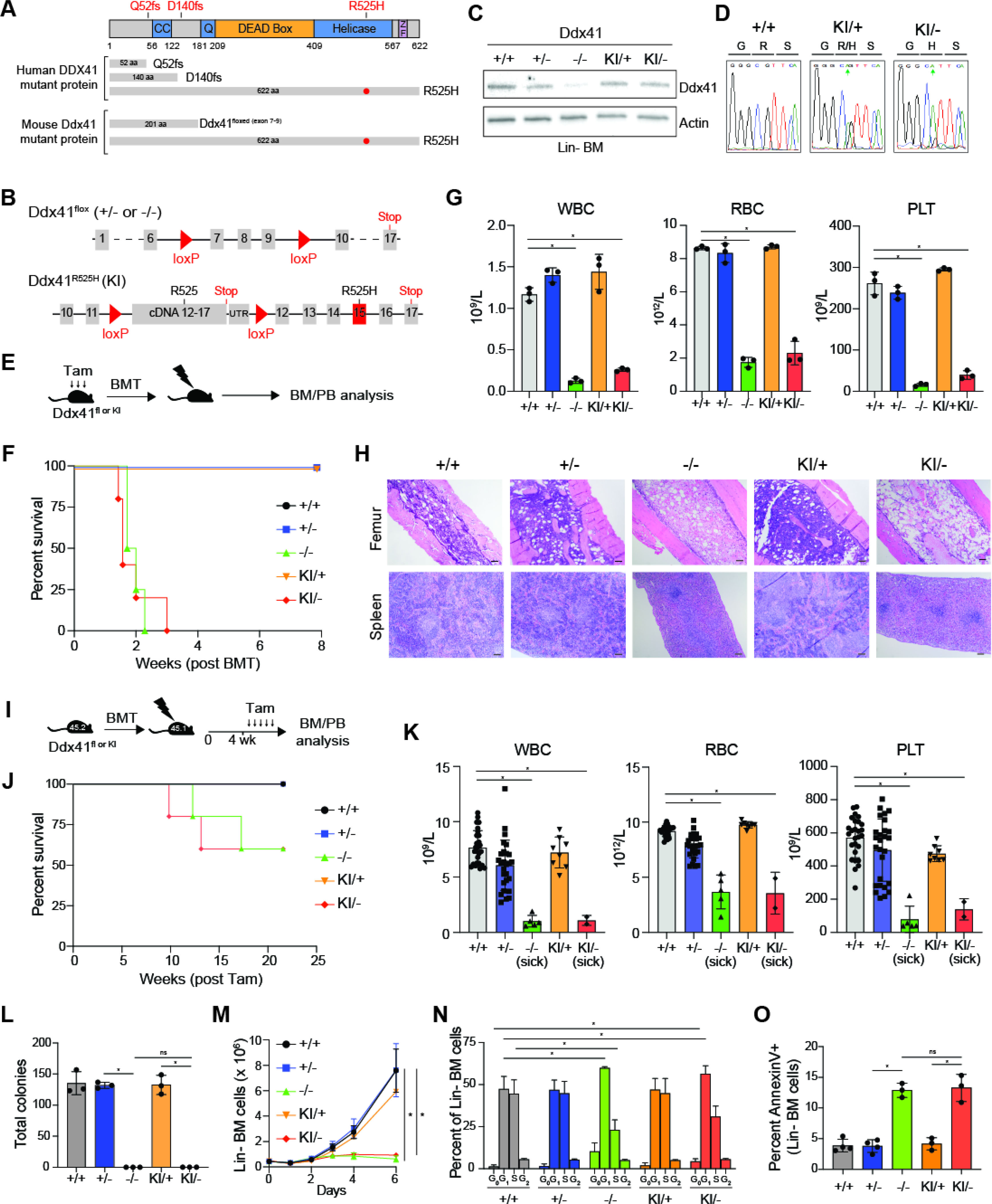
(A) Structure of mouse and human DDX41. (B) Schematic of Ddx41 floxed and Ddx41R525H conditional knock-in (KI) alleles. (C) Protein expression in Lin− BM cells treated with 4-OH-tamoxifen. (D) Sequencing of cDNA from 4-OH-tamoxifen-treated Lin− BM cells. (E) Tamoxifen injection schedule prior to BM transplant. (F) Kaplan-Meier analysis of mice transplanted with BM cells from the indicated mice (4 mice per group). (G-H) Blood counts (*P < 0.0001), H&E-stained femur and spleen, and Wright-Giemsa–stained blood smears of mice 15 days post-transplant or at time of sacrifice. Scale bars, 100 μm. (I) Tamoxifen injection schedule after BM transplant. (J) Kaplan-Meier analysis of mice transplanted with BM cells from the indicated mice (5 mice per group). (K) Blood counts of transplanted mice 12 weeks post-tamoxifen or at time of sacrifice (*P < 0.0001). (L) Myeloid colony formation of Lin- BM cells (*P < 0.0001). (M) Viable cell counts Lin− BM cells in liquid culture (*P < 0.05). (N-O) Cell cycle analysis (*P < 0.0001) and Annexin V staining (*P < 0.05) of Lin− BM cells 72 hours post-tamoxifen (n = 3 per group).
To investigate the reason for the rapid BMF following deletion of DDX41, we next assessed hematopoietic progenitor cell (HPC) function in vitro by isolating Lin− BM cells. Wild-type, Ddx41+/−, or Ddx41KI/+ HPCs formed equivalent number of colonies in methylcellulose (Figure 1L) and grew in liquid culture at similar rates (Figure 1M). In contrast, Ddx41−/− and Ddx41KI/− Lin− BM cells were incapable of forming colonies and failed to expand in liquid culture (Figure 1L,M). The impaired function and proliferation of Ddx41−/− and Ddx41KI/− HPCs were due to significant G1 cell cycle arrest (Figure 1N) and increased apoptosis (Figure 1O). To assess whether the requirement for Ddx41 extends to leukemic cells, we immortalized Lin− cells from wild-type and Ddx41-mutant mice by retroviral expression of the KMT2A/MLL1-AF9 fusion gene, which induces a Hox gene expression program commonly implicated in AML, referred herein as leukemic stem/progenitor cells (LSPC)(Supplemental Figure 1F). Consistent with the requirement of Ddx41 in normal HSPCs, we found that Ddx41 is also required for LSPC proliferation and survival (Supplemental Figure 1G–I). Upon loss of Ddx41 or expression of Ddx41R525H, the LSPC underwent significant cell cycle arrest and apoptosis (Supplemental Figure 1H,I). The cell viability observed at 48 hours post-tamoxifen was comparable between the wild-type and Ddx41-mutant LSPC (Supplemental Figure 1I), whereas at 72 hours the viability of Ddx41−/− and Ddx41KI/− LSPC significantly decreased (Supplemental Figure 1I). These findings suggest that the function of DDX41 is required for proliferating HSPCs. Ddx41+/− LSPC were indistinguishable from wild-type LSPC in these assays, indicating that a single copy of DDX41 is sufficient to maintain the function of these cells. To confirm these observations, we expressed wild-type or DDX41R525H in Ddx41−/− LSPC (from Supplemental Figure 1J). We found that Ddx41−/− LSPC growth was rescued by expression of wild-type DDX41 but not the R525H mutant (Supplemental Figure 1K). Lastly, to evaluate the requirement of DDX41 in human AML, we expressed shRNAs targeting DDX41 (shDDX41) in a human AML cell line (THP1) (Supplemental Figure 1L). We found that THP1 cells expressing shDDX41 failed to proliferate and had increased apoptosis compared to non-targeting control shRNA-expressing cells (Supplemental Figure 1M,N). These findings indicate that DDX41 expression and activity is required for the viability, proliferation, and function of normal and leukemic hematopoietic cells.
Monoallelic mutations of DDX41 confer a HSPC competitive advantage
To determine the consequences of the monoallelic and biallelic Ddx41 mutations on HSPC function, we performed competitive BM transplants by engrafting equal numbers of CD45.2+ Ddx41flox (+/− or −/−) and Ddx41KI (KI/+ or KI/−);Rosa-Cre-ERT cells with CD45.1+ competitor BM cells into lethally irradiated CD45.1+ recipient mice (Figure 2A). Four weeks post engraftment, chimeric mice were administered tamoxifen to induce recombination of the floxed alleles and then examined for 16 weeks (Figure 2A). The chimerism of biallelic Ddx41-mutant (−/− and KI/−) peripheral blood (PB) cells diminished from ~50% to 15% after 16 weeks (Figure 2B,C). In the BM, the chimerism of Ddx41−/− and Ddx41KI/− mononuclear cells (MNCs) and Lin−cKit+Sca1+ (LSK) HSPCs were significantly reduced at 16 weeks (Figure 2D). In addition, the proportions of LT-HSC, HPC-1, HPC-2, and MPP populations were also reduced in chimeric mice reconstituted with Ddx41−/− and Ddx41KI/− BM cells as compared to Ddx41+/+ chimeric mice (Figure 2E). These findings suggest that DDX41 expression and activity is required for HSPC function in vivo. In contrast, the chimerism of monoallelic Ddx41-mutant (+/− and KI/+) PB cells progressively increased from ~50% to 70% after 16 weeks (Figure 2C). Moreover, the BM chimerism of Ddx41+/− and Ddx41KI/+ HSPCs (LSK, HPC-1, and MPP), and HSCs (LT-HSC) was modestly increased at 16 weeks (Figure 2D,E). In a secondary BM transplant, the chimerism of Ddx41+/− and Ddx41KI/+ MNCs in the PB and BM, and HSPCs (LSK) within the BM remained elevated as compared with wild-type cells (Figure 2F–G). Together, these data indicate that Ddx41 is required for HSPC function, and that loss of a single allele of Ddx41 confers a competitive HSPC advantage.
Figure 2. DDX41 haploinsufficiency confers HSPC competitive advantage.
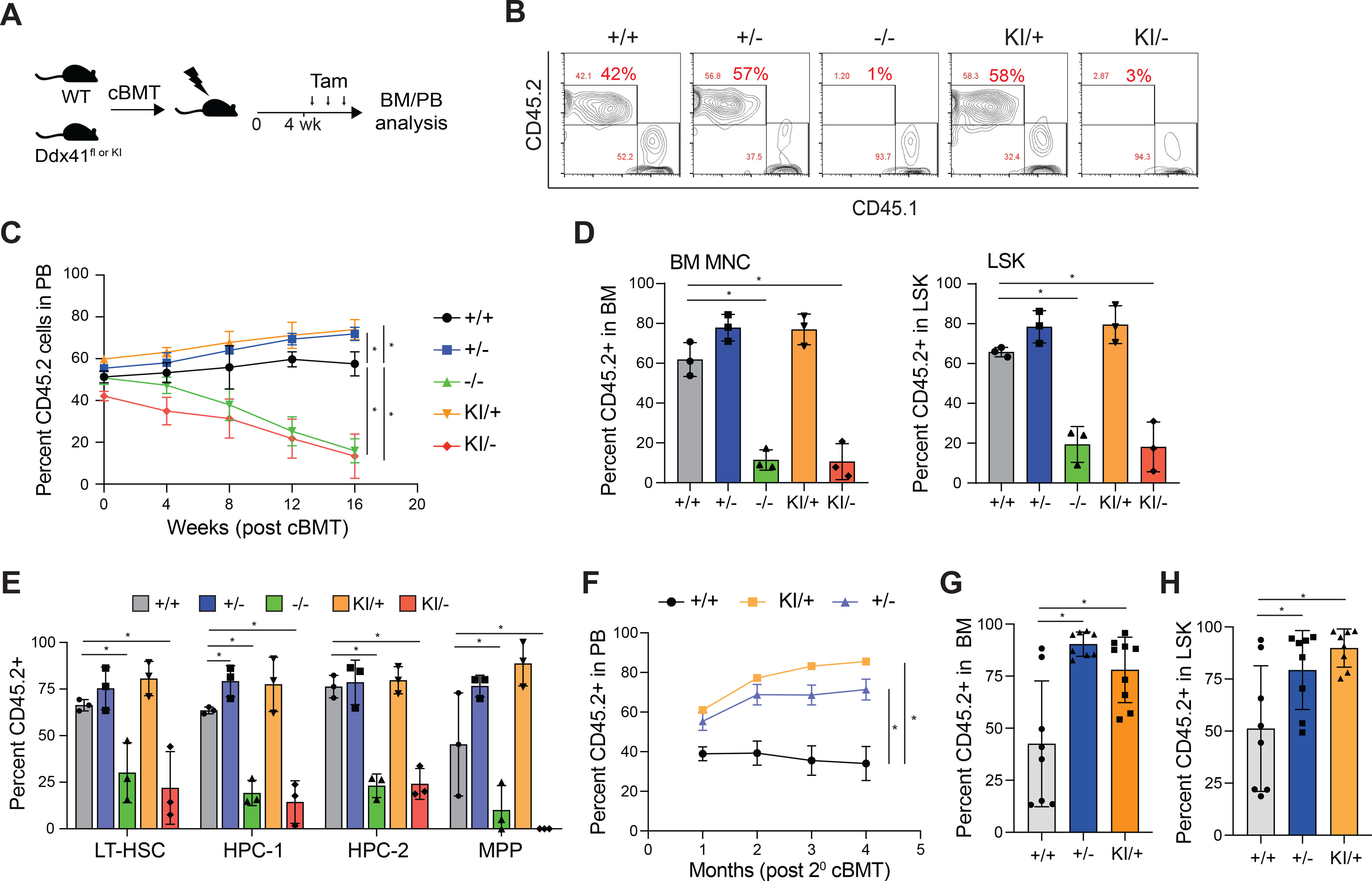
(A) Tamoxifen injection schedule of competitive BM transplants. (B) Representative flow cytometry plots of blood. (C) Percentage of CD45.2+ cells in the blood of competitive transplant recipients over 16 weeks (*P < 0.05; n = 7 mice per group). (D) Percent CD45.2+ in BM mononuclear cells (MNCs) and LSK of competitive transplant recipient mice 16 weeks post-tamoxifen. (*P < 0.01; n = 7 mice per group) (E) Percent CD45.2+ in BM LT-HSC (LSK CD48−CD150+), HPC-1 (LSK CD48+CD150−), HPC-2 (LSK CD48+CD150+), and MPPs (LSK CD48−CD150−) of competitive transplant recipient mice 16 weeks post-tamoxifen (*P < 0.05). (F) Percentage of CD45.2+ cells in the blood of secondary transplant recipients over 16 weeks (*P < 0.01; n = 3 mice per group). (G) Percent CD45.2+ in BM MNCs of secondary competitive transplant recipient mice 16 weeks post-tamoxifen (*P < 0.01). (H) Percent CD45.2+ in the LSK gate of secondary competitive transplant recipient mice 16 weeks post-tamoxifen (*P < 0.05).
Monoallelic mutations of DDX41 causes age-dependent hematopoietic defects
Individuals with heterozygous germline DDX41 mutations typically develop disease in adulthood; therefore, we asked if mice with monoallelic-Ddx41 mutant BM cells develop hematopoietic defects upon aging. BM cells isolated from wild-type, Ddx41+/f (+/−), and Ddx41KI/+ (KI/+) mice administered with tamoxifen daily for three days were transplanted into lethally-irradiated recipient mice (as in Figure 1l) and then examined for 15 months. Although there was no survival difference between the groups, characterization of the mice at the termination of the experiment revealed hematopoietic defects in mice engrafted with Ddx41+/− BM cells. Anemia, thrombocytopenia and a myeloid cell bias were observed in Ddx41+/− recipients as compared to the Ddx41+/+ recipients (Figure 3A,B). The Ddx41+/− group also had reduced BM cellularity and enlarged spleens (Figure 3C,D). We did not observe a significant difference in the relative proportions of HSPCs or mature cells in the BM between each group (Figure 3E–G). Mice engrafted with Ddx41+/− BM cells developed a hypocellular BM with evidence of dysplastic neutrophils (Figure 3H). These mice also exhibited extramedullary hematopoiesis in the spleen associated with myeloid expansion as compared to mice engrafted with wild-type or Ddx41KI/+ BM cells (Figure 3H). We did not observe evidence of leukemic blasts in the mice engrafted with either Ddx41+/− or Ddx41KI/+ BM cells (Figure 3H). These observations suggest that mice engrafted with Ddx41+/− BM cells upon aging exhibit impaired hematopoiesis, with features resembling human ICUS and MDS, but without evidence of overt AML. Interestingly, the mice engrafted with Ddx41KI/+ BM cells did not have the same hematopoietic defects as compared to mice engrafted with Ddx41+/− BM cells, suggesting that the R525H mutation is not functionally equivalent to the frameshift loss-of-function allele.
Figure 3. Monoallelic Ddx41 mutations causes age-dependent hematopoietic defects.
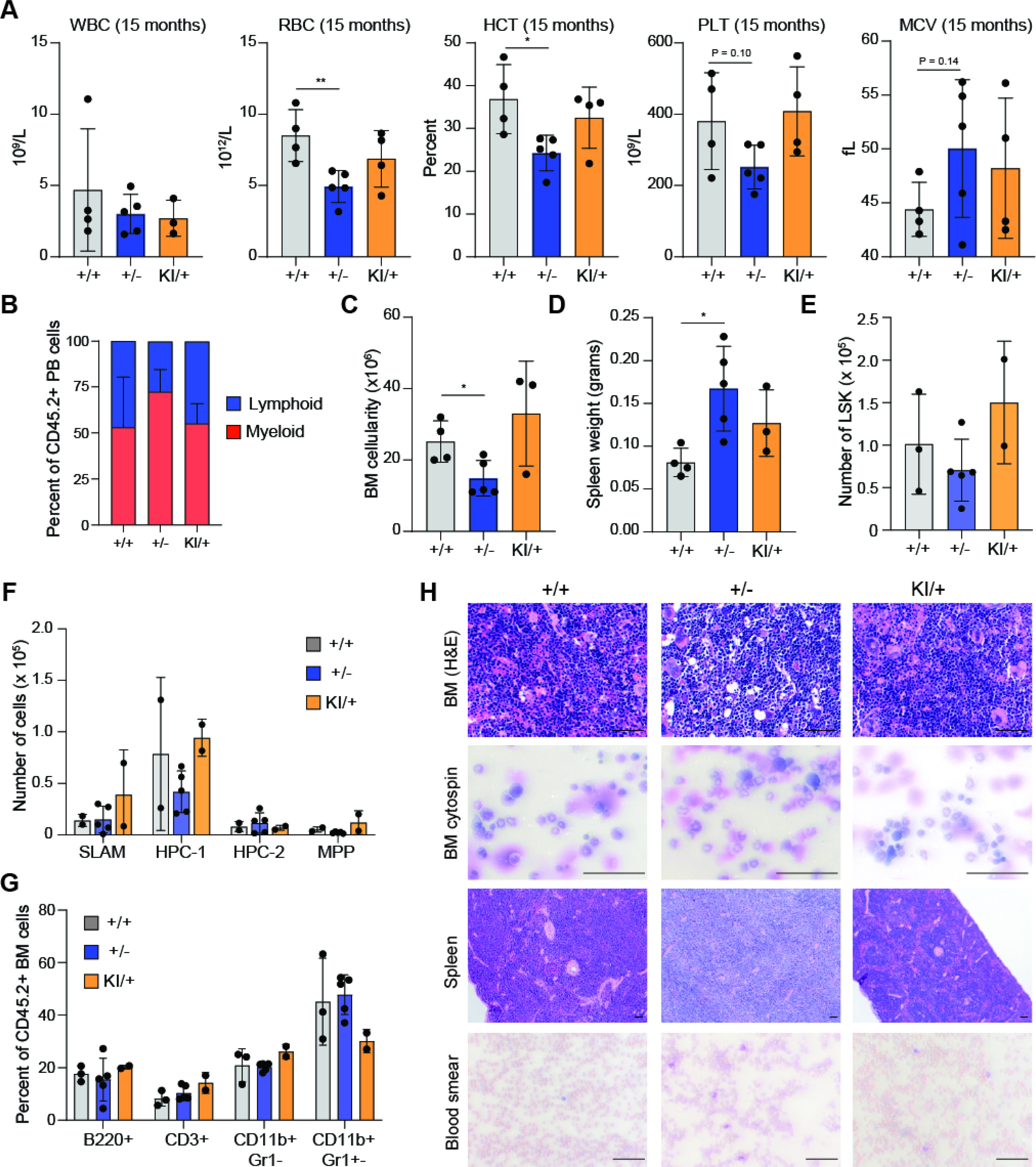
(A) Blood counts at 15 months post-transplant (**P < 0.01; *P < 0.05). (B) Proportion of myeloid and lymphoid MNCs in blood of transplant recipient mice. (C) Cell counts of 2 tibias and 1 femur at 15 months post-transplant (*P < 0.05). (D) Spleen weight at 15 months post-transplant (*P < 0.05). (E) Number of LSK cells in the BM of mice at 15 months post-transplant. (F) Number of HSPCs in the BM of mice at 15 months post-transplant. (G) Percent of CD45.2+ mature BM cells at 15 months post-transplant. (H) Histology on mice at 15 months post-transplant. BM and spleen sections were H&E stained. Blood and cytospins were stained with Wright-Giemsa. Scale bars, 100 μm.
To exclude the possibility that the timing of Ddx41 mutant expression impacts hematopoietic defects and disease development in mice, we also evaluated mice engrafted with BM cells in which Cre-mediated recombination of the Ddx41 allele was induced following BM engraftment. BM cells isolated from wild-type (+/+) and Ddx41+/f (+/−) mice were transplanted into lethally-irradiated recipient mice, and then after 4 weeks were administered with tamoxifen (Supplemebntal Figure 2A). A subset (6/16) of the Ddx41+/− mice developed variable hematopoietic abnormalities in the first 150 days and then another group became moribund after one year post-transplant (Supplemental Figure 2B). Examination of all remaining mice 15 months post-tamoxifen revealed that the Ddx41+/− group were anemic and thrombocytopenic, exhibited reduced BM cellularity, an enlarged spleen, and variable expansion of neutrophils as compared to Ddx41+/+ mice (Supplemental Figure 2C–E). The spleen showed increased percentage of myeloid cells in Ddx41+/− mice as compared Ddx41+/+ mice (Supplemental figure 2F). Histological examination revealed dysplastic myeloid cells in the BM and fewer circulating white blood cells (Supplemental Figure 2G). These studies indicate that monoallelic DDX41 loss-of-function mutations can contribute to age-dependent hematopoietic defects, leading to phenotypes that resemble human disease.
DDX41 is required for snoRNA processing in HSPCs
To determine the molecular basis for the functional decline of DDX41-mutant HSPCs, RNA-sequencing was performed on Ddx41+/+ and Ddx41−/− LSPC (48 hours post-treatment with tamoxifen). We identified 638 differentially expressed genes between Ddx41+/+ and Ddx41−/− LSPC (2.0 fold; P < 0.05)(Supplemental Table 1). We found that small nucleolar RNAs (snoRNAs) were significantly overexpressed in Ddx41−/− LSPC (Figure 4A, Supplemental Figure 3A). SnoRNAs are short non-polyadenylated non-coding RNAs that guide small nuclear ribonucleoprotein complexes (snRNPs) to catalyze chemical modifications of ribosomal RNA (rRNA) and transfer RNA (tRNA) (Sloan et al., 2017, Kufel and Grzechnik, 2019). We assessed the expression of all 110 snoRNAs in the mouse genome and found that nearly all of them had increased expression in Ddx41−/− LSPC compared to Ddx41+/+ LSPC with 33 of them being statistically significant (Figure 4B, Supplemental Figure 3B). We also found enrichment of a Reactome snoRNA gene signature in Ddx41−/− LSPC compared to Ddx41+/+ LSPC (Figure 4C). We confirmed increased expression of Snora16a, Snora7a, and Snora70 by qRT-PCR in Lin− BM cells isolated from Ddx41−/− and Ddx41KI/− mice (Figure 4D, Supplemental Figure 3C). A similar increase in snoRNA expression was observed in Ddx41−/− LSPC and in human AML cells expressing shRNAs targeting DDX41 (Supplemental Figure 3D,E). In eukaryotic cells, snoRNAs are predominantly encoded within introns of host genes. In most cases, snoRNAs are released from excised, debranched introns by exonucleolytic trimming and/or endonucleolytic cleavage of flanking intronic RNA (Kufel and Grzechnik, 2019) (Figure 4E). We compared the expression of each snoRNA and its host gene in Ddx41−/− and Ddx41+/+ HSPCs, and found that expression of the affected snoRNAs in Ddx41−/− HSPCs were increased relative to the respective host genes, suggesting that defective snoRNA processing contributes to the increased abundance of snoRNAs in DDX41-mutant cells and is not simply due to increased expression of the respective host genes (Figure 4F).
Figure 4. DDX41 is required for snoRNA processing.
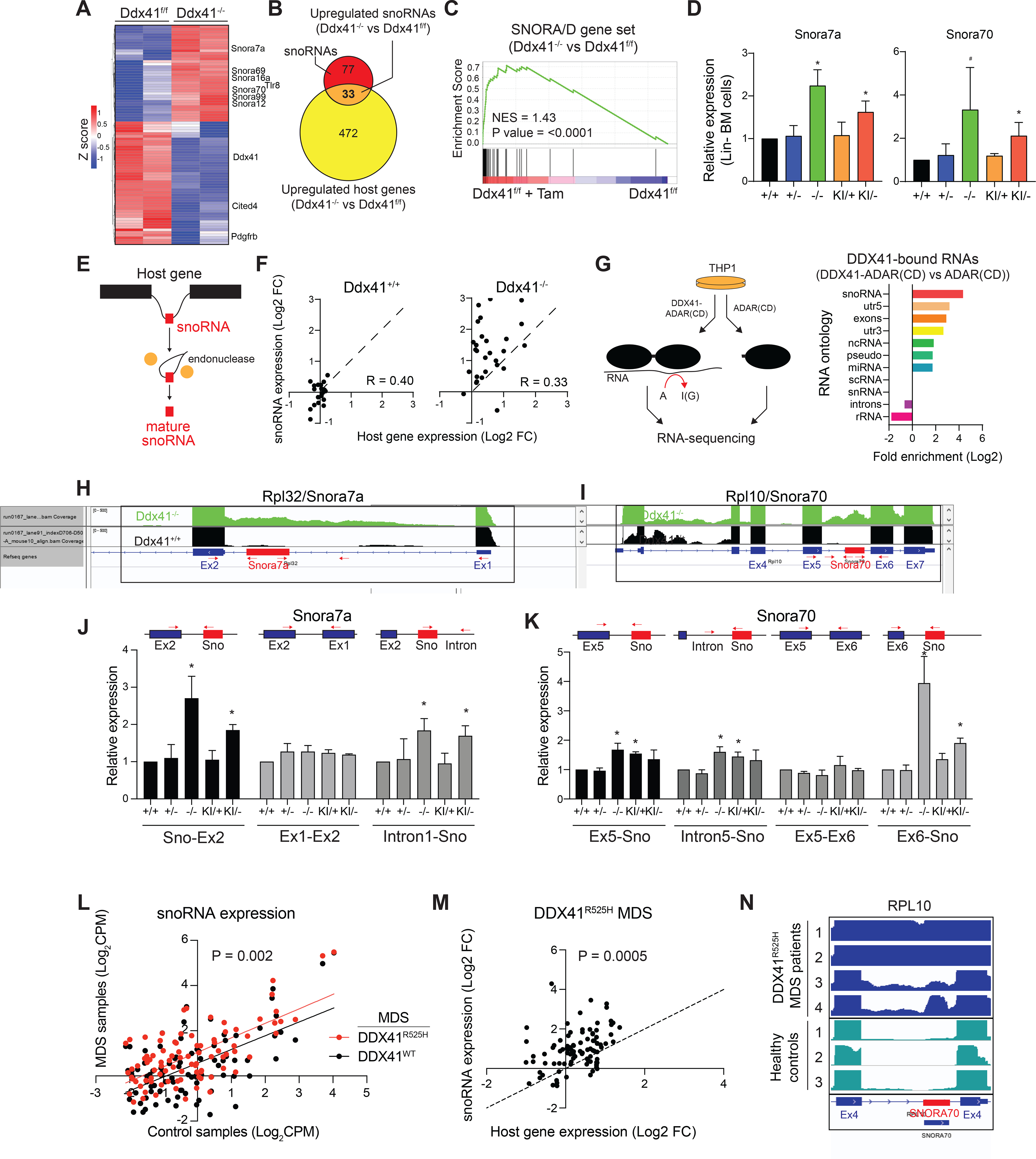
(A) Differentially expressed genes in Ddx41−/− LSPC (Ddx41f/f + TAM) compared to Ddx41f/f. (B) Venn diagram of upregulated genes in Ddx41−/− LSPC and the 110 snoRNAs in the mouse genome. (C) Gene set enrichment analysis for SNORA/D genes in Ddx41−/− LSPC vs Ddx41f/f. (D) Expression of snoRNAs in Lin− BM cells (*P < 0.05; n = 3 per group from independent biological replicates). (E) Schematic of snoRNA processing from host gene introns by splicing and exonuclease digestion to form mature snoRNA. (F) Differential expression of snoRNAs as compared to their host genes in Ddx41−/− and Ddx41+/+ LSPC. (G) Schematic of HyperTRIBE analysis for protein-RNA interactions. Enrichment of each RNA ontology in DDX41 HyperTRIBE analysis is shown. (H-I) Integrative Genomics Viewer (IGV) plots of RNA-seq reads for Snora7a and Snora70 in LSPC. (J-K) Relative expression of exon-snoRNA, intron-snoRNA, and exon-exon transcripts at snoRNA host genes in Lin− BM cells as determined by qRT-PCR. Red arrows indicate the primer locations (*P < 0.05). (L) Expression of snoRNA genes in MDS patient samples with DDX41R525H (n = 4) or without DDX41 mutations (n = 482) as compared to healthy donors (n = 63). Linear regression lines for DDX41-R525H samples (red line) and MDS samples without DDX41 mutations (black line) are shown (P = 0.002). (M) Differential expression of snoRNA genes relative to their host genes in MDS patient samples with DDX41R525H (n = 4) as compared to healthy donors (n = 63). P-value represents likelihood that the slope of the linear regression is equal to 1. (N) IGV plots of RNA-Seq reads at loci for SNORA70 in R525H mutant patients and healthy controls.
To explore the function of DDX41 that contributes to increased abundance of snoRNAs, we assessed global DDX41 binding to RNAs by employing HyperTRIBE (Rahman et al., 2018). Expression of DDX41 fused to the catalytic domain (CD) of ADAR will result in modification of DDX41-bound RNAs from adenosine (A) to iosine (I) (Figure 4G). DDX41-ADAR(CD) fusion or just the ADAR(CD) alone (control) were stably expressed in THP1 cells, and then A-I modifications were identified by RNA-sequencing (Rahman et al., 2018). We identified 1196 unique edited sites after eliminating modified sites in control cells and repeat genomic sequences (Supplemental Table 2). An analysis of all uniquely modified RNAs in DDX41-ADAR(CD) expressing cells revealed that snoRNAs were the most significantly enriched class of DDX41-bound RNAs (P value = 3.9 × 10−4)(Figure 4G). SNORA7A, SNORA74A, and SNORD55 were the top snoRNAs modified in DDX41-ADAR expressing cells (Supplemental Table 2). The A-I modification occurred within the mature snoRNA suggesting that DDX41 preferentially binds to snoRNA-containing RNAs (Figure 4G).
To determine the basis of increased snoRNA abundance observed in Ddx41−/− LSPC, we examined the read density at snoRNA loci and found that the flanking intronic regions were also increased in Ddx41−/− LSPCs as compared to Ddx41+/+ LSPCs (Figure 4H,I). Since the RNA-sequencing samples were selected for polyadenylated mRNAs yet mature snoRNAs are non-polyadenylated, we posited that unprocessed snoRNAs containing intronic reads resulted from inefficient processing of the snoRNA-containing introns in DDX41-mutant HSPCs. To determine if increased snoRNA expression in DDX41-mutant HSPCs can be explained by incomplete snoRNA processing in the host gene RNA, we performed targeted qPCR amplification of sequences flanking Snora7a within the host gene Rpl32 and Snora70 within the host gene Rpl10. Based on this analysis, we found that Ddx41−/− and Ddx41KI/− both exhibited increased “snoRNA-exon” and “snoRNA-intron” amplicons (Figure 4J,K). There was no observed increase in the exon-exon host gene amplicon, indicating that changes in host gene expression are not responsible for changes in the expression of the snoRNA. Importantly, the host gene PCR products were significantly more abundant than the snoRNA and snoRNA-exon products. This observation suggests that any defect in snoRNA processing at these genes does not affect overall host gene expression. Since DDX41 has been implicated in RNA splicing, we explored whether the snoRNA processing defect is the result of global changes in RNA splicing upon loss of DDX41. We found negligible changes in the number of exon exclusion and inclusion events in Ddx41−/− or Ddx41+/− LSPCs as compared to Ddx41+/+ LSPCs. However, Ddx41−/− LSPCs did exhibit an increase in retention events at 295 introns (Supplemental Figure 3F). This finding suggests that intron excision is perturbed at select sites in DDX41-deficient cells, but the changes are not indicative of genome-wide splicing defects upon loss of DDX41. We conclude that reduced function of DDX41 results in a defect in processing of snoRNAs, likely due to incomplete removal of the intron from the downstream host gene exon.
To determine if a defect in processing of intronic snoRNAs also occurs in human disease, we compared RNA-sequencing data from MDS patients with DDX41-R525H mutations to patients without alterations in DDX41. We found significantly increased expression of snoRNA-containing RNAs in DDX41-R525H mutant MDS cells as compared to MDS cells without alterations in DDX41 (Figure 4L). This increase in snoRNA expression was independent from the expression of the host gene expression (Figure 4M). Examination of the sequencing reads surrounding the upregulated snoRNA genes showed that flanking intron sequence reads were increased for select snoRNAs in DDX41-R525H mutant MDS cells as compared to healthy hematopoietic cells (Figure 4N). For example, aberrant SNORA70 processing in DDX41-R525H mutant MDS was similar to that observed in biallelic DDX41-deficient mouse LSPC (Figure 4I). These data suggest that snoRNA processing is dysregulated in BM cells of DDX41-mutant MDS patients.
Loss of DDX41-mediated snoRNA processing leads to ribosome defects and reduced protein synthesis.
We next determined if the snoRNA processing defects in DDX41-deficient cells results in impaired mature snoRNA expression and function. In the small RNA fraction, we found that abundance of mature snoRNAs was decreased in Ddx41−/− and Ddx41KI/− Lin− BM cells as determined by qRT-PCR for Snora7a, Snora16a, and Snora70 (Figure 5A) and by Northern blot analysis of mature Snora7a (Figure 5B). These findings indicate that loss of DDX41 contributes to impaired mature snoRNA processing. We next sought to determine whether decreased mature snoRNA processing is sufficient to contribute to the functional defects observed in Ddx41-deficient HSPCs. We expressed shRNAs targeting five independent snoRNAs in wild-type mouse Lin− BM cells. Knockdown of individual snoRNAs was sufficient to impair cell proliferation (Figure 5C, Supplemental Figure 4A), induce G1 cell cycle arrest (Figure 5D, Supplemental Figure 4B), decrease cell viability (Figure 5E, Supplemental Figure 4C), and reduce hematopoietic differentiation of HSPCs (Supplemental Figure 4D). These defects upon loss of individual mature snoRNAs in HSPCs are similar to the phenotype observed following loss of DDX41 (Figure 1M–O). This data suggests that decreased expression of mature snoRNAs, as in Ddx41-deficient cells, likely cause cell cycle arrest and apoptosis of proliferative HSPCs.
Figure 5. Loss of DDX41-mediated snoRNA processing leads to ribosomal RNA processing defects.
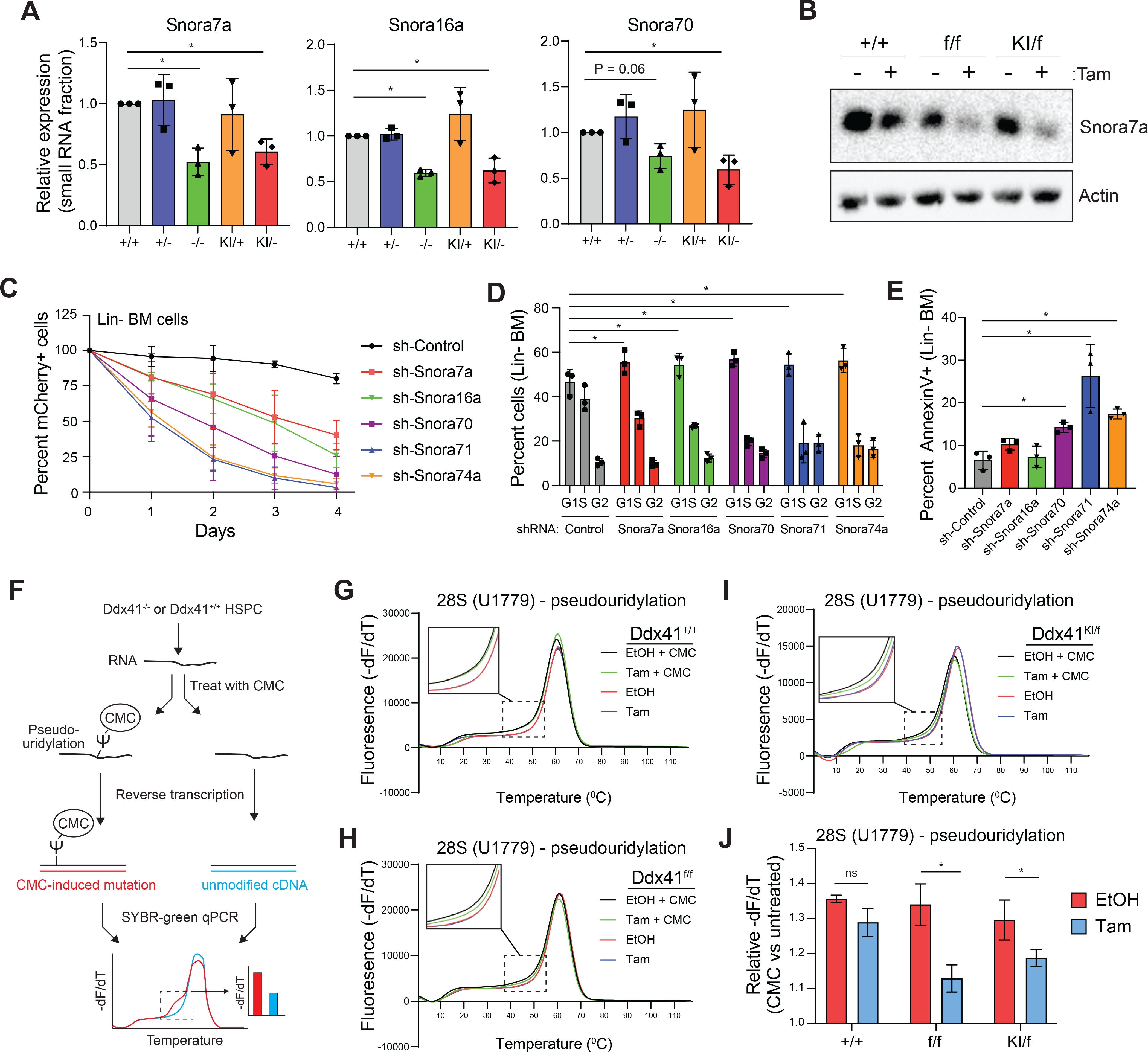
(A) Expression of mature snoRNAs in Lin− BM cells (*P < 0.05). (B) Northern blot for expression of Snora7a in LSPC (+TAM vs. −TAM). (C) Relative counts of mCherry+ cells in mouse Lin− BM cultures transduced with shRNAs targeting snoRNAs. (D-E) Cell cycle and AnnexinV on mCherry+ cells in mouse Lin− BM cultures transduced with shRNAs targeting snoRNAs (*P < 0.05; n = 3 independent biological replicates). (F) qRT-PCR assay to quantitate abundance of pseudouridine in rRNA. CMC covalently binds pseudouridine in RNA and induces a mutation during cDNA synthesis, which alters the melting temperature of the qPCR product. (G-I) Melting curves for pseudouridine analysis of U1779 in 28S rRNA in LSPC (+TAM vs. −TAM). (J) Quantification of the relative difference in the dF/dT for CMC-treated and untreated RNA at 47°C for PCR products encompassing U1779 (*P < 0.05; n = 3 independent biological replicates).
snoRNAs can catalyze sequence-specific chemical modifications in rRNAs, which are essential for ribosome biogenesis and protein synthesis (Sloan et al., 2017, Taoka et al., 2018). The majority of the dysregulated snoRNA identified in DDX41-deficient HSPCs were from the H/ACA family of snoRNAs (SNORA), which catalyze rRNA pseudouridylation (Zhao and He, 2015). To determine whether the abnormal processing of SNORA genes in DDX41-deficient HSPCs correlates with decreased pseudouridine modification in rRNA, we utilized a semi-quantitative approach that measures the abundance of pseudouridine after treatment of RNA with the chemical N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMC). CMC covalently binds to pseudouridine that is then detected by a decrease in the melting curve of the qPCR product (Lei and Yi, 2017) (Figure 5F). We examined pseudouridinylation at the uridines orthologous to human rRNA positions U1779 of 28S rRNA modified by Snora7a, position U3741 of 28S rRNA modified by Snora74a, and U406 of 18S rRNAs modified by Snora71. In wild-type cells, we observed a significant shift in the melting curve of the amplicons targeting U1779 upon treatment with CMC, indicating the presence of pseudouridine at position U1779 of 28S rRNA (Figure 5G,H). In contrast, the CMC-induced melting curve shift in in Ddx41−/− and Ddx41KI/− LSPCs was less pronounced as compared to control cells (ethanol-treated), suggesting that position U1779 of 28S rRNA does not undergo efficient pseudouridinylation in the absence of wild-type DDX41 (Figure 5G–J). Similarly, we found reduced pseudouridinylation at position U406 of 18S rRNA and U3741 of 28S rRNA in DDX41-deficient LSPC (Supplemental Figure 4E–G). We also conducted primer extension assays on CMC-treated RNA from Ddx41+/+ and Ddx41KI/f LSPC using fluorescently-tagged primers that initiate transcription near U406 and U3741. The CMC-adduct causes transcriptional pausing, resulting in an accumulation of reverse transcription products. The abundance of primer pausing at that position indicates the relative abundance of pseudouridine at that location. As expected, we found that Ddx41KI/− cells had less pseudouridine at these sites as compared to wild-type cells (Supplemental Figure 4H). These data suggest that the function of Snora7A and Snora71 is impaired in biallelic Ddx41-mutant cells, as evidenced by reduced rRNA pseudouridine modifications.
rRNA modifications are necessary for efficient processing of the full-length rRNA transcript into its processed forms (Atzorn et al., 2004, Sloan et al., 2017, Kiss et al., 2004). To determine whether the observed processing defect of snoRNAs and aberrant rRNA modification results in impaired formation of rRNAs, we assessed the multi-step processing of rRNAs in Ddx41-mutant LSPC by performing a Northern blot analysis with probes that recognize the transcribed spacer regions upstream of and between the mature rRNA sequences (Srivastava et al., 2010, Henras et al., 2015) (Supplemental Figure 4I). We found Ddx41−/− LSPC exhibited increased abundance of full-length 45S rRNA and decreased abundance of 19S and 12S intermediate rRNAs, indicating a defect of rRNA processing in DDX41-mutant HSPCs (Supplemental Figure 4J,K). These findings establish a role for DDX41 in rRNA processing, which likely explains its essentiality in proliferating HPCs.
Since rRNA processing is essential for efficient ribosome formation and mRNA translation, we posited that protein synthesis would be impaired in DDX41-deficient HSPCs. Biallelic Ddx41-mutant Lin− BM cells had reduced protein synthesis rates, indicated by decreased incorporation of the methionine analog L-homopropargylglycine (HPG), as compared to wild-type cells (Figure 6A,B). In contrast, we did not observe a significant decrease in HPG incorporation in monoallelic Ddx41-mutant Lin− BM cells (Figure 6A,B). Biallelic Ddx41-mutant LSPC and human AML cells expressing shRNAs targeting DDX41 also exhibited a reduction in protein synthesis (Supplemental Figure 5A,B). The observed reduced protein synthesis rates in biallelic DDX41-mutant HSPCs is not merely caused by a non-specific deficit in metabolic function, as mitochondrial function was similar in DDX41-proficient and -deficient HSPCs (Supplemental Figure 5C,D). Since snoRNA-mediated processing of rRNA is critical for ribosome genesis, we next assessed the assembly and function of ribosomes in DDX41-deficient HSPCs. Ddx41−/− and Ddx41KI/− LSPC exhibited a reduction in polysomes and an accompanying increase in monosomes (Figure 6C), which is indicative of a ribosomal defect (Chassé et al., 2017). Although Ddx41KI/− LSPC develop a defect in polysome formation, the accumulation of the monosomal peak was not as profound as compared to Ddx41−/− LSPC (Figure 5C). This may be indicative of a hypomorphic function of the R525H mutation rather than complete loss of function. We confirmed that loss of snoRNA function causes a similar decrease in protein synthesis by inhibiting individual snoRNAs with shRNAs (Figure 6D, Supplemental Figure 5E). To determine if the reduced translation rate in biallelic DDX41-mutant cells is responsible for the susceptibility of the cells to death, we treated LSPCs with low levels of the translation inhibitor puromycin. Indeed, Ddx41−/− and Ddx41KI/− LSPC are more sensitive to puromycin as compared to control LSPC (Figure 6E). In contrast, these DDX41-mutant cells were not more sensitive to the chemotherapy agent methotrexate and even displayed a relative protection at higher doses (Supplemental Figure 5F).
Figure 6. DDX41 loss causes ribosome defects, reduced protein synthesis, and impaired HSPCs.
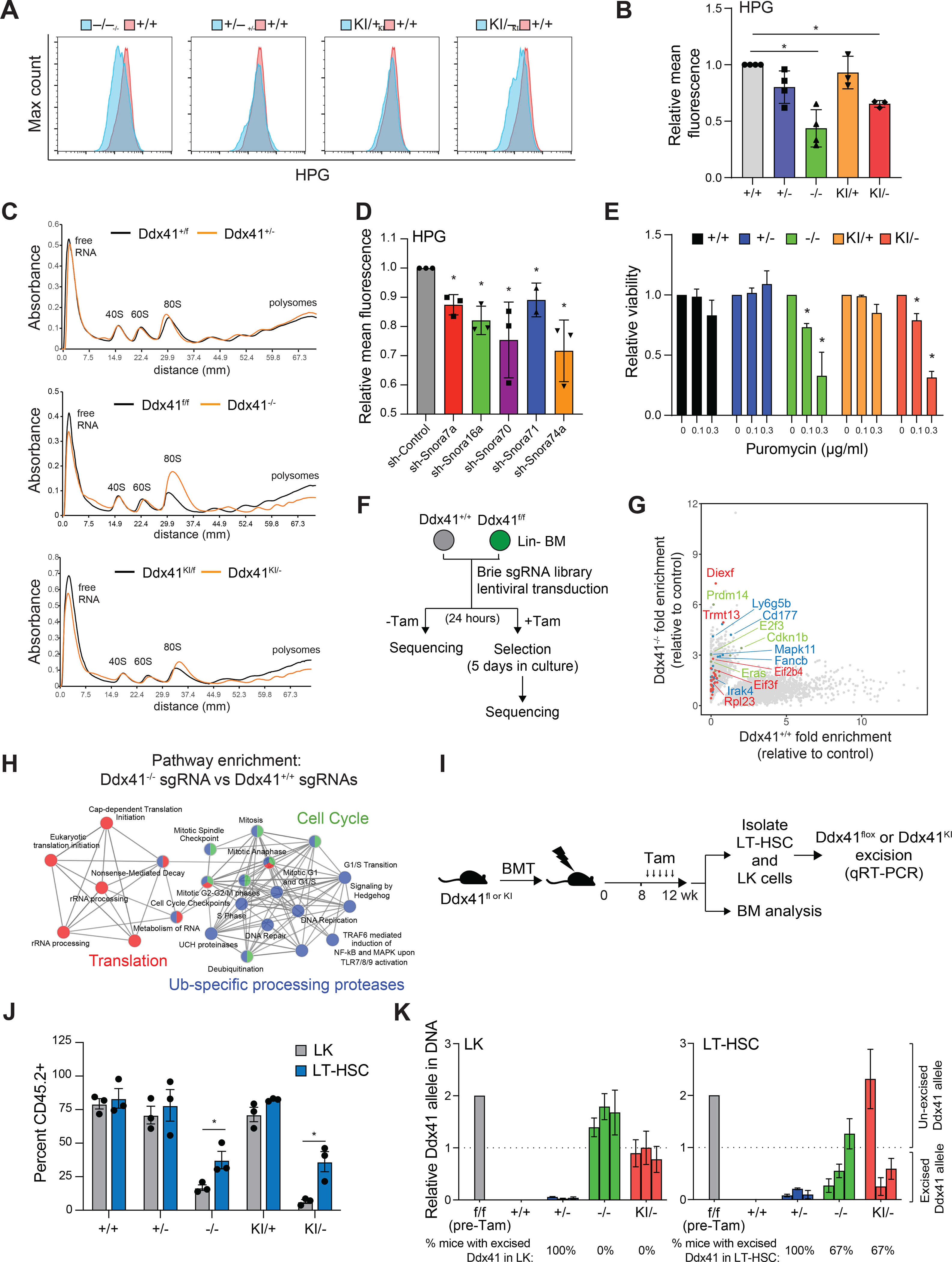
(A-B) Click-IT HPG analysis of protein synthesis rates in Lin− BM cells (*P < 0.05; n = 3 independent biological replicates). (C) Polysome analysis of LSPC (+TAM vs. −TAM) for 48 hrs. (D) Click-IT HPG analysis of protein synthesis rates in Lin− BM cells transduced with shRNAs targeting snoRNAs (*P < 0.05; n = 3 independent biological replicates). (E) Relative viability of LSPC (+TAM vs. −TAM) treated with puromycin determined by CellTiter-Glo (*P < 0.05; n = 3 independent biological replicates). (F) CRISPR knockout screen for gRNA that promote the survival of DDX41-deficient Lin− BM cells. (G) Guide RNAs that were enriched in Ddx41−/− Lin− BM cells relative to control-transduced cells. Color-coding of highlighted genes corresponds to H. (H) Pathway enrichment for gRNAs that are enriched in Ddx41−/− Lin− cultures compared to Ddx41+/+. (I) Tamoxifen injection schedule after BM transplant. (J) Percent of CD45.2+ LK cells and LT-HSC (LSK, CD150+, CD48−) 4-weeks post-tamoxifen (*P < 0.05; n = 3 mice per group). (K) Relative amount of unexcised Ddx41 allele remaining in LK and LT-HSC in the BM 4-weeks post-tamoxifen. The dotted line is equivalent to one unexcised allele.
To determine the pathways and processes involved in the cellular defect of DDX41-deficient HSPCs, we conducted a genome-wide CRISPR-Cas9 rescue screen. Lin− BM cells from Ddx41f/f;Rosa-CreERT;Rosa-Cas9 mice were transduced with the lentiviral Brie sgRNA library, which contains 80,000 guides targeting 20,000 murine genes with 4 independent guides per gene (Doench et al., 2016). The Lin− BM cells transduced with the CRISPR library were grown for 5 days in the presence of tamoxifen, and then harvested for DNA sequencing to identify enriched sgRNAs by next generation sequencing from three independent biological replicates. After normalizing the read counts to cells harvested 24 hours after transduction, we identified 3823 sgRNA with significantly increased read count (P < 0.1) in the Ddx41−/− compared to Ddx41+/+ Lin− BM cells (Figure 6G). To identify genes that were significantly enriched based on analysis of all four sgRNAs, we utilized MAgeCK (Li et al., 2014). Using a cutoff of beta>0.5 and P<0.05, we identified 499 genes that were significantly enriched in Ddx41−/− cells compared to Ddx41+/+ Lin− BM cells (Supplemental Table 3). Pathway analysis of these genes revealed significant enrichment for ribosome and protein synthesis pathways as well as pathways required for HSPC proliferation, such as Toll-like receptor signaling (Figure 6H). This result suggests that reduction in protein synthesis demands, either directly or through reduced cell proliferation, is the prevailing mode of survival upon complete loss of Ddx41. For example, Diexf encodes an RNA helicase that is involved in rRNA processing, and its loss is expected to slow cell growth (Charette and Baserga, 2010). These findings support the theory that complete loss of DDX41 is tolerated when protein synthesis and proliferative demands of a cell are low, such as in quiescent cells.
Acquisition of biallelic DDX41-mutant BM subclones is a frequent event in DDX41-mutant germline patients, as such we sought to determine if HSCs, which have lower protein synthesis and cell cycle demands, survive in vivo without functional DDX41 alleles. Competitive BM transplant experiments were established by engrafting equal numbers of CD45.2+ Ddx41flox (+/− or −/−) and Ddx41KI (KI/+ or KI/−);Rosa-Cre-ERT cells with CD45.1+ competitor BM cells into lethally irradiated CD45.1+ recipient mice (as in Figure 2A). We waited 8 weeks post engraftment to allow HSC to re-enter quiescence before inducing recombination of the floxed alleles (Nakagawa et al., 2018). As expected, the chimerism of biallelic Ddx41-mutant (−/− and KI/−) BM progenitor cells (LK) was significantly diminished at 16 weeks (Figure 6J). However, the chimerism of biallelic Ddx41-mutant BM HSCs (CD48-CD150+LSK) was significantly higher as compared to the BM LK populations, and only modestly reduced as compared to wild-type and monoallelic Ddx41-mutant HSCs (Figure 6J), suggesting that HSCs can persist with biallelic Ddx41 mutations. Moreover, we assessed the presence of unexcised Ddx41 alleles to confirm efficient recombination of the floxed alleles in HSCs and LK progenitors. As expected, the Ddx41 allele was excised in monoallelic Ddx41-mutant LK and HSCs (Figure 6K). However, we observed that all of the Ddx41−/− and Ddx41KI/− LK cells had an unexcised Ddx41 allele (Figure 6K), indicating that biallelic DDX41 loss of function mutations are not compatible with progenitor cells. In contrast, the Ddx41 allele was efficiently excised in Ddx41−/− and Ddx41KI/− HSCs of 67% (2/3) of mice examined (Figure 6K). These findings indicate that HSCs with biallelic DDX41 mutations can survive, and that DDX41 is divergently required for proliferative versus quiescent hematopoietic cell populations.
Ineffective hematopoiesis occurs in younger mice upon co-transplantation of biallelic and monoallelic DDX41-mutant BM cells
Although a somatic DDX41 mutation is typically acquired in DDX41-mutant germline patients, our mouse model data suggest that a biallelic DDX41-mutant BM subclone will have a competitive disadvantage. To explore the role of the acquired R525H mutation in MDS/AML patients, we performed a meta-analysis of publicly available data and found that the variant allele frequency (VAF) of the acquired R525H mutation is ~10% (median frequency) at diagnosis, indicative that <20% of the BM cells possess biallelic DDX41 mutations in patients with objective disease (Quesada et al., 2019, Qu et al., 2020, Polprasert et al., 2015, Sébert et al., 2019) (Figure 7A). Analysis of independent validation patient cohorts revealed a similarly low VAF for the R525H mutation (Figure 7B–C). There was no significant difference in the complete blood counts of DDX41-mutant patients with low VAF compared to higher VAF, indicating that a low abundance of the biallelic DDX41-mutant cells (low VAF) has a similar effect on disease severity as in patients with a higher proportion of the biallelic DDX41-mutant cells (high VAF) (Supplemental Figure 6A). These findings pointed to a model of disease development in which a minor biallelic DDX41-mutant BM clone contributes to ineffective hematopoiesis in the BM of germline DDX41-mutant patients. As such, we set out to model the low VAF of the biallelic DDX41-mutant cells by establishing mice in which 80% of the donor BM cells are Ddx41+/−, representing the patient monoallelic germline mutation, and 20% of the BM cells are Ddx41KI/−, representing the cells with the acquired R525H mutation (Figure 7D). To establish the 80:20 chimeric mice, lethally-irradiated CD45.1+ recipient mice were engrafted with CD45.2+ 8 × 105 Ddx41f/+;Rosa-Cre-ERT cells with 2 × 105 BM cells from Ddx41f/KI;Rosa-Cre-ERT mice (Figure 7D). As negative controls, mice were also engrafted with 80:20 ratios of BM cells from Ddx41+/+;Rosa-Cre-ERT and Ddx41f/KI;Rosa-Cre-ERT mice, or Ddx41f/+;Rosa-Cre-ERT and Ddx41+/+;Rosa-Cre-ERT mice (Figure 7E). Four weeks post engraftment, the mice were administered tamoxifen and then examined for hematopoietic chimerism for 8 weeks. Mice engrafted with an 80:20 ratio of Ddx41+/− and Ddx41KI/− BM cells, exhibited significantly reduced white blood cells (WBC), RBCs, and hematocrit (HCT) as compared to the control mice engrafted with 80:20 ratios of Ddx41+/+ and Ddx41KI/− or Ddx41+/− and Ddx41+/+ BM cells (Figure 7F, Supplemental Figure 6B). The reduction in WBC populations was primarily due to lower levels of lymphocytes, while the neutrophils and monocyte counts were variable between the three groups (Supplemental Figure 6B). It is important to note that mice engrafted with monoallelic DDX41-mutant BM cells (Ddx41+/−) do not exhibit changes in PB at these time points (Supplemental Figure 1B). These findings suggest that cytopenias are exacerbated and occur earlier in mice containing the biallelic Ddx41KI/− BM cells when co-transplanted with the monoallelic Ddx41+/− mutant BM cells. In contrast, we did not observe cytopenias in mice co-transplanted with biallelic Ddx41KI/− and wild-type BM cells (Figure 7F). The observed cytopenias in mice engrafted with Ddx41+/− and Ddx41KI/− BM cells were not accompanied by a significant change in abundance of BM HSPC populations (Supplemental Figure 6C–D). Given that proliferating hematopoietic cells with biallelic DDX41 mutations undergo apoptosis, we reasoned that the Ddx41KI/− BM cells would undergo premature cell death, which would contribute to hematopoietic defects in mice co-transplanted with the monoallelic DDX41-mutant BM cells. Indeed, we detected an increase in cleaved-caspase positive cells in mice co-transplanted with the 80:20 chimeric mixture of monoallelic and biallelic DDX41-mutant BM cells (Figure 7G–H). We also observed increased percentage of myeloid cells in the BM and spleen of these mice (Figure 7I, Supplemental Figure 6E–F) and evidence of neutrophil dysplasia (Figure 7G). These findings suggest that the biallelic DDX41-mutant cells are a disease-modifying minor clone in the context of monallelic DDX41-mutant BM cells.
Figure 7. The minor biallelic DDX41-mutant cells contribute to an MDS-like phenotype in DDX41-heterogyzgous mouse.
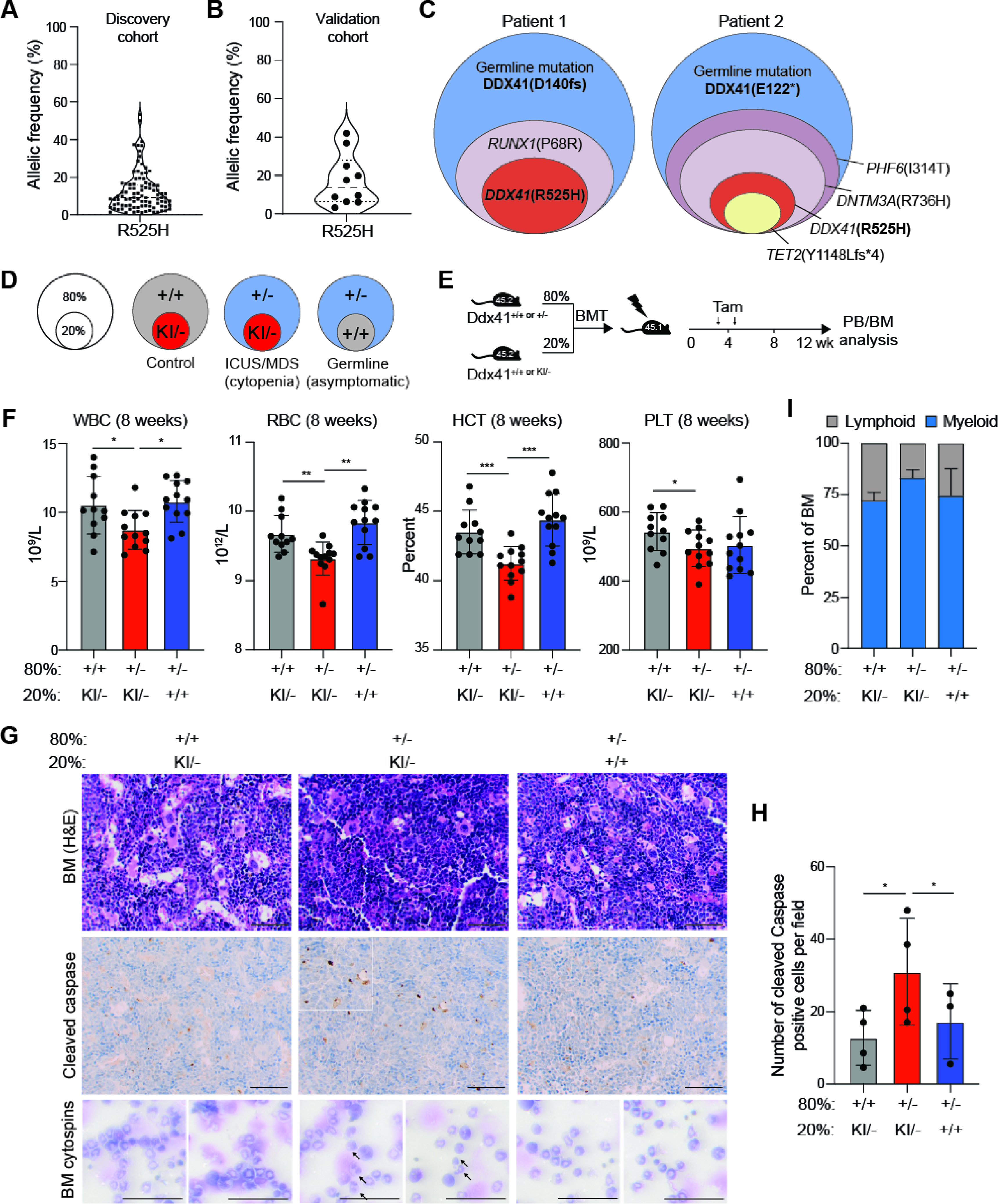
(A) Variant allele frequency for R525H in germline DDX41 mutant patients from public data sets. (B) Variant allele frequency for R525H in germline DDX41 mutant patients. (C) Sample variant allele proportions in germline DDX41 mutant patients. (D) Schematic of mixed transplant to model minor clone with biallelic DDX41 mutations. (E) Tamoxifen injection schedule for mixed BM transplants. (F) Blood counts of transplant recipients 8-weeks post-tamoxifen (*P < 0.05; **P < 0.01; ***P < 0.0001). (G) BM sections from transplant recipients 8-weeks post-tamoxifen stained with H&E and IHC for cleaved caspase. Arrows indicate abnormal/dysplastic neutrophils, identified by segmented nuclei. Scale bars, 100 μm. (H) Quantification of cleaved-caspase staining in G (*P < 0.05; n = 3 independent mice). (I) Percent of myeloid and lymphoid BM MNCs 8-weeks post-tamoxifen.
Discussion
Here we report that DDX41 is critical for ribosome biogenesis through snoRNA processing and essential for hematopoiesis. Biallelic DDX41-mutant BM cells exhibit impaired snoRNA processing and ribosome function, which leads to cell death of proliferating hematopoietic cells, but not LT-HSCs. Furthermore, we describe a mechanism whereby minor HSPC clones harboring biallelic DDX41 mutations contribute to disease pathogenesis by exacerbating hematopoietic defects in heterozygous DDX41-mutant BM cells. We posit that the increased cell death of the proliferating and differentiating BM cells derived from the minor biallelic DDX41-mutant clone creates an environment which contributes to hematopoietic defects. We propose that crosstalk between heterozygous DDX41-mutant HSPCs and the minor biallelic DDX41-mutant BM cells contributes to MDS in DDX41-mutated patients. Previous studies have identified increased inflammation in MDS HSPCs with deficient ribosome function (Schneider et al., 2016, Bibikova et al., 2014). In addition, cell death can result in secretion of inflammatory signaling into the BM milieu (Bergsbaken et al., 2009). Thus, dysregulation of innate immune pathways and inflammatory signals may also play a role in DDX41-mutant MDS (Barreyro et al., 2018, Trowbridge and Starczynowski, 2021). Nevertheless, the mechanism by which the minor biallelic DDX41-mutant BM cells contribute to ineffective hematopoiesis in the context of heterozygous DDX41-mutant BM cells remains unknown. One can speculate that the activity of DDX41 as an innate immune sensor might play a role in the altered response of monoallelic DDX41-mutant HSPCs to damage associated molecular patterns (DAMPs) in the BM (Jiang et al., 2017, Maciejewski et al., 2017). Thus, one potential mechanism is that increased DAMPs from the apoptotic biallelic DDX41-mutant BM cells can induce innate immune signaling preferentially in the heterozygous DDX41-mutant BM cells as compared to wild-type BM cells. It is also conceivable that MDS arises through the combined effect of age-related dyshematopoiesis of monoallelic DDX41-mutant BM and reduced hematopoietic output from the HSC pool due to the persistence of biallelic mutant LT-HSC.
Defects in ribosome biogenesis are known to contribute to MDS as evidenced by recurrent mutations in ribosome-related genes. Germline mutations in genes involved in ribosome structure, translation initiation, and rRNA pseudouridylation cause congenital disorders associated with BM failure and MDS (Bannon and DiNardo, 2016). In addition, haploinsufficiency of the small ribosomal subunit protein RPS14 plays an important role in acquired del(5q) MDS pathogenesis (Nakhoul et al., 2014, Ebert et al., 2008). Our data suggests that DDX41 mutations also promote MDS through ribosome translation defects but through aberrant snoRNA processing. snoRNAs have been implicated in hematopoiesis and cancer, including AML (Su et al., 2014, Liang et al., 2019, Zhou et al., 2017, Pauli et al., 2020). Moreover, loss of snoRNA function causes cell cycle impairment by several mechanisms, including p53 activation or decreased PI3K/AKT and Wnt/β-catenin signaling (Su et al., 2014, Liang et al., 2019).
HSCs have low protein synthesis and therefore reduced protein translation demands (Signer et al., 2014). In contrast, rapidly proliferating HPCs have high levels of protein synthesis, especially erythroid progenitors, which are particularly sensitive to translation deficits (Magee and Signer, 2021, Schneider et al., 2016, Dutt et al., 2011). Thus, HSC are relatively protected from ribosome defects while proliferating progenitors are sensitive to such defects. Since the second-hit DDX41 mutation is not selected against in the HSC compartment due to low protein synthesis, it is likely that these biallelic DDX41-mutant HSCs can persist and contribute to cell-intrinsic and -extrinsic defects during hematopoiesis. The low variant allele frequency of second-hit DDX41 mutations observed in patients supports this model. The specific preference for acquired missense mutations in the helicase domain (i.e., R525H) rather than frameshift mutations might indicate that complete loss of DDX41 function is not compatible with HSC survival and that R525H is hypomorphic for the critical DDX41 function. The residual DDX41 activity in the R525H mutant protein may allow for HSC to survive and even be selected for due to low translation rates. In support of this idea, we observed that biallelic Ddx41KI/− HSPCs have milder phenotypes as compared to Ddx41−/− HSPCs.
Recent clinical reports suggest that many germline DDX41 MDS patients present initially with ICUS (Choi et al., 2021). This is consistent with the progressive age-dependent anemia and thrombocytopenia that we observed in Ddx41+/− mouse models. The interaction between the aged BM and defective HSCs bearing the acquired DDX41 mutation is one potential mechanism that promotes overt MDS. It is possible that the non-hematopoietic BM microenvironment cells, which are also heterozygous for DDX41 in germline patients, play a role in the disease. We have monitored full-body Ddx41+/− mice up to 18 months of age and have not observed significant hematopoietic defects, suggesting that stress on the BM, such as from BM transplantation or from crosstalk with the biallelic DDX41-mutant BM cells, might be required for hematopoietic defects to arise during the short murine lifespan.
The age-dependent defects in Ddx41+/− BM cells were especially pronounced in the erythroid compartment. The erythroid cell lineage is particularly sensitive to ribosome defects (Da Costa et al., 2020). Although we did not observe significant defects in protein synthesis or snoRNA processing in Ddx41+/− HSPCs, we speculate that subtle defects in this pathway contributes to age-dependent erythroid defects. Perhaps this snoRNA/ribosome defect in heterozygous DDX41-mutant cells only arises after aging or is below the level of detection of our assays. Alternatively, loss of a single copy of DDX41 could affect other cellular pathways. Nonetheless, modest defects associated with loss of a single copy of DDX41 is sufficient to contribute to hematopoietic defects upon aging. Association of TP53 mutations in DDX41-mutant patients also supports a role for ribosome defects as p53 activation plays a central role in anemia caused by ribosome stress (Fok et al., 2017, Danilova et al., 2008, Dutt et al., 2011, Quesada et al., 2019). Further experimentation in this area is necessary to determine the precise mechanism of age-dependent hematopoietic inefficiency in heterozygous DDX41 patients.
Limitations of the studies
Patients with DDX41 germline mutations exhibit loss of one DDX41 allele in all cells, however our studies primarily focused on the hematopoietic system. Future studies will be necessary to explore the contribution of a DDX41-mutant BM niche to hematopoiesis. Monoallelic DDX41 mutations result in a competitive advantage upon serial transplantation, however the physiological conditions under which this advantage occurs in patients is unclear. Moreover, the mechanistic basis for the competitive advantage of monoallelic DDX41-mutant cells remains unknown. We also demonstrated that rare biallelic DDX41-mutant HSPCs contribute to disease by exacerbating hematopoietic defects in heterozygous DDX41-mutant BM cells, which may occur through cell-extrinsic factors. However, the signal(s) from the rare biallelic DDX41-mutant BM cells that impairs the heterozygous DDX41-mutant BM cells, but not WT BM cells requires further investigation. Lastly, the variant allele frequency of the acquired DDX41 R525H mutation is uniformly low at diagnosis. Since the allele frequency was determined at a single time point, we are unable to comment on the stability of the biallelic mutant clone during the course of disease.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Daniel Starczynowski (Daniel.Starczynowski@cchmc.org).
Materials Availability
Plasmids and mouse lines generated for this study are available upon request and will be fulfilled by the lead contact, Daniel Starczynowski (Daniel.Starczynowski@cchmc.org).
Data and Code Availability
Data
Next Generation Sequencing data, including data for mouse RNA-Seq, HyperTRIBE, and CRISPR knockout screen, have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Patient sequencing data is available by request to Torsten Haferlach, Munich Leukemia Laboratory, Munich, Germany (torsten.haferlach@mll.com).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-DDX41 antibody | Abcam | Ab210809 |
| Rabbit monoclonal anti-Vinculin antibody (E1E9V) | Cell Signaling Technology | AB_2728768 |
| Rabbit polyclonal Pan-Actin Antibody | Cell Signaling Technology | AB_2313904 |
| Rabbit monoclonal anti-GAPDH antibody (D16H11) | Cell Signaling Technology | AB_10622025 |
| Anti-B220 APC | eBioscience | AB_469395 |
| Anti-CD11b PE-Cy7 | eBioscience | AB_469587 |
| Anti-CD45.1 BV510 | BioLegend | AB_2563378 |
| Anti-CD45.2 APC-eFluor780 | eBioscience | AB_1272175 |
| Anti-CD48 FITC | eBiosceince | AB_465078 |
| Anti-CD117 APC | eBioscience | AB_469429 |
| Anti-CD150 PE-Cy7 | BioLegend | AB_439797 |
| Anti- Ly-6G (Gr-1) eFluor 450 | eBioscience | AB_1548788 |
| Anti-Ly-6A/E (Sca-1) PE | eBioscience | AB_466086 |
| Streptavidin eFluor® 450 | eBioscience | AB_10359737 |
| Mouse Hematopoietic Lineage Biotin Panel | eBioscience | AB_476399 |
| Anti-CD3e FITC | eBioscience | AB_464882 |
| Anti-Cleaved Caspase 3 | ||
| AnnexinV APC | eBioscience | AB_2575166 |
| Bacterial and virus strains | ||
| OneShot TOP10 Competent Cells | ThermoFisher | C404010 |
| Chemicals, peptides, and recombinant proteins | ||
| Mouse Flt3-ligand cytokine | PeproTech | Cat# 250-31 |
| Mouse IL-3 cytokine | PeproTech | Cat# 213-13 |
| Human IL-6 cytokine | PeproTech | Cat# 200-06 |
| Mouse SCF cytokine | PeproTech | Cat# 250-03 |
| Human TPO cytokine | PeproTech | |
| Methocult GF M3434 | Stem Cell Technologies | Cat# 03434 |
| Dulbecco’s PBS without Ca and Mg | VWR | Cat# 45000-446 |
| A/G Protein PLUS-Agarose | Santa Cruz | Cat# sc-2003 |
| RPMI | Fisher | Cat# SH30027.01 |
| RPMI, no methionine | ThermoFisher | Cat# A1451701 |
| IMDM | Corning | Cat# 10-016-CV |
| DMEM | Fisher | Cat# SH30022FS |
| TransIT-LT1 transfection reagent | Mirus | Cat# MIR2306 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11550 |
| Penicillin/Steptomycin | ThermoFisher | Cat# SV30010 |
| Superscript II | ThermoFisher | Cat# 18064014 |
| RnaseOUT RecombinantRibonuclease Inhibitor | ThermoFisher | Cat# 10777019 |
| N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMC) | ThermoFisher | Cat# C106402-1G |
| MnCl2 | Sigma | Cat# M1787 |
| Bicine | Sigma | Cat# B3876 |
| Tamoxifen | Sigma | Cat# T-5648 |
| 4-Hydroxytamoxifen | Sigma | Cat# H-7904 |
| Critical commercial assays | ||
| CellTiterGlo kit | Promega | Cat# G7570. As in Melgar et al. Sci Transl Med. 2019. |
| Mouse hematopoietic progenitor cell enrichment kit | Stem Cell Technologies | Cat# 19856 |
| Pierce Biotin 3’ End DNA Labeling Kit | ThermoFisher | Cat# 89818 |
| NEBNext Magnesium RNA Fragmentation Module | ThermoFIsher | Cat# E6150S |
| POWRUP SYBR green Master Mix | ThermoFisher | Cat# A25776 |
| Click-IT L-Homopropargylglycine | ThermoFisher | Cat# C10186 |
| NorthernMAX Kit | ThermoFisher | Cat# AM1940 |
| mirVana miRNA Isolation Kit | ThermoFisher | Cat# AM1561 |
| GeneAmp Fast PCR master mix 2x | ThermoFisher | Cat# 4359187 |
| Click-IT Edu Alexa Fluor 647 | ThermoFisher | Cat# C10424 |
| Chemiluminescent Nucleic Acid Detection Module Kit | ThermoFisher | Cat# 89880 |
| Zymo Quick-RNA Miniprep Kit | Zymo Research | Cat# R1050 |
| Deposited data | ||
| LSPC RNA-Seq | This study | GSE178895 |
| HyperTRIBE | This study | GSE178895 |
| CRISPR Screen | This study | GSE178895 |
| Experimental models: Cell lines | ||
| Mouse MLL-AF9 immortalized Rosa-CreERT+ lineage-negative cells ( LSPC). Gentoypes: Ddx41+/+, Ddx41+/f, Ddx41f/f, Ddx41KI/+, Ddx41KI/f | This paper | N/A |
| THP-1 cells | ATCC | TIB-202 |
| HEK293T cells | ATCC | ATC CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: C57Bl/6N Ddx41tm1a | KOMP | MMRRC_047340-UCD |
| Mouse: C57Bl/6N Ddx41conditionalR525H | Ozgene | N/A |
| Mouse: Rosa26-CreERT2 (B6.129-Gt(ROSA)26Sortm1 (cre/ERT2)Tyj/J) | Jackson | RRID:IMSR_JAX:008463 |
| Mouse: Rosa26-Cas9 (B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J) | Jackson | RRID:IMSR_JAX:026179 |
| Mouse: Rosa26::Flpe (B6.129S4-Gt(ROSA)26Sortm1 (FLP1)Dym/RainJ) | Jackson | RRID:IMSR_JAX:009086 |
| Oligonucleotides | ||
| Primers listed in Supplemental Table 4 | This study | N/A |
| Recombinant DNA | ||
| MSCV MLL-AF9 pGK-GFP | Gift from Ashish Kumar laboratory | N/A |
| pCDLN packaging vector | Gift from Gang Huang laboratory | N/A |
| pMD.2 VSV-G envelope vector | Gift from Gang Huang laboratory | N/A |
| M57 retroviral gag/pol plasmid | Gift from Gang Huang laboratory | N/A |
| pLKO.1 scrambled control mCherry | This study | Cloned from TRCN SHC202 |
| pLKO.1 shSnora7a-1 mCherry | This study | N/A |
| pLKO.1 shSnora7a-2 mCherry | This study | N/A |
| pLKO.1 shSnora16a-1 mCherry | This study | N/A |
| pLKO.1 shSnora70-1 mCherry | This study | N/A |
| pLKO.1 shSnora70-2 mCherry | This study | N/A |
| pLKO.1 shSnora71-1 mCherry | This study | N/A |
| pLKO.1 shSnora74a-1 mCherry | This study | N/A |
| pLKO GFP shDDX41 #1 | This study | Cloned from TRCN0000001268 |
| pLKO GFP shDDX41 #2 | This study | Cloned from TRCN0000001270 |
| pLKO scrambled shRNA GFP | This study | Cloned from TRCN SHC202 |
| pGK-GFP DDX41-ADARcd | This study | N/A |
| pGK-GFP ADARcd | Rahman et al. Nat Protoc. 2018 | N/A |
| MSCV-HA-DDX41-mCherry | This study | |
| MSCV-HA-DDX41(R525H)-mCherry | This study | |
| MSCV-IRES-mCherry | This study | Addgene, Cat# 52114 |
| Software and algorithms | ||
| iGeak | Choi et al. BMC Genomics. 2019 | N/A |
| AltAnalyze | Emig, et al. Nucleic Acids Res. 2010 | N/A |
| HyperTRIBE | Rahman et al. Nat Protoc. 2018 | N/A |
| MAGeCK | Li et al. Genome Biology, 2014 | N/A |
| Other | ||
Code
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animals were bred and housed in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of Cincinnati Children’s Hospital Medical Center. All mouse strains were kept on C57BL/6N background. Ddx41flox mice were derived from Ddx41tm1a mice purchased from the UC Davis KOMP Repository by breeding to Rosa26::Flpe mice to remove lacz-Neo cassette bracketed by Frt sites. Ddx41R525H mice were produced by Ozgene (Australia) by targeting of the endogenous Ddx41 locus in ES cells, which were transmitted through the germline in chimeric mice. These mice were bred to Rosa26-CreERT2 (Jackson) mice to allow tamoxifen-inducible excision of floxed regions. For CRISPR screen, Ddx41flox;Rosa26-CreERT2 mice were bred to Rosa26-Cas9 C57BL/6 mice (Jackson). For bone marrow transplant experiments, aged and gender matched mice between 8 and 12 weeks of age were used as donors. For competitive transplant experiments (Figure 2), the donor mice were female. For all other transplants, the donors were male. 1e6 bone marrow mononuclear cells (BMNC) suspended in 200μL PBS were injected into the tail vein of lethally-irradiated recipient mice. 2e6 total bone marrow cells (1e6 of each strain) were injected for competitive transplants. All recipient mice were age-matched female C57Bl/6 BoyJ mice between 6 and 12 weeks of age. All mice were housed in identical conditions in the Rodent Barrier Facility, four mice per cage. The time of analysis as it relates to time post-transplant and post-tamoxifen treatment are included in the figures and figure legends for each experiment.
For tamoxifen injections, 1mg of tamoxifen dissolved in 50μl corn oil was injected intraperitoneal daily for 5 consecutive days. Injections were repeated one week later. Mice were bled monthly by submandibular puncture to obtain 50–100μl of blood for CBC and flow cytometry analysis. When mice were moribund, they were sacrificed by CO2 asphyxiation, and bones and spleen were harvested immediately for analysis.
To isolate BMNC, bone marrow cells were isolated from mice by crushing with mortar and pestle, and red blood cells were lysed in BD Pharm Lyse (BD Biosciences, 555899). Lineage-negative cells were isolated with EasySep Mouse Hematopoietic Progenitor Isolation Kit (StemCell Technologies). Lineage negative cells were cultured in IMDM with 10% FBS, 1% penicillin-streptomycin, and 50ng/mL of hIL-6, mSCF, mIL-3, TPO and FLT3 ligand.
Cell lines
Mouse LSPC were derived from Ddx41flox and Ddx41R525H;Rosa-CreERT lineage-negative bone marrow cells. Lineage-negative cells were purified as above and then they were cultured overnight in in IMDM with 10% FBS, 1% penicillin-streptomycin, and 50ng/mL of hIL-6, mSCF, and mIL-3. The following day, they were transduced with MSCV-MLL-AF9-GFP virus to achieve 10–20% transduction(Niederkorn et al., 2020). The cells were plated in methycellulose colony assays (M3434, StemCell Technologies) and transferred to a new plate every 7 days for a total of 3 platings. The third plating was then transferred to liquid culture and the cells were passaged as cell lines. LSPC lines were cultured in IMDM with 10% FBS, 1% penicillin-streptomycin, and 10ng/mL of hIL-6, mSCF, and mIL-3 with passaging every 2–3 days. To induce Cre activity, cells were treated for 24h with 1μM 4-OH-tamoxifen (Sigma, H-7904) followed by a media change to remove the tamoxifen. THP-1 cell lines were cultured in RPMI 1640 medium with 10% FBS and 1% Penicillin-streptomycin. HEK293T cell lines used for virus production were cultured in DMEM with 10% FBS and 1% Penicillin-streptomycin.
METHOD DETAILS
RNA-Sequencing of mouse LSPC
For RNA-sequencing, RNA was isolated from triplicate samples of wildtype, Ddx41+/f, and Ddx41f/f LSPC 48h post- treatment with 4-hydroxytamoxifen or ethanol (1:1000) using Quick RNA MiniPrep Kit (Zymo Research, R1055). RNA libraries were prepared with polyA selection using the TruSeq RNA Library Prep Kit v2 (Illumina) and then sequenced on a HiSeq2500. We obtained 30–40M 75bp paired-end reads for each sample. Reads were mapped to the mouse genome (mm10), and then analyzed for differential gene expression by iGeak (Choi and Ratner, 2019). For splicing analysis of the RNA-Seq data, alternative splicing events were predicted using AltAnalyze (http://www.altanalyze.org, v2.1.3 ) with mouse mm10 Ensembl database (v72) (Emig et al., 2010). Briefly, alternative gene/exon statistics were calculated using a unpaired moderated t-test option and default cutoffs (dabg_p (P value corresponds to the detection above background (DABG)): 1.0, junction expression threshold: 5.0, exon_exp_threshold: 5.0, gene_exp_threshold: 200.0, exon_rpkm_threshold: 0.5, gene_rpkm_threshold: 1.0). To identify differential alternative exon usages and alternative splicing events, the MultiPath-PSI splicing algorithm was chosen with p-value cutoff < 0.05 (alt_exon_fold_variable: 0.1, gene_expression_cutoff: 10.0.
HyperTRIBE
For HyperTRIBE (https://github.com/rosbashlab/HyperTRIBE/, v1.0.0), a single RNA sample was isolated from THP-1 cells transduced with ADARcd or DDX41-ADARcd constructs and control (non-transduced) cells (Rahman et al., 2018). RNA was depleted of ribosomal RNA and then libraries were prepared by TruSeq Stranded Total RNA Kit (Illumina) and sequenced on a NovaSeq. 20–30M 100bp paired-end reads were obtained. The reads trimmed using trimmomatic (https://github.com/timflutre/trimmomatic/, v0.33) were mapped to human hg38 genome using bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/, v2.2.6) and to the transcriptome using STAR (https://github.com/alexdobin/STAR/ v2.7.1a). Aligned reads were processed to remove PCR duplicates using Picard (https://broadinstitute.github.io/picard/, v2.18.22), sorted by SAMtools (http://samtools.sourceforge.net/, v1.3), then analyzed with a 1% cutoff.
Patient sample sequencing
The cohort comprised bone marrow from 565 AML, 486 MDS cases and 63 healthy bone marrow samples. Cases were diagnosed by the Munich Leukemia Laboratory between 09/2005 and 04/2017. The diagnoses were established by a combination of cytomorphology, immunophenotyping, cytogenetics, and molecular genetics techniques adhering to WHO 2016 guidelines. All patients provided their written informed consent for scientific evaluations in accordance with the Internal Review Board-approve protocol and this study adhered to the tenets of the Declaration of Helsinki. To identify samples with R525H mutations, DNA was extracted from whole bone marrow and prepared for sequencing using the Illumina Truseq PCR free library prep kit. The libraries were sequenced on the NovaSeq 6000 or the HiSeqX with a median of 100x coverage. Paired-end reads were mapped to the hg19 genome using Isaac3. Streka2 was used for variant calling to identify R525H mutations. Two MDS and two AML patients harboring the R525H mutations were identified. For RNA-Sequencing, 250ng of total RNA was extracted and prepared for high throughput sequencing using the Illumina TruSeq Total Stranded RNA library preparation kit. The samples were sequenced on the NovaSeq 6000 producing paired-end reads of 100bp with a median sequencing depth of 50 million reads per sample. Samples were separated by bcl2fastq 1.8.4 and aligned with STAR 2.5.0 to the hg19 reference genome. Gene counts for mRNAs and snoRNAs were generated using Cufflinks 2.2.1 and normalized using trimmed mean of M-values (TMM) normalization (Robinson et al.) to produce log2 CPM for each sample. DNA and RNA sequences are available by request to Torsten Haferlach, Munich Leukemia Laboratory, Munich, Germany.
Polysome analysis
LSPC were treated with 4-Hydroxytamoxifen (1μM) or ethanol (1:1000) for 48hrs. 15–20M live cells were treated for 5min at 37°C and 5% CO2 with cycloheximide (100ug/mL). Cells were washed twice with 1xPBS containing cycloheximide (100ug/mL). Cells were resuspended in Hypotonic buffer (5mM Tris-HCl (pH 7.5), 2.5mM MgCl2, 1.5mM KCl, 1x protease inhibitor cocktail (EDTA-Free)) with 80U of RNase inhibitor and vortexed for 5sec, then incubated on ice for 5min. Hypotonic lysis buffer (3 parts hypotonic buffer, 1 part 10% Sodium deoxycholate, 1 part 10% Triton-X 100) containing 2.5mM DTT and 100ug/mL cycloheximide was added 1:1 to the cells in hypotonic buffer. Samples were vortexed for 1min in 5min pulses and then centrifuged at 16,000xg for 7min at 4°C. OD was measured at 254nm using a Nanodrop One and samples were adjusted to the same OD in a final volume of 200μl. A Gradient Master and Piston Fractionator Combo Unit (BioComp Instruments, New Brunswick, Canada) was used to both make gradients and fractionate. Sucrose gradients made from vacuum filtered 5% and 50% sucrose in DEPC treated water were formed with the Gradient Master. Samples were layered onto each gradient and spun at 35,000rpms for 3hrs in an SW41 ultracentrifuge rotor (Beckman Coulter). Gradients were fractionated with the Piston Fractionator and measured with a UV detector at 254nm. Gradient Profiler Software (BioComp Instruments) was used to capture and export all measurements.
Plasmids and transduction
Short hairpin RNAs (shRNAs) were obtained from the Open Biosystems TRC lentiviral shRNA library in the pLKO.1 vector. The puromycin resistance cassette was replaced with GFP or mCherry by subcloning using BamHI and KpnI sites. ShRNA clones were as follows: shDDX41 #1 (TRCN0000001268), shDDX41 #2 (TRCN0000001270), and control shRNA (TRCN SHC202). pLKO.1 vectors with anti-snoRNA shRNAs were generated following the addgene pLKO.1 cloning protocol (https://www.addgene.org/protocols/plko/). Briefly, complementary oligos containing the desired hairpin sequence were annealed together and then cloned into the EcoRI and AgeI sites of the pLKO.1-TRC cloning vector (Addgene), which had been modified as above to express mCherry in place of the puromycin resistance gene. Oligo sequences are listed in Supplementary Table 4. For the MSCV-HA-DDX41 plasmids, wild-type DDX41 was PCR cloned using primers containing an N-terminal HA-tag sequence and restriction enzyme sites, and then the cDNA was cloned into the MSCV-IRES-mCherry vector (Addgene, #52114) at the EcoRI and BamHI sites. For the HyperTRIBE plasmids, the DDX41-ADARcd or ADARcd alone cDNA sequences were generated as plasmid inserts by Genewiz and then were cloned into a pMSCK-PGK-GFP retroviral vector using EcoRI and BglII sites. Lentiviral supernatants (pLKO) were made by transfecting HEK293T cells using Trans-LT (Mirus) transfection reagent with lentiviral plasmid, pCDLN packaging vector, and pMD.2 VSV-G envelope vector and harvesting the culture medium 48hrs post-transfection. Retroviral supernatants were made by the same process using M57 (packaging) and RD114 (envelope) helper plasmids. Cells were transduced with viral supernatant containing 0.8 μg/ml polybrene for 24hrs. Expression of GFP/mCherry was determined using flow cytometry.
Proliferation assay
THP-1 shDDX41 cell lines were plated 72hrs after transduction and counted daily for four days with Typan blue using a Countess II FL (Fisher). LSPCs were plated at 1e5 cells/ml, treated with 4-Hydroxy tamoxifen (1μM) or ethanol (1:1000), and counted daily for four days with trypan blue using a Countess II FL (Fisher).
Colony assay
Lineage-negative cells were treated with 4-Hydroxytamoxifen (1μM) or ethanol (1:1000) for 48 hrs and were then plated in triplicate in MethoCult GF M3434 and incubated for 12 days. Colonies were counted using Stemvision (StemCell Technologies).
Immunoblotting
Whole cell lysates were made with RIPA buffer (20 mM Tris HCL pH 7.4, 37 mM NaCL, 2mM EDTA, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% NaDeoxycholate) in the presence of PMSF (10 mM final), complete Mini Protease Inhibitor Cocktail (Roche), and Phosphatase Inhibitor Cocktail 2 and 3 (Sigma Aldrich). Lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted. Immunoblotting was performed with the following antibodies: DDX41 (Abcam, ab210809), Vinculin (E1E9V) XP® Rabbit mAb (Cell Signaling, 13901), Pan-Actin (Cell Signaling, 4968) and GAPDH (D16H11) XP™ Rabbit mAb (Cell Signaling, 5174).
CellTiter-Glo luminescent cell viability assay
LSPC cells were treated with 4-Hydroxytamoxifen (1μM) or ethanol (1:1000) for 24hrs. Cells were collected and plated in a 96 well plate with or without puromycin (0.1ug, 0.3ug, and 0.5ug). CellTiter-Glo® Luminescent Cell Viability Assay (Promega) analysis was performed at 24hrs and 48hrs post puromycin treatment as previously described (Melgar et al., 2019).
Flow cytometry
THP-1 shDDX41 cells were collected six days after transduction and stained for Annexin V (1:100) (eBiosciences 88–8007-74), TMRE (Abcam, ab113852), and HPG (Life Technologies, C10186). LSPC and Lineage negative cells were treated with 4-OHT Hydroxy-tamoxifen (1μM) or ethanol (1:1000) for 48hr or 72hr and then were stained for Annexin V, EdU (Click-IT EdU flow kit, Life Technologies, C10634), TMRE (Molecular Probes, T669), and HPG (Click-IT HPG kit, Life Technologies, C10429). Mouse bone marrow and blood mononuclear cells were stained for cell surface markers in PBS containing 2% FCS at recommended dilutions. Antibodies are contained in the Key Resources Table. All flow analyses were performed on LSRII (BD Biosiences) and data were analyzed using FlowJo (Muto et al., 2020).
Pseudouridine analysis
RNA was isolated using the Quick RNA MiniPrep (Zymo Research). 10μg of RNA was fragmented using the NEBNext Magnesium RNA Fragmentation Module (New England Biolabs). Fragmented RNA was precipitated by adding 3M Sodium Acetate followed by 100% ice cold Ethanol. Samples were incubated for 30 minutes at −20°C then pelleted for 30min at 4°C. Pellets were washed once with ice cold 75% ethanol and centrifuged for 10min at 4°C. Pellets were air dried at room temperature for 10 mins then resuspended in 5mM EDTA and incubated for 5 minutes at 80°C. Denatured RNA (9uL) was added to 91μl BEU buffer (500mM Bicine pH 8.5, 0.5M EDTA, 8M Urea) with or without 0.2M N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMC) (Sigma) and incubated for 20 minutes at 37°C. RNA was precipitated as above and resuspended in 50uL of sodium carbonate solution (50mM Sodium Bicarbonate pH 10.4, 0.5M EDTA) then incubated for 6 hours at 37°C. RNA was precipitated as above and dried pellets were dissolved in 10μl RNase free water. For cDNA synthesis, each sample was added with 1uL of 100 μM Random Hexamer primer (TaKaRa) and incubated for 5min at 65°C, and then on ice for 1min. 8uL of freshly prepared RT buffer(125mM Tris pH 8.0, 15mM MnCl2, 187.5mM KCl, 1.25mM dNTPs, 25mM DTT) was added and incubated for 2min at 25°C. 1uL of Superscript II reverse transcriptase (200U/uL) (Life Technologies) was added and samples were loaded into a thermocycler (25°C for 10 min, 42°C for 3h and heat-inactivated at 70°C for 15 min). cDNA was diluted 1:10 for qPCR analysis. The primer extension assay for quantification of pseudouridine was conducted as described previously(McCann et al., 2020).
Genotyping
DNA was isolated from mouse tails with 50mM NaOH for 1hr at 95°C. 100mM Tris HCL was added and samples were vortexed until tails dissolved. Tail debris was pelleted by centrifuging samples for 2min at 15,000xg. 1–2uL of DNA was genotyped for DDX41 (CSD-Ddx41-ttR2, CSD-Ddx41-F2), Rosa-CreERT (CreER, Rosa26-R, Rosa26-F2) using GeneAmp Fast PCR master mix 2x (ThermoFisher). Genotyping for DDX41 R525H was performed using Primetime gene expression master mix (IDT) and IDT primer probes (Lo5WT, GoConK, GoK, and TERT). DNA from blood or bone marrow mononuclear cells (BMNC) was genotyped for DDX41 excision (CSD-Ddx41-R, CSD-Ddx41-F2) using GeneAmp Fast PCR master mix 2x (ThermoFisher). POWERUP SYBR green Master mix was used for quantitative DDX41 excision (Ddx41 ex9 F and LoxP R) in DNA from blood or BMNC.
Quantitative PCR
RNA was isolated from cells with the Quick RNA MiniPrep Kit (Zymo Research, R1055). 1 ug of RNA was used for cDNA synthesis using the High Capacity RNA to cDNA Kit (ThermoFisher, 4387406). cDNA was diluted 1:10 and samples were run on a 96-well Fast Thermal Cycling plate (Invitrogen, 4346907) in triplicate using PowerUp SYBR Green Master Mix (ThermoFisher, A25776). Small RNA fractions were isolated using the mirVana miRNA isolation kit (ThermoFisher, AM1560) according to manufacturer’s instructions. List of primers is included in Supplemental Table 4.
Northern Blot
RNA was isolated from cells with the Quick RNA MiniPrep Kit (Zymo Research, R1055). The NorthernMax Kit (ThermoFisher, AM1940) was utilized following manufacturer’s instructions. 10 ug of RNA was loaded per sample into agarose gel. Pierce Biotin 3’ End DNA Kabeling Kit (ThermoFisher, 89818) was used to label probes. Probes were diluted 1:100 for blotting. The Chemiluminescent Nucleic Acid Detection Module Kit (ThermoFisher, 89880) was utilized for detection. Pictures of the developed blots were taken using BioRad Chemidoc.
CRISPR-Cas9 screen
Lin− BM cells were harvested from three Ddx41+/+;Rosa-Cas9 and three Ddx41f/f;Rosa-Cas9 mice and cultured as above for 24h. They were transduced with the Brie sgRNA library on retronectin coated plates for 24hr (Doench et al., 2016). One quarter of each wild-type culture was harvested as a pool for the control sample DNA at this time. The remaining cells were then washed and replated in culture medium containing 1μM Tamoxifen for 4 days. The cells were then harvested for DNA, and the sgRNA sequence was amplified and attached to Illumnina sequencing adaptors as described previously (Doench et al., 2016). Samples were sequenced on a NextSeq5000 for 75bp paired-end reads. We used the MAGeCK pipeline for data processing and analysis ((v0.5.9, https://sourceforge.net/p/mageck). First, original paired-end reads were trimmed using cutadapt (v1.9.1, https://cutadapt.readthedocs.io) using the following parameter sets: “-g GGACGAAACACCG -a TtttagagctaG -A CGGTGTTTCGTCC -G CTAGCTCTAAAA -e 0.1 -m 20 -l 20”. Next, MAGeCK-count was used to calculate sgRNA read counts using trimmed reads and mouse CRISPR knockout pooled library (Brie, https://www.addgene.org/pooled-library/broadgpp-mouse-knockout-brie/). Finally, MAGeCK-MLE was used to predict gene essentiality from the resulting count matrix after considering a control sample (Li et al., 2014). This method reports beta-scores to call gene essentialities (>0: positive selected genes, <0: negatively selected genes) and we chose genes passing two cutoffs: beta-score > 0.5 and p < 0.05. Enrichment analyses using resulting 499 target genes were performed using Enrichr (https://maayanlab.cloud/Enrichr) using default options.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, results are depicted as the mean ± standard deviation. Statistical details for each experiment are provided in the figure legends. Statistical analyses were performed using Student’s t-test for direct comparisons or one-way ANOVA test for multiple-groups comparisons. For correlation analysis, Pearson correlation coefficient (r) was calculated. To determine if the slopes or intercepts were different between two correlative comparisons, simple linear regression analysis was conducted. All graphs and analysis were generated using GraphPad Prism software.
Supplementary Material
Table S1. Differentially expressed genes in RNA-Seq of Ddx41f/f LSPC (+TAM vs control), Related to Figure 4.
Table S2. HyperTRIBE data for DDX41-bound RNAs in THP-1 cells, Related to Figure 4G.
Table S3. Significantly enriched genes in genome-wide CRISPR screen in Ddx41−/− Lin− BM cells, Related to Figure 6G.
Table S4. Primer sequences used in this study, Related to STAR Methods.
Highlights.
Monoallelic DDX41 mutations result in age-dependent hematopoietic defects
DDX41 regulates snoRNA processing and protein synthesis
Biallelic DDX41 mutations are not compatible with proliferating hematopoietic cells.
Biallelic DDX41-mutant cells are disease-modifying and accelerate hematopoietic defects
Acknowledgements
This work was supported by the National Institute of Health (R35HL135787, R01DK102759, R01DK113639 to DTS; K01DK121733 and F32HL143993 to TMC; R01HL132071 to RAP and JPM; R35HL135795 to JPM; F31HL131140 to CEH), Cancer Free Kids (DTS), American Society of Hematology (TMC), the Edward P. Evans Foundation (TMC), and the Vera and Joseph Dresner Foundation (RAP). We thank Jeff Bailey and Victoria Summey for assistance (Comprehensive Mouse and Cancer Core at CCHMC). We thank the Marie Dominique-Filippi and the Starczynowski lab for suggestions and feedback. We thank Dr. Lynn Lee for advice for the HyperTRIBE analysis.
Footnotes
Declaration of Interests: DTS serves on the scientific advisory board at Kurome Therapeutics. All other authors have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- ATZORN V, FRAGAPANE P & KISS T 2004. U17/snR30 is a ubiquitous snoRNA with two conserved sequence motifs essential for 18S rRNA production. Mol Cell Biol, 24, 1769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANNON S & DINARDO C 2016. Hereditary Predispositions to Myelodysplastic Syndrome. International Journal of Molecular Sciences, 17, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARREYRO L, CHLON TM & STARCZYNOWSKI DT 2018. Chronic immune response dysregulation in MDS pathogenesis. Blood, 132, 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGSBAKEN T, FINK SL & COOKSON BT 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol, 7, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIBIKOVA E, YOUN MY, DANILOVA N, ONO-URUGA Y, KONTO-GHIORGHI Y, OCHOA R, NARLA A, GLADER B, LIN S & SAKAMOTO KM 2014. TNF-mediated inflammation represses GATA1 and activates p38 MAP kinase in RPS19-deficient hematopoietic progenitors. Blood, 124, 3791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDOSO SR, RYAN G, WALNE AJ, ELLISON A, LOWE R, TUMMALA H, RIO-MACHIN A, COLLOPY L, AL SERAIHI A, WALLIS Y, PAGE P, AKIKI S, FITZGIBBON J, VULLIAMY T & DOKAL I 2016. Germline heterozygous DDX41 variants in a subset of familial myelodysplasia and acute myeloid leukemia. Leukemia, 30, 2083–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHARETTE JM & BASERGA SJ 2010. The DEAD-box RNA helicase-like Utp25 is an SSU processome component. RNA, 16, 2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHASSÉ H, BOULBEN S, COSTACHE V, CORMIER P & MORALES J 2017. Analysis of translation using polysome profiling. Nucleic acids research, 45, e15–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEAH JJC, HAHN CN, HIWASE DK, SCOTT HS & BROWN AL 2017. Myeloid neoplasms with germline DDX41 mutation. International Journal of Hematology, 106, 163–174. [DOI] [PubMed] [Google Scholar]
- CHOI EJ, CHO YU, HUR EH, JANG S, KIM N, PARK HS, LEE JH, LEE KH, KIM SH, HWANG SH, SEO EJ, PARK CJ & LEE JH 2021. Unique ethnic features of DDX41 mutations in patients with idiopathic cytopenia of undetermined significance, myelodysplastic syndrome, or acute myeloid leukemia. Haematologica. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K & RATNER N 2019. iGEAK: an interactive gene expression analysis kit for seamless workflow using the R/shiny platform. BMC Genomics, 20, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DA COSTA L, LEBLANC T & MOHANDAS N 2020. Diamond-Blackfan anemia. Blood, 136, 1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANILOVA N, SAKAMOTO KM & LIN S 2008. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood, 112, 5228–5237. [DOI] [PubMed] [Google Scholar]
- DOENCH JG, FUSI N, SULLENDER M, HEGDE M, VAIMBERG EW, DONOVAN KF, SMITH I, TOTHOVA Z, WILEN C, ORCHARD R, VIRGIN HW, LISTGARTEN J & ROOT DE 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol, 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUTT S, NARLA A, LIN K, MULLALLY A, ABAYASEKARA N, MEGERDICHIAN C, WILSON FH, CURRIE T, KHANNA-GUPTA A, BERLINER N, KUTOK JL & EBERT BL 2011. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood, 117, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBERT BL, PRETZ J, BOSCO J, CHANG CY, TAMAYO P, GALILI N, RAZA A, ROOT DE, ATTAR E, ELLIS SR & GOLUB TR 2008. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature, 451, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMIG D, SALOMONIS N, BAUMBACH J, LENGAUER T, CONKLIN BR & ALBRECHT M 2010. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res, 38, W755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOK WC, NIERO ELO, DEGE C, BRENNER KA, STURGEON CM & BATISTA LFZ 2017. p53 Mediates Failure of Human Definitive Hematopoiesis in Dyskeratosis Congenita. Stem Cell Reports, 9, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANGAT N, PATNAIK MM & TEFFERI A 2016. Myelodysplastic syndromes: Contemporary review and how we treat. American Journal of Hematology, 91, 76–89. [DOI] [PubMed] [Google Scholar]
- HENRAS AK, PLISSON-CHASTANG C, O’DONOHUE MF, CHAKRABORTY A & GLEIZES PE 2015. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA, 6, 225–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG Y, ZHU Y, LIU Z-J & OUYANG S 2017. The emerging roles of the DDX41 protein in immunity and diseases. Protein & Cell, 8, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KADONO M, KANAI A, NAGAMACHI A, SHINRIKI S, KAWATA J, IWATO K, KYO T, OSHIMA K, YOKOYAMA A, KAWAMURA T, NAGASE R, INOUE D, KITAMURA T, INABA T, ICHINOHE T & MATSUI H 2016. Biological implications of somatic DDX41 p.R525H mutation in acute myeloid leukemia. Exp Hematol, 44, 745–754.e4. [DOI] [PubMed] [Google Scholar]
- KISS AM, JÁDY BE, BERTRAND E & KISS T 2004. Human Box H/ACA Pseudouridylation Guide RNA Machinery. Molecular and Cellular Biology, 24, 5797–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUFEL J & GRZECHNIK P 2019. Small Nucleolar RNAs Tell a Different Tale. Trends in Genetics, 35, 104–117. [DOI] [PubMed] [Google Scholar]
- LEE KG, KIM SS, KUI L, VOON DC, MAUDUIT M, BIST P, BI X, PEREIRA NA, LIU C, SUKUMARAN B, RENIA L, ITO Y & LAM KP 2015. Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep, 10, 1055–65. [DOI] [PubMed] [Google Scholar]
- LEI Z & YI C 2017. A Radiolabeling-Free, qPCR-Based Method for Locus-Specific Pseudouridine Detection. Angewandte Chemie International Edition, 56, 14878–14882. [DOI] [PubMed] [Google Scholar]
- LEWINSOHN M, BROWN AL, WEINEL LM, PHUNG C, RAFIDI G, LEE MK, SCHREIBER AW, FENG J, BABIC M, CHONG C-E, LEE Y, YONG A, SUTHERS GK, POPLAWSKI N, ALTREE M, PHILLIPS K, JAENSCH L, FINE M, D’ANDREA RJ, LEWIS ID, MEDEIROS BC, POLLYEA DA, KING M-C, WALSH T, KEEL S, SHIMAMURA A, GODLEY LA, HAHN CN, CHURPEK JE & SCOTT HS 2016. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood, 127, 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI R, SOBREIRA N, WITMER PD, PRATZ KW & BRAUNSTEIN EM 2016. Two novel germline DDX41 mutations in a family with inherited myelodysplasia/acute myeloid leukemia. Haematologica, 101, e228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI W, XU H, XIAO T, CONG L, LOVE MI, ZHANG F, IRIZARRY RA, LIU JS, BROWN M & LIU XS 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biology, 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG J, WEN J, HUANG Z, CHEN X-P, ZHANG B-X & CHU L 2019. Small Nucleolar RNAs: Insight Into Their Function in Cancer. Frontiers in oncology, 9, 587–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACIEJEWSKI JP, PADGETT RA, BROWN AL & MULLER-TIDOW C 2017. DDX41-related myeloid neoplasia. Semin Hematol, 54, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGEE JA & SIGNER RAJ 2021. Developmental Stage-Specific Changes in Protein Synthesis Differentially Sensitize Hematopoietic Stem Cells and Erythroid Progenitors to Impaired Ribosome Biogenesis. Stem Cell Reports, 16, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCANN KL, KAVARI SL, BURKHOLDER AB, PHILLIPS BT & HALL TMT 2020. H/ACA snoRNA levels are regulated during stem cell differentiation. Nucleic Acids Res, 48, 8686–8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELGAR K, WALKER MM, JONES LM, BOLANOS LC, HUENEMAN K, WUNDERLICH M, JIANG JK, WILSON KM, ZHANG X, SUTTER P, WANG A, XU X, CHOI K, TAWA G, LORIMER D, ABENDROTH J, O’BRIEN E, HOYT SB, BERMAN E, FAMULARE CA, MULLOY JC, LEVINE RL, PERENTESIS JP, THOMAS CJ & STARCZYNOWSKI DT 2019. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUTO T, WALKER CS, CHOI K, HUENEMAN K, SMITH MA, GUL Z, GARCIA-MANERO G, MA A, ZHENG Y & STARCZYNOWSKI DT 2020. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat Immunol, 21, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAGAWA MM, DAVIS H & RATHINAM CV 2018. A20 deficiency in multipotent progenitors perturbs quiescence of hematopoietic stem cells. Stem Cell Research, 33, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKHOUL H, KE J, ZHOU X, LIAO W, ZENG SX & LU H 2014. Ribosomopathies: mechanisms of disease. Clinical medicine insights. Blood disorders, 7, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIEDERKORN M, HUENEMAN K, CHOI K, VARNEY ME, ROMANO L, PUJATO MA, GREIS KD, INOUE JI, MEETEI R & STARCZYNOWSKI DT 2020. TIFAB Regulates USP15-Mediated p53 Signaling during Stressed and Malignant Hematopoiesis. Cell Rep, 30, 2776–2790.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARVATIYAR K, ZHANG Z, TELES RM, OUYANG S, JIANG Y, IYER SS, ZAVER SA, SCHENK M, ZENG S, ZHONG W, LIU Z-J, MODLIN RL, LIU Y-J & CHENG G 2012. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nature Immunology, 13, 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAULI C, LIU Y, ROHDE C, CUI C, FIJALKOWSKA D, GERLOFF D, WALTER C, KRIJGSVELD J, DUGAS M, EDEMIR B, PABST C, MÜLLER LP, ZHOU F & MÜLLER-TIDOW C 2020. Site-specific methylation of 18S ribosomal RNA by SNORD42A is required for acute myeloid leukemia cell proliferation. Blood, 135, 2059–2070. [DOI] [PubMed] [Google Scholar]
- POLPRASERT C, SCHULZE I, SEKERES MA, MAKISHIMA H, PRZYCHODZEN B, HOSONO N, SINGH J, PADGETT RA, GU X, PHILLIPS JG, CLEMENTE M, PARKER Y, LINDNER D, DIENES B, JANKOWSKY E, SAUNTHARARAJAH Y, DU Y, OAKLEY K, NGUYEN N, MUKHERJEE S, PABST C, GODLEY LA, CHURPEK JE, POLLYEA DA, KRUG U, BERDEL WE, KLEIN HU, DUGAS M, SHIRAISHI Y, CHIBA K, TANAKA H, MIYANO S, YOSHIDA K, OGAWA S, MULLER-TIDOW C & MACIEJEWSKI JP 2015. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell, 27, 658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QU S, LI B, QIN T, XU Z, PAN L, HU N, HUANG G, PETER GALE R & XIAO Z 2020. Molecular and clinical features of myeloid neoplasms with somatic DDX41 mutations. Br J Haematol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUESADA AE, ROUTBORT MJ, DINARDO CD, BUESO-RAMOS CE, KANAGAL-SHAMANNA R, KHOURY JD, THAKRAL B, ZUO Z, YIN CC, LOGHAVI S, OK CY, WANG SA, TANG Z, BANNON SA, BENTON CB, GARCIA-MANERO G, KANTARJIAN H, LUTHRA R, MEDEIROS LJ & PATEL KP 2019. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am J Hematol, 94, 757–766. [DOI] [PubMed] [Google Scholar]
- RAHMAN R, XU W, JIN H & ROSBASH M 2018. Identification of RNA-binding protein targets with HyperTRIBE. Nature Protocols, 13, 1829–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNEIDER RK, SCHENONE M, FERREIRA MV, KRAMANN R, JOYCE CE, HARTIGAN C, BEIER F, BRÜMMENDORF TH, GERMING U, PLATZBECKER U, BÜSCHE G, KNÜCHEL R, CHEN MC, WATERS CS, CHEN E, CHU LP, NOVINA CD, LINDSLEY RC, CARR SA & EBERT BL 2016. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med, 22, 288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SÉBERT M, PASSET M, RAIMBAULT A, RAHMÉ R, RAFFOUX E, SICRE DE FONTBRUNE F, CERRANO M, QUENTIN S, VASQUEZ N, DA COSTA M, BOISSEL N, DOMBRET H, PEFFAULT DE LATOUR R, SOCIÉ G, ITZYKSON R, FENAUX P, SOULIER J, ADÈS L & CLAPPIER E 2019. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood, 134, 1441–1444. [DOI] [PubMed] [Google Scholar]
- SIGNER RA, MAGEE JA, SALIC A & MORRISON SJ 2014. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature, 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLOAN KE, WARDA AS, SHARMA S, ENTIAN K-D, LAFONTAINE DLJ & BOHNSACK MT 2017. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA biology, 14, 1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPERLING AS, GIBSON CJ & EBERT BL 2016. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nature Reviews Cancer, 17, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SRIVASTAVA L, LAPIK YR, WANG M & PESTOV DG 2010. Mammalian DEAD Box Protein Ddx51 Acts in 3′ End Maturation of 28S rRNA by Promoting the Release of U8 snoRNA. Molecular and Cellular Biology, 30, 2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SU H, XU T, GANAPATHY S, SHADFAN M, LONG M, HUANG THM, THOMPSON I & YUAN ZM 2014. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene, 33, 1348–1358. [DOI] [PubMed] [Google Scholar]
- TAOKA M, NOBE Y, YAMAKI Y, SATO K, ISHIKAWA H, IZUMIKAWA K, YAMAUCHI Y, HIROTA K, NAKAYAMA H, TAKAHASHI N & ISOBE T 2018. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res, 46, 9289–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROWBRIDGE JJ & STARCZYNOWSKI DT 2021. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINREB JT, GHAZALE N, PRADHAN K, GUPTA V, POTTS KS, TRICOMI B, DANIELS NJ, PADGETT RA, DE OLIVEIRA S, VERMA A & BOWMAN TV 2021. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev Cell, 56, 627–640.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Z, YUAN B, BAO M, LU N, KIM T & LIU YJ 2011. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol, 12, 959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO BS & HE C 2015. Pseudouridine in a new era of RNA modifications. Cell research, 25, 153–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU F, LIU Y, ROHDE C, PAULI C, GERLOFF D, KÖHN M, MISIAK D, BÄUMER N, CUI C, GÖLLNER S, OELLERICH T, SERVE H, GARCIA-CUELLAR MP, SLANY R, MACIEJEWSKI JP, PRZYCHODZEN B, SELIGER B, KLEIN HU, BARTENHAGEN C, BERDEL WE, DUGAS M, TAKETO MM, FAROUQ D, SCHWARTZ S, REGEV A, HÉBERT J, SAUVAGEAU G, PABST C, HÜTTELMAIER S & MÜLLER-TIDOW C 2017. AML1-ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat Cell Biol, 19, 844–855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes in RNA-Seq of Ddx41f/f LSPC (+TAM vs control), Related to Figure 4.
Table S2. HyperTRIBE data for DDX41-bound RNAs in THP-1 cells, Related to Figure 4G.
Table S3. Significantly enriched genes in genome-wide CRISPR screen in Ddx41−/− Lin− BM cells, Related to Figure 6G.
Table S4. Primer sequences used in this study, Related to STAR Methods.
Data Availability Statement
Data
Next Generation Sequencing data, including data for mouse RNA-Seq, HyperTRIBE, and CRISPR knockout screen, have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Patient sequencing data is available by request to Torsten Haferlach, Munich Leukemia Laboratory, Munich, Germany (torsten.haferlach@mll.com).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-DDX41 antibody | Abcam | Ab210809 |
| Rabbit monoclonal anti-Vinculin antibody (E1E9V) | Cell Signaling Technology | AB_2728768 |
| Rabbit polyclonal Pan-Actin Antibody | Cell Signaling Technology | AB_2313904 |
| Rabbit monoclonal anti-GAPDH antibody (D16H11) | Cell Signaling Technology | AB_10622025 |
| Anti-B220 APC | eBioscience | AB_469395 |
| Anti-CD11b PE-Cy7 | eBioscience | AB_469587 |
| Anti-CD45.1 BV510 | BioLegend | AB_2563378 |
| Anti-CD45.2 APC-eFluor780 | eBioscience | AB_1272175 |
| Anti-CD48 FITC | eBiosceince | AB_465078 |
| Anti-CD117 APC | eBioscience | AB_469429 |
| Anti-CD150 PE-Cy7 | BioLegend | AB_439797 |
| Anti- Ly-6G (Gr-1) eFluor 450 | eBioscience | AB_1548788 |
| Anti-Ly-6A/E (Sca-1) PE | eBioscience | AB_466086 |
| Streptavidin eFluor® 450 | eBioscience | AB_10359737 |
| Mouse Hematopoietic Lineage Biotin Panel | eBioscience | AB_476399 |
| Anti-CD3e FITC | eBioscience | AB_464882 |
| Anti-Cleaved Caspase 3 | ||
| AnnexinV APC | eBioscience | AB_2575166 |
| Bacterial and virus strains | ||
| OneShot TOP10 Competent Cells | ThermoFisher | C404010 |
| Chemicals, peptides, and recombinant proteins | ||
| Mouse Flt3-ligand cytokine | PeproTech | Cat# 250-31 |
| Mouse IL-3 cytokine | PeproTech | Cat# 213-13 |
| Human IL-6 cytokine | PeproTech | Cat# 200-06 |
| Mouse SCF cytokine | PeproTech | Cat# 250-03 |
| Human TPO cytokine | PeproTech | |
| Methocult GF M3434 | Stem Cell Technologies | Cat# 03434 |
| Dulbecco’s PBS without Ca and Mg | VWR | Cat# 45000-446 |
| A/G Protein PLUS-Agarose | Santa Cruz | Cat# sc-2003 |
| RPMI | Fisher | Cat# SH30027.01 |
| RPMI, no methionine | ThermoFisher | Cat# A1451701 |
| IMDM | Corning | Cat# 10-016-CV |
| DMEM | Fisher | Cat# SH30022FS |
| TransIT-LT1 transfection reagent | Mirus | Cat# MIR2306 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11550 |
| Penicillin/Steptomycin | ThermoFisher | Cat# SV30010 |
| Superscript II | ThermoFisher | Cat# 18064014 |
| RnaseOUT RecombinantRibonuclease Inhibitor | ThermoFisher | Cat# 10777019 |
| N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMC) | ThermoFisher | Cat# C106402-1G |
| MnCl2 | Sigma | Cat# M1787 |
| Bicine | Sigma | Cat# B3876 |
| Tamoxifen | Sigma | Cat# T-5648 |
| 4-Hydroxytamoxifen | Sigma | Cat# H-7904 |
| Critical commercial assays | ||
| CellTiterGlo kit | Promega | Cat# G7570. As in Melgar et al. Sci Transl Med. 2019. |
| Mouse hematopoietic progenitor cell enrichment kit | Stem Cell Technologies | Cat# 19856 |
| Pierce Biotin 3’ End DNA Labeling Kit | ThermoFisher | Cat# 89818 |
| NEBNext Magnesium RNA Fragmentation Module | ThermoFIsher | Cat# E6150S |
| POWRUP SYBR green Master Mix | ThermoFisher | Cat# A25776 |
| Click-IT L-Homopropargylglycine | ThermoFisher | Cat# C10186 |
| NorthernMAX Kit | ThermoFisher | Cat# AM1940 |
| mirVana miRNA Isolation Kit | ThermoFisher | Cat# AM1561 |
| GeneAmp Fast PCR master mix 2x | ThermoFisher | Cat# 4359187 |
| Click-IT Edu Alexa Fluor 647 | ThermoFisher | Cat# C10424 |
| Chemiluminescent Nucleic Acid Detection Module Kit | ThermoFisher | Cat# 89880 |
| Zymo Quick-RNA Miniprep Kit | Zymo Research | Cat# R1050 |
| Deposited data | ||
| LSPC RNA-Seq | This study | GSE178895 |
| HyperTRIBE | This study | GSE178895 |
| CRISPR Screen | This study | GSE178895 |
| Experimental models: Cell lines | ||
| Mouse MLL-AF9 immortalized Rosa-CreERT+ lineage-negative cells ( LSPC). Gentoypes: Ddx41+/+, Ddx41+/f, Ddx41f/f, Ddx41KI/+, Ddx41KI/f | This paper | N/A |
| THP-1 cells | ATCC | TIB-202 |
| HEK293T cells | ATCC | ATC CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: C57Bl/6N Ddx41tm1a | KOMP | MMRRC_047340-UCD |
| Mouse: C57Bl/6N Ddx41conditionalR525H | Ozgene | N/A |
| Mouse: Rosa26-CreERT2 (B6.129-Gt(ROSA)26Sortm1 (cre/ERT2)Tyj/J) | Jackson | RRID:IMSR_JAX:008463 |
| Mouse: Rosa26-Cas9 (B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J) | Jackson | RRID:IMSR_JAX:026179 |
| Mouse: Rosa26::Flpe (B6.129S4-Gt(ROSA)26Sortm1 (FLP1)Dym/RainJ) | Jackson | RRID:IMSR_JAX:009086 |
| Oligonucleotides | ||
| Primers listed in Supplemental Table 4 | This study | N/A |
| Recombinant DNA | ||
| MSCV MLL-AF9 pGK-GFP | Gift from Ashish Kumar laboratory | N/A |
| pCDLN packaging vector | Gift from Gang Huang laboratory | N/A |
| pMD.2 VSV-G envelope vector | Gift from Gang Huang laboratory | N/A |
| M57 retroviral gag/pol plasmid | Gift from Gang Huang laboratory | N/A |
| pLKO.1 scrambled control mCherry | This study | Cloned from TRCN SHC202 |
| pLKO.1 shSnora7a-1 mCherry | This study | N/A |
| pLKO.1 shSnora7a-2 mCherry | This study | N/A |
| pLKO.1 shSnora16a-1 mCherry | This study | N/A |
| pLKO.1 shSnora70-1 mCherry | This study | N/A |
| pLKO.1 shSnora70-2 mCherry | This study | N/A |
| pLKO.1 shSnora71-1 mCherry | This study | N/A |
| pLKO.1 shSnora74a-1 mCherry | This study | N/A |
| pLKO GFP shDDX41 #1 | This study | Cloned from TRCN0000001268 |
| pLKO GFP shDDX41 #2 | This study | Cloned from TRCN0000001270 |
| pLKO scrambled shRNA GFP | This study | Cloned from TRCN SHC202 |
| pGK-GFP DDX41-ADARcd | This study | N/A |
| pGK-GFP ADARcd | Rahman et al. Nat Protoc. 2018 | N/A |
| MSCV-HA-DDX41-mCherry | This study | |
| MSCV-HA-DDX41(R525H)-mCherry | This study | |
| MSCV-IRES-mCherry | This study | Addgene, Cat# 52114 |
| Software and algorithms | ||
| iGeak | Choi et al. BMC Genomics. 2019 | N/A |
| AltAnalyze | Emig, et al. Nucleic Acids Res. 2010 | N/A |
| HyperTRIBE | Rahman et al. Nat Protoc. 2018 | N/A |
| MAGeCK | Li et al. Genome Biology, 2014 | N/A |
| Other | ||
Code
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.