

Abstract
Alpha-1 antitrypsin deficiency (AATD) is most commonly caused by the Z mutation, a single-base substitution that leads to AAT protein misfolding and associated liver and lung disease. In this study, we apply adenine base editors to correct the Z mutation in patient induced pluripotent stem cells (iPSCs) and iPSC-derived hepatocytes (iHeps). We demonstrate that correction of the Z mutation in patient iPSCs reduces aberrant AAT accumulation and increases its secretion. Adenine base editing (ABE) of differentiated iHeps decreases ER stress in edited cells, as demonstrated by single-cell RNA sequencing. We find ABE to be highly efficient in iPSCs and do not identify off-target genomic mutations by whole-genome sequencing. These results reveal the feasibility and utility of base editing to correct the Z mutation in AATD patient cells.
Keywords: adenine base editor, alpha-1 antitrypsin deficiency, base edit, human induced pluripotent stem cells, iPSC, hepatocyte, iHep, single cell RNA sequencing, whole genome sequencing
Graphical abstract
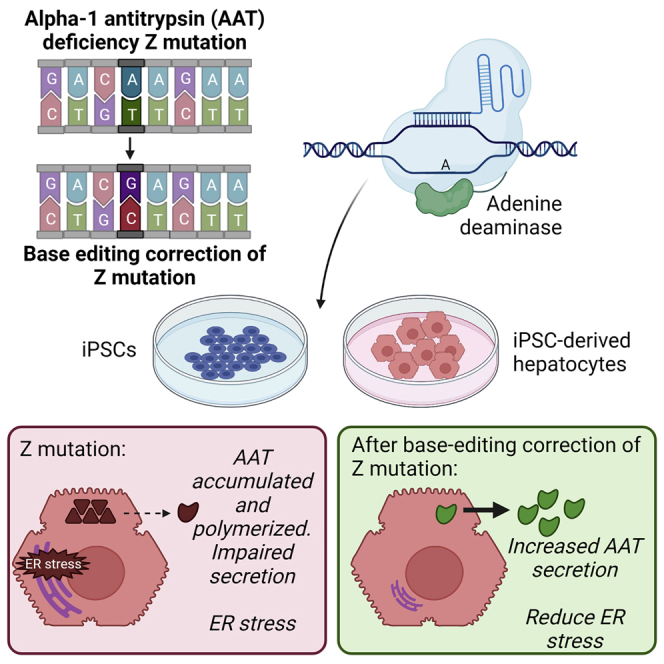
In vivo gene editing of hepatocytes represents a potential curative therapy for patients with the monogenic disease alpha-1 antitrypsin deficiency. Werder et al. apply adenine base editors to correct the disease-inducing mutation in patient iPSCs and iHeps and demonstrate efficient editing with amelioration of disease consequences in edited cells.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a common heritable cause of both lung and liver disease. AATD results from mutations in the SERPINA1 gene, which encodes the antiprotease alpha-1 antitrypsin (AAT). AAT is produced in high abundance by hepatocytes1 and functions primarily to neutralize neutrophil elastase. The most prevalent disease-causative mutation is a single-base substitution from guanine (G) to adenine (A), which results in a glutamic acid-to-lysine substitution (Glu342Lys) and production of a mutant protein prone to misfolding and aggregation, termed “Z-AAT.”2 Reduced Z-AAT secretion and associated diminished circulating AAT levels3 result in a protease/antiprotease imbalance in the lungs that over time predisposes affected individuals to injury, most commonly manifested as emphysema.4 In addition, accumulation of polymerized Z-AAT in the liver can result in toxic gain-of-function effects in hepatocytes, leading to liver disease in both neonates and adults.5,6
Although infusion of pooled human AAT protein (“augmentation therapy”) has been shown to slow progression of lung disease in AATD patients,7 no specific treatments are available for AATD-associated liver disease. To address both lung and liver disease in AATD, emerging treatment strategies have focused on correction of the Z mutation. Among these, early human clinical trials delivering a normal copy of the SERPINA1 gene to skeletal muscle have achieved long-term but sub-therapeutic production of normal “M-AAT” protein.8,9 Although promising, this strategy does not rectify toxic gain-of-function effects in Z-AAT-expressing hepatocytes. Attractive alternatives include direct correction of the SERPINA1 mutation in patient cells in vivo or delivery of engraftable, patient-specific cells containing a normal copy of SERPINA1. ZFN, TALEN, and CRISPR editing strategies have previously been used to correct the Z mutation in human iPSCs in vitro and mice in vivo10, 11, 12, 13 but share important limitations, including the induction of double-strand breaks (DSBs) in targeted cells and associated potential toxicity.14 An editing approach that avoids DSBs would be preferable in correcting the single-base pair Z mutation in AATD.
Base-editing technology allows the precise editing of single-point mutations without generating DSBs. In this system, a catalytically inactive Cas9 is fused to a deaminase enzyme that, upon binding to its target DNA sequence through the direction of a guide RNA (gRNA), directly modifies the base of interest. Thus far, cytosine base editors (mediating C→T) and adenine base editors (ABEs) (mediating A→G) have been described.15,16 Here, we applied ABEs to correct the disease-causing G-to-A point mutation in the SERPINA1 gene in patient induced pluripotent stem cells (iPSCs). We developed novel base editors to specifically target the Z mutation and demonstrated efficient editing in both iPSCs and differentiated iPSC-derived hepatocytes (iHeps) that we have previously found to recapitulate AATD disease phenotypes in vitro.12,17 Successful correction of the Z mutation abrogated the aberrant accumulation of intracellular AAT protein and promoted AAT secretion. Furthermore, single-cell RNA sequencing (scRNA-seq) revealed that base-edited iHeps were protected from disease-associated endoplasmic reticulum (ER) stress. These studies suggest the feasible application of ABEs to ameliorate Z-AAT-induced proteotoxicity in patient hepatocytes.
Results
Design of base editing reagents to correct the Z mutation
A base editing correction strategy for the Z mutation in AATD would require the use of an ABE (Figure 1A). However, the local sequence context of the Z mutation poses two major challenges: (1) the absence of a canonical Cas9 protospacer adjacent motif (PAM) that would place the target adenine within the editing window and (2) the presence of adjacent bystander adenines that could undergo editing in addition to the target adenine. To overcome the former challenge, we elected to use an spCas9 variant that would accept a noncanonical NGC PAM. Application of this PAM would place the target adenine at position 7 and would result in a single additional bystander adenine within the active base editing window at position 5. In initial experiments, spCas9-NG (ngcABEvar1)18 and spCas9-MQKSER (ngcABEvar2)19 yielded low levels correction of the PiZ mutation, confirming that an NGC PAM variant and the selected sgRNA could yield the desired outcomes (Figures S1A and S1B). To further optimize this spCas9-NGC PAM variant, we shuffled mutations from spCas9-NG and MQKSER to yield ngcABEvar3. We then incorporated ABE8 mutations into the TadA deaminase predicted to increase editing of the target adenine.20 These perturbations initially yielded ngcABEvar5, which exhibited 25% editing of the target adenine and minimal bystander editing in Z-AAT-expressing human fibroblasts and ultimately resulted in ngcABEvar9, which accomplished 45% editing of the target adenine but with associated bystander editing of the adjacent adenine (Figure S1B). To further refine our editing results, we tested synthetic gRNAs21 with various protospacer lengths (sequences in Table S7). We found that a protospacer length of 19 nt retained significant editing of the target adenine, while decreasing the bystander editing (Figure S1C). Subsequent experiments were performed with this gRNA together with ngcABEvar5 and ngcABEvar9.
Figure 1.

Base editing corrects the Z mutation in AATD iPSCs
(A) Z allele (Lys342) and desired A > G edit to M allele. (B) Schematic representation of workflow. iPSCs (PiZZ1 line) were nucleofected with adenine base editor (ngcABEvar5) and guide RNA (gRNA), then immediately plated as single cells. Cells were allowed to clonally expand, and some colonies were passaged again to single cells at D6 post-nucleofection. Clones were collected and analyzed using next-generation sequencing (NGS). (C) Editing efficiency of base editor to correct Z mutation. Fifty-six clones, each clone represented by a circle, were screened by NGS for on-target editing at the Z mutation (A > G) and for bystander edits within the editing window. (D) Following additional subcloning, pure populations of MZ and MM iPSCs were obtained and analyzed using Sanger sequencing. (E) NGS of pure ZZ, MZ, and MM iPSCs showing percentage of reads at each position. (F) Whole-genome sequencing was performed with 60× coverage. Passage-matched samples (ZZ, ZZ cells that were nucleofected without editors, MZ, and MM) were compared with earlier passage ZZ cells (the same passage used to commence base editing). Variants were detected using Mutect2 and VarScan, then these variants were overlayed with predicted off-target mutation sites (determined with COSMID), with no mutations overlapping with off-target mutation sites.
Base editing corrects the Z mutation in AATD iPSCs
After confirming efficient mRNA delivery to iPSCs (Figure S2A), we nucleofected an iPSC line (PiZZ1)12 derived from an AATD patient homozygous (“ZZ”) for the Z mutation with ngcABEvar5 and gRNA (Figure 1B). We manually isolated single-cell derived clones ∼10 or 16 days (including a single passage at 6 days) post-nucleofection to allow completion of editing before picking emergent clones after an additional 10 days. Colonies were passaged and gDNA collected simultaneously to perform next-generation sequencing (NGS). We found that 55 of 56 clones screened had undergone A > G editing at the Z mutation site (Figure 1C; Table S1).
A majority of clones (66%) were observed to have undergone additional bystander editing of nearby adenine nucleotides within the editing window (Figure 1C). One clone remained unedited at the Z mutation site but underwent editing in the adjacent codon (Asp341Gly). The remaining 17 clones were found only to have on-target editing at the Z mutation site. Regardless of isolation strategy, we found that all colonies screened contained mosaic mixtures that varied in proportion of edited cell populations (22% mosaic wild-type/heterozygous mix, 4% heterozygous/ homozygous mix, 6% pure heterozygous). In future experiments, delaying the isolation of clones would likely allow completion of base editing and result in fewer mosaic colonies.
We selected two mosaic colonies of interest for subcloning to isolate pure cell populations. Using this method, we generated heterozygous (MZ) or homozygous (MM) base-edited iPSCs without bystander edits, confirmed by both Sanger sequencing (Figure 1D) and NGS (Figure 1E; Table S1). Newly generated MZ and MM iPSC lines were confirmed to be karyotypically normal before proceeding to additional experiments (Figure S2B). Next, we applied whole-genome sequencing (WGS) to investigate whether off-target editing had occurred during the editing process. We performed WGS with at least 60× coverage on passage-matched parental ZZ and base-edited MZ and MM iPSC lines and determined the frequency of mutations by integrating two somatic mutation tools (VarScan and Mutect2) (Figure 1F; Table S8) or the LoFreq somatic mutation tool (Figure S2C). In contrast with findings that predated current, feeder-free iPSC culture techniques,22 we found that serial passaging of iPSCs resulted in only minimal accumulation of somatic mutations (approximately two mutations per passage); however, mutations were acquired in cells that underwent nucleofection, resulting in similar numbers in ZZ samples not exposed to editors, as were observed in edited MZ and MM samples (Figure 1F). Although our analysis does not capture potential low-frequency, guide-dependent off-target events that would appear in a population of many treated cells, we did not identify any A > G mutations within in silico predicted guide-dependent off-target sites (Figure 1F; Figure S2C). More generally, we did not see evidence that adenine base editing treatment induces A > G somatic mutations at a rate greater than that observed in a passage-matched control. In light of these analyses, the observed somatic mutations appear to be consistent with genetic variation introduced during the nucleofection process.
Base-edited MZ and MM iPSC-derived hepatocytes are protected from a Z-AAT-driven disease signature
After generating MZ and MM iPSCs, we next derived iHeps to test the effects of editing in SERPINA1-expressing cells. Using an established protocol that we have found to generate hepatic cells with a transcriptional profile overlapping significantly with primary hepatocyte controls,12,17,23 we differentiated parental ZZ and edited MZ and MM daughter iPSCs in biological triplicate. All three syngeneic lines efficiently generated iHeps, quantified by expression of alpha-fetoprotein (AFP), a marker of fetal hepatocytes (Figures 2A and 2B; Figure S3A). As we have shown previously,12,17 ZZ iHeps recapitulate salient disease features, including retention of AAT intracellularly with associated low levels of AAT secretion. ngcABE correction of the Z mutation on one (MZ) or both (MM) SERPINA1 alleles significantly reduced intracellular AAT retention (Figures 2A–2C; Figure S3A) and resulted in a concomitant increase in total secreted AAT in cell supernatants (Figure 2D). Z-AAT protein levels were reduced in MZ relative to ZZ cell supernatants and completely absent from MM supernatants, as expected (Figure 2E). Correction of the Z mutation did not alter other functions of iHeps, such as albumin secretion (Figure S3B) or CYP expression (Figure S3C).
Figure 2.

Base-edited MZ and MM iPSC-derived hepatocytes are protected from disease signature
(A) Flow cytometry analysis for intracellular (B) alpha-fetoprotein (AFP) and (C) alpha-1 antitrypsin (AAT) expression. (D) Total AAT expression and (E) Z-AAT in culture supernatants. n = 3 independent differentiations; error bars represent SD. Statistical significance was determined using a one way-ANOVA with a Tukey multiple-comparison test; ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.001.
Base editing of differentiating iHeps corrects the Z mutation
We next sought to correct the Z mutation in iHeps, a cell type with more biological relevance to patient hepatocytes in vivo. Previous literature has indicated the ability of base editors to edit post-mitotic cells, a feature not shared by editing strategies that require homology-directed repair, such as TALENs and CRISPR.24,25 We quantified EdU incorporation in differentiating iHeps and found that proliferation diminished significantly by day (D) 15 of the differentiation protocol (Figure S4A), a time point at which cells have reached a hepatic progenitor stage and express SERPINA1.17 We next optimized delivery of mRNA using both forward and reverse transfection approaches (for details of reverse transfection, see Materials and methods) at a series of time points (Figure 3A) and found that reverse transfection enhanced delivery with peak efficiency on D16 of the differentiation protocol, as evidenced by GFP-mRNA+ cells (Figure S4B). On the basis of these results, we hypothesized that a reverse transfection strategy would enhance cargo delivery and thus the efficiency of editing. We therefore applied this strategy to deliver ngcABEvar5 and gRNA to D15 iHeps and observed efficient A > G editing at the Z mutation site in ∼20% of cells (Figure 3B), compared with lower efficiency with forward transfection (∼5%) (Figure S4C; Table S2). In addition, we tested ngcABEvar9, which resulted in the highest A > G editing of the editors tested (Figure S1B), and observed significantly improved on-target editing compared with ngcABEvar5 (Figure 3B) but with an associated increase in bystander editing of the adjacent adenine (Figure 3C; Table S2), consistent with our previous experiments (Figure S1). This bystander edit results in a single amino acid change (Asp341Gly) that we have previously found to have no adverse impact on either AAT secretion or elastase inhibition when in linkage with precise correction of the Z mutation (M.S. Packer, 2020, Am. Soc. Gene Cell Ther., conference).
Figure 3.

Base editing in differentiating iHeps partially corrects Z mutation
(A) iPSC-derived hepatocyte (iHeps) directed differentiation overview. (B) iHeps were forward (fwd) or reverse (rev) transfected with ngcABEvar5 or ngcABEvar9 base editor on D15 of the directed differentiation protocol. Editing efficiency and (C) bystander editing was assessed using NGS to quantify the correction of the Z mutation from A > G. n = 2–5 independent differentiations; error bars represent SD. Statistical significance was determined using a one way-ANOVA with a Tukey multiple-comparison test; ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.001.
Base editing in differentiating iHeps reduces intracellular accumulation of Z-AAT
To assess how ABE correction of the Z mutation affects the disease signature of iHeps, we reverse-transfected D15 iHeps with ngcABEs on D15 in three separate experiments. ngcABEvar5 and ngcABEvar9 successfully corrected the Z mutation with ∼25% and ∼50% efficiency, respectively (Figure S5A), with an increase in bystander editing again noted with ngcABEvar9 (Figure S5A; Table S3). Editing with either ngcABEvar5 or ngcABEvar9 did not affect the efficiency of hepatic differentiation, quantified by AFP protein expression, but significantly reduced intracellular AAT levels quantified by flow cytometry (Figures 4A and 4B; Figure S5D). There was no change in either albumin secretion (Figure S5B) or CYP expression (Figure S5C) in edited cell populations. ELISA of cell supernatants using a pan-AAT antibody did not identify an increase in total secreted AAT following this base-editing approach that yields a heterogeneous iHep population (Figure 4C). Additional analysis using an antibody specific to Z-AAT,26 however, did detect a significant decline in secreted Z-AAT following iHep editing with either ngcABEvar5 or ngcABEvar9 (Figure 4D). To further investigate the effects of editing on cellular Z-AAT processing, we applied the 2C1 antibody27 to specifically quantify polymerized AAT. We observed a reduction in AAT polymers in edited cells by immunostaining, which was quantitatively significant when additionally assessed by flow cytometry (Figures 4E and 4F; Figure S5E).
Figure 4.

Base editing in differentiating iHeps partially rescues disease signature.
(A) Flow cytometry analysis for intracellular alpha-fetoprotein (AFP) and (B) alpha-1 antitrypsin (AAT) expression. (C) Total AAT expression and (D) Z-AAT in culture supernatants. (E) Immunofluorescent staining for hepatocyte nuclear factor 4 α (HNF4α) (red), 2C1 (polymerized AAT) (green), and Hoechst (blue). Representative images from one of three experiments are shown; scale bar, 20 μm. (F) Flow cytometry analysis for 2C1 (polymerized AAT). n = 5 independent differentiations; error bars represent SD. Statistical significance was determined using a one way-ANOVA with a Tukey multiple-comparison test; ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.001.
Base-edited iHeps are protected from ER stress
To understand the effects of editing on the global transcriptome of differentiated ZZ iHeps at single-cell resolution, we next performed scRNA-seq in edited cells. ngcABEs were delivered to D15 iHeps that were then collected at D21 for single-cell capture. Unedited ZZ iHeps, ngcABEvar5-edited iHeps, ngcABEvar9-edited iHeps, and previously edited PiZZ1 MZ and MM iHeps (Figure 1) were captured for scRNA-seq using the 10x Genomics Chromium platform (Figure 5A; Figure S6A). We performed NGS on D21 to define editing efficiency and resulting cell genotypes and found that approximately 15% of the Z alleles in each ngcABE-treated sample were successfully edited (Figure 5B; Table S4). We further confirmed cell genotypes using a single-cell genotyping tool (Vartrix; https://github.com/10xgenomics/vartrix) and identified 12% MZ and 2% MM iHeps in the ncgABEvar5-edited population, compared with 12% MZ and 4% MM in iHeps edited with ncgABE-var9 (Figure 5B). Bystander edits at the 5A position (Asp341Gly), almost exclusively identified in ngcABEvar9-edited cells, did not affect clustering and biallelic bystander edits were associated with biallelic editing of the Z mutation (i.e., MM cells) (Figures S6C–S6F), as expected. We used uniform manifold approximation and projection (UMAP) dimensionality reduction to visualize all ZZ, MZ, and MM cells (Figure 5C) and applied Louvain clustering, which identified four distinct clusters (Figure 5D). We next performed gene set enrichment analysis (GSEA) on differentially expressed genes (DEGs) for each cluster and annotated clusters using the top Gene Ontology (GO) term (Figure 5E; Figures S7 and S8). Archetypal hepatic processes characterized cluster 0, which was enriched for protein synthesis, and cluster 1, which was enriched for protein-lipid organization. In contrast, cluster 2 was strongly associated with extracellular matrix organization, while cluster 3 was identified by ER stress and unfolded protein response (UPR) (Figure S7).
Figure 5.

Base-edited iHeps are protected from ER stress.
(A) Schematic representation of 10x Genomics single-cell RNA sequencing (scRNA-seq) experiment. (B) Editing efficiency was determined using DNA next-generation sequencing (left) or Vartrix Single-Cell Genotyping tool (10x Genomics) (right). (C) UMAP representation of ZZ, MZ, and MM genotyped cells. (D) UMAP visualization of Louvain clusters at a resolution of 0.17. (E) Gene set enrichment analysis of differentially expressed genes was determined for each Louvain cluster with the top GO biological process (BP) term shown. (F) Proportion of each Louvain cluster by ZZ, MZ, and MM genotypes, normalized by total number of cells. (G) UMAP projections for each sample showing ZZ, MZ, and MM genotyped cells. (H) Proportion of each genotype in Louvain clusters in each sample. (I) Immunofluorescent staining for binding immunoglobulin protein (BiP; also called HSPA5 or GRP78) (red), 2C1 (polymerized AAT) (green), and Hoechst (blue). Representative images from one of three experiments are shown; scale bar, 20 μm. (J) BiP mean fluorescent intensity (MFI) was quantified using flow cytometry; n = 5 independent differentiations; error bars represent SD, ∗∗∗∗ p ≤ 0.0001.
Previous work has demonstrated that Z-AAT expression causes ER stress and in some instances induces a UPR in human hepatic and immune cells.17,28, 29, 30 Consistent with these findings, cluster 3 contained proportionally more ZZ than MZ or MM cells, suggesting that this cellular response was induced by elevated levels of misfolded Z-AAT protein (Figure 5F). We determined the cluster distribution of ZZ, MZ, and MM cells in each sample and found fewer ABE-edited MZ and MM cells in cluster 3 relative to unedited ZZ cells from those same samples (Figures 5G and 5H), suggesting that editing the Z mutation may rescue iHeps from Z-AAT-induced ER stress. To confirm this finding, we performed immunostaining for the chaperone protein BiP (HSPA5/GRP78), a top DEG in cluster 3 (Figure S8), together with 2C1 staining for polymerized AAT. In ZZ iHeps, we found that BiP often co-localized with 2C1; however, in base-edited cells, we observed minimal overlap of BiP and 2C1 and lower BiP intensity by flow cytometry (Figures 5I and 5J).
Discussion
The single-base pair Z mutation in the SERPINA1 gene causes chronic, progressive lung and liver disease in AATD. In this study we sought to correct this mutation using base-editing technology in ZZ patient iPSCs and iHeps. In both strategies, monoallelic or biallelic correction of the mutation rectified accumulation of intracellular AAT in differentiated hepatocytes. We also used scRNA-seq to transcriptomically profile the mixed population of base-edited iHeps and found that base-edited cells were rescued from undergoing ER stress.
The differentiated progeny of patient derived iPSCs have been found to faithfully replicate cellular features of human disease phenotypes, including AATD.11,12,17 In this study, we observed accumulated intracellular AAT protein with associated polymerization and low levels of secretion that were corrected following base editing. Profiling of ZZ iHeps by scRNA sequencing identified a cluster of cells that exhibited an ER-stress and UPR signature. This cluster was predominated by ZZ cells, suggesting this cellular response might be induced by high levels of misfolded Z protein. Of note, this cluster comprised only 5% of all cells, which could explain why previous studies evaluating the global hepatocellular response to protein misfolding in AATD either did not observe a UPR31 or did so only at a specific stage of hepatocyte differentiation.17
The use of programmable endonucleases (TALENs and CRISPR) to edit human cells is immensely promising for gene and cell therapy.32, 33, 34 Previously, we applied CRISPR to correct the Z mutation by co-nucleofecting iPSCs with a plasmid encoding Cas9 together with a single-stranded oligodeoxynucleotide (ssODN) repair template and then sorting for Cas9+ cells.12 This approach results in biallelic correction of the Z mutation in 6% of sorted emergent clones on average, approximately 0.3% of the total starting iPSC number. In contrast, in this report we found that without cell sorting, 95% of emergent iPSC clones had been corrected at the Z mutation site, with the limitation being that almost all colonies were mosaic and had to be further subcloned to produce pure populations of monoallelic and biallelic edited cells. In future experiments, delaying the selection of clones would likely allow completion of base-editing and result in fewer mosaic colonies. Another drawback of TALENs and CRISPR is their restricted application to cycling cells, as homologous recombination is suppressed in G1 phase.35 Although both primary hepatocytes36 and differentiated iHeps exhibit minimal cell turnover in the absence of injury, base editors are able to edit efficiently in post-mitotic cells25 and effectively edited the Z mutation in our studies. This finding suggests that base editors may prove useful in editing other quiescent cell populations.
A major concern for gene-editing technologies is the potential for off-target editing. Although the functional consequence of such “bystander” editing of additional adenine residues in the editing window together with the Z mutation do not appear to adversely affect AAT function, we did observe this phenomenon in our studies, particularly with ngcABEvar9. WGS of base-edited iPSCs in our studies did not reveal any editing at predicted off-target sites in the genome. To comprehensively characterize potential unguided off-target DNA base editing would require additional WGS of multiple single cell clones, although previous work has not identified the introduction of such edits by ABEs.20,37,38 However, our data suggest that the mutations we did observe in MZ and MM edited cells were accumulated over time in culture or during the nucleofection process, consistent with previous observations made in iPSCs.11,39,40 Finally, it has recently been shown that base editors can edit RNA in addition to DNA in immortalized cell lines,41 which we cannot exclude as a possibility in our study and warrants further investigation and/or optimization of ngcABEs.42
In conclusion, we have applied adenine base editing in ZZ patient iPSCs and iHeps to repair the single-base pair Z mutation in SERPINA1 responsible for chronic, progressive lung and liver disease in AATD. Two distinct editors accomplished monoallelic or biallelic correction of this mutation and rectified accumulation of intracellular AAT in differentiated hepatocytes. Transcriptional profiling of base-edited iHeps by scRNA-seq demonstrated that correction of the Z mutation in edited cells rescued them from Z-AAT-associated ER stress. Taken together, our results suggest that base editing of the Z mutation is an efficient process with minimal genome-wide effects, supporting its future application gene or cell-based therapy in AATD.
Materials and methods
Derivation and maintenance of iPSC lines
All experiments involving the differentiation of human iPSC lines were performed with the approval of the Institutional Review Board of Boston University (protocol H33122). The PiZZ1 AATD iPSC line was obtained from our previous study.12 iPSCs were maintained in feeder-free conditions in mTeSR1 medium (StemCell Technologies) on growth factor reduced Matrigel (Corning), using Gentle Cell reagent for passaging (StemCell Technologies). All iPSCs displayed a normal karyotype when analyzed by G-banding (Cell Line Genetics). Additional details pertaining to iPSC derivation, characterization, and culture are available for free download at http://www.bu.edu/dbin/stemcells/protocols.php.
Generation of ABE mRNAs
ABEs were cloned into mRNA template plasmids encoding a T7 promoter, followed by a 5′ UTR, Kozak Sequence, editor open reading frame (ORF), 3′ UTR, a polyA tail, and a BbsI restriction site (sequences available in Table S6). Plasmids were purified using commercial midiprep kits (Zymo Research) and linearized using BbsI-HF (NEB). Linearized plasmid template was purified on a DNA Clean and Concentrate Column (Zymo Research). The NEB HiScribe High-Yield Kit was used as per the instruction manual but with full substitution of N1-methyl-pseudouridine for uridine and co-transcriptional capping with CleanCap AG (TriLink). Isolation of mRNA from transcription reactions was achieved by lithium chloride precipitation.
Base editing in primary PiZZ fibroblasts
PiZZ primary fibroblasts (GM11423, Coriell) were cultured at 37°C and 5% CO2 in EMEM 15% fetal bovine serum (FBS) plus Earle’s salts and nonessential amino acids as directed by the supplier. Transfections were performed using a Neon Electroporation device (Thermo Fisher Scientific) as follows. Each electroporation was performed with 80,000 cells, 100 ng mRNA, and 50 ng gRNA (sequences available in Table S7) using a 10 μL Neon tip in buffer R with a single pulse at 1,000 V for 40 ms. Cells were grown in 24-well tissue culture-treated plates for 48 h after electroporation. Genomic DNA was harvested in 125 μL of quick lysis buffer per well (10 mM Tris, pH 8.0, 0.05% SDS, 20 μg/mL Proteinase K), followed by incubation at 37°C for 1 h and heat inactivation at 85°C for 15 min. These lysates were then used in subsequent targeted amplicon sequencing.
SERPINA1 targeted amplicon sequencing and data analysis
Amplicon sequencing was performed as previously described.20 Genomic loci were amplified in 25 μL PCRs using Q5 2x Hot Start Master Mix (NEB), 0.5 μM each primer (BEAM54 BEAM1704; Table S5), and 1 μL of genomic DNA template. Barcoded amplicons were generated in 25 μL PCRs using Q5 2x Hot Start Master Mix (NEB), 0.5 μM of barcode primers, and 1 μL of the prior PCR. Barcoded amplicons were combined and purified via DNA agarose gel extraction (Zymo Research). The resulting DNA library was quantified by NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific) and sequenced on an Illumina MiSeq Instrument. Data analysis was performed as previously described.20 Briefly this process can be summarized in four steps: (1) demultiplexing of sequencing reads into fastq files, (2) read trimming and quality score filtering, (3) alignment of all reads to the expected amplicon sequence, and (4) quantification of editing rates and allele frequencies. The accession number of the amplicon sequencing data reported in this paper is SRA: PRJNA733506.
Whole-genome sequencing
PiZZ1-ZZ, ZZ-nucleofected, and MZ and MM base-edited iPSCs were collected at the same passage (P42), or PiZZ1-ZZ cells were collected at the passage used for base editing (P34) and gDNA was extracted using a QIAamp extraction kit (QIAGEN). WGS with 60× coverage was performed with an Illumina NovaSeq 6000. Base-edited samples were aligned to the reference genome (PiZZ1-ZZ). Somatic mutations were identified using VarScan version 2.3.9 and the gatk version 4.1.2 tool Mutect2,43, 44, 45 or in separate analyses somatic mutations were determined using the LoFreq tool.46 The PiZZ1-ZZ sample was treated as the “normal” sample, and ZZ-nucleofected, MZ and MM were treated as “tumor” samples for somatic mutation discovery. For Mutect2 the default parameters were used for identifying somatic mutations, and for VarScan parameters were determined following recommendations47 (see statistics in Table S8). Mutect2 outputs were additionally filtered using the TLOD (tumor log odds) score. There was an observed drop-off in variant quality (assessed by STRANDQ, quality of strand bias artifact; SEQQ, quality that alternative alleles are not sequencing errors; and GERMQ, quality that alternative alleles are not germline variants) at a TLOD score of 30 (for the ZZ versus MM and ZZ versus MZ comparisons) and a TLOD score of 15 (for the ZZ comparisons at different passages). These were used as lower bounds for accepting variants (variants with higher TLOD scores were accepted). Predicted off-target sites were determined in silico with the web-based tool COSMID48 using hg38 as a reference and a criterion of a maximum of three allowed mismatches, one allowed deletion, and one allowed insertion, or with Cas-OFFinder49 using a criterion of NGN PAM, up to six mismatches, or two mismatches and 1 bp RNA/DNA bulge. One hundred fifty-seven potential off-target sites were identified with COSMID, and 51,214 potential off-target sites were identified with Cas-OFFinder. These off-target sites were then compared with the somatic variants identified jointly by VarScan and Mutect2 using the BEDTools intersect function,50 which reports any overlapping features. We considered a variant to overlap an off-target site if it was found within any of the exact off-target sites identified. The accession number of the WGS data is dbGap: phs002471.v1.p1.
Base editing iPSCs
PiZZ1-ZZ iPSCs were treated for 3 h with 10 μm Y-27632 (Tocris, catalog #1254), then dissociated to a single-cell suspension using Gentle Cell reagent (StemCell Technologies). iPSCs (1 × 106) were resuspended in P3 Primary Cell Nucleofector Solution + Supplement 1, as per manufacturer’s instructions (Lonza) with 2,000 ng GFP mRNA or 2,000 ng mRNA ngcABEvar5 and 1,000 ng gRNA, having determined that a ratio of 2:1 of mRNA to gRNA performed efficiently (data not shown). Cells were quickly transferred to a Nucleocuvette (Lonza), nucleofected (Lonza 4D-Nucleofector X Unit; program CB150 or DC100), then replated in mTeSR1 medium with 10 μm Y-27632 at serial dilutions to obtain single-cell colonies. The efficiency of nucleofection was determined by quantifying GFP+ cells by flow cytometry (Stratedigm S1000EON) and analysis by a Keyence BZ-X700 fluorescence microscope. iPSCs receiving ngcABEvar5 + gRNA were monitored for the formation of single-cell derived clones. Some cells were passaged to single-cell again 6 days post-nucleofection (when most base editing should be completed). Single-cell derived colonies were manually picked, replated, and simultaneously analyzed using NGS for editing efficiency. Approximately half of the colonies assessed underwent single-cell passaging 6 days post nucleofection. Promising clones based on MiSeq analysis were further subcloned to obtain pure populations of MZ and MM edited iPSCs. The editing was again assessed using MiSeq and through Sanger Sequencing (GeneWiz).
Differentiation of iPSCs to iHeps
iPSCs were differentiated to iHeps using previously established protocols.17,23 Differentiating iHeps were maintained in a 5% CO2, 5% O2, 90% N2 environment. iPSCs were dissociated to a single-cell suspension using Gentle Cell and replated at a density of 1 × 106 cells. Twenty-four hours later, cells were differentiated to definitive endoderm for 4 days using the StemDiff Definitive Endoderm Kit (StemCell Technologies, catalog #05110), as per manufacturer’s instructions. On D5 of the protocol, definitive endoderm cells were dissociated with Gentle Cell to determine endoderm efficiency by flow cytometry and replated in hepatocyte differentiation media using stage-specific growth factors for directed differentiation to hepatocytes.17 A detailed protocol for the hepatocyte differentiation is available for free download at: http://www.bu.edu/dbin/stemcells/protocols.php.
Base editing iHeps
In initial experiments, we optimized delivery of mRNA to differentiating iHeps at D13, D15, D16, and D19 of the protocol. GFP mRNA (600 ng) was mixed with Lipofectamine MessengerMax (Thermo Fisher Scientific) in OptiMem (GIBCO) as per manufacturer’s instructions. Approximately 250,000 iHeps per condition were forward transfected by adding mRNA/lipofectamine complex to the cells for 6 h, before replacing with fresh media (appropriate for the day of differentiation). Reverse transfection of iHeps was achieved by incubating the cells with 0.25% trypsin (Life Technologies) for 5–8 min, washing and centrifuging the cells, then resuspending in media with 10 μm Y-27632 and mRNA/lipofectamine complex. These iHeps were then plated on fresh Matrigel (maintaining the same well size as prior to trypsin), and media was replaced 24 h later to remove Y-27632. To base-edit iHeps, cells growing in 12W plates were reverse transfected on D15 with 1,800 ng mRNA ngcABE and 600 ng gRNA with lipofectamine.
Flow cytometry
Cells were stained with cell surface antibodies against the following antigens: CXCR4 (PE-conjugated; Life Technologies, MHCXCR404), cKit (APC-conjugated; Life Technologies, CD11705). For intracellular staining, cells were fixed with 1.6% paraformaldehyde at 37°C for 20 min, then permeabilized in saponin buffer (BioLegend). Intracellular antigens were probed with the following antibodies: AAT (Santa Cruz, sc-59438), AFP (Abcam, ab169552), 2C1 (a kind gift from David Lomas and Elena Miranda), and/or BiP (Invitrogen, PA1-014A), then incubated with goat anti-mouse AF647 (Jackson ImmunoResearch, 115-605-003) and donkey anti-rabbit AF488 (Jackson ImmunoResearch, 711-545-152). To assess proliferation by EdU incorporation, cells were incubated for 24 h with 10 μM EdU, then stained using the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Life Technologies, C10635) as per manufacturer’s instructions. For all flow cytometry experiments, gating was based on isotype-stained controls. Stained cells were quantified using a Stratedigm S1000EON, and data were analyzed using FlowJo (Tree Star).
ELISA
Extracellular AAT in cell culture supernatant was quantified using the human AAT ELISA kit (GenWay Biotech), as per the manufacturer’s instructions. Z-AAT was detected by modifying this protocol to use a Z-specific antibody (a kind gift from Chris Mueller).26
Immunohistochemistry
On the day of base editing, hepatic progenitors were replated in chamber slides (Thermo Fisher Scientific, 177380PK). Upon completion of the differentiation, cells were fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were permeabilized with 0.3% Triton, then blocked with 5% normal donkey serum, before incubation with primary antibodies overnight at 4°C. Antibodies used were HNF4A (Abcam, ab201460), 2C1 (a kind gift from David Lomas and Elena Miranda), and BiP (Invitrogen, PA1-014A). Following incubation, cells were washed and incubated with appropriate secondary antibodies (Invitrogen, A21202, A21247, and A10042). Finally, nuclei were stained with Hoechst 33342 (Invitrogen, H3570). Cells were imaged using a Leica SP5 confocal microscope, and images were processed in ImageJ and Fiji.
scRNA-seq
iHeps were dissociated to single cell at D21 of the differentiation with trypsin, and live cells were sorted using Calcein Blue (Invitrogen, C1429) on a MoFlo Astrios EQ (Beckman Coulter) at the Boston University Flow Cytometry Core Facility (FCCF). MZ and MM cells were pooled in a 1:1 ratio prior to capture. Live cells were captured and libraries prepared as per the 10x Genomics scRNA-seq (V3) protocol. Libraries were sequenced using an Illumina NextSeq 2000 instrument. The sequencing run generated reads with 93% ≥ Q30. The Cell Ranger software pipeline produced the FASTQ and Counts matrix files. The ZZ sample captured 2,082 cells with 61,793 mean reads per cell and 3,957 median genes per cell; the ABE-var5 sample captured 1,633 cells with 78,154 mean reads per cell and 4,590 median genes per cell; the ABE-var9 sample captured 1,655 cells with 71,935 mean reads per cell and 4,654 median genes per cell; the MZ/MM sample captured 1,993 cells with 72,196 mean reads per cell and 4,426 median genes per cell. Seurat (version 3.2.0) was used to further process and analyze data. Data were normalized using the regularized negative binomial regression method with cell degradation (e.g., mitochondrial percentage) regressed out. The Vartrix Single-Cell Genotyping tool (10x Genomics) was applied to assign a genotype of ZZ, MZ, or MM to each cell. For further analyses, cells without a genotype assigned, or an incorrect genotype in known samples (i.e., in the ZZ, MZ, or MM samples) were filtered out. Dimensionality reduction methods was visualized using UMAP. The Louvain method was applied for clustering. Differential gene expression was determined by a log fold change of 0.25 with a Wilcoxon rank-sum test, and GSEA was performed using hypeR. The accession number of the scRNA-seq data reported in this paper is GEO: GSE164417.
Statistics
Statistical analyses for each figure are detailed in the figure legend. Briefly, unpaired two-tailed Student’s t tests were used to compare two groups, or one-way ANOVA with a Tukey multiple-comparisons test was used to compare three or more groups. A p value of < 0.05 was determined to indicate statistical significance, and p value annotations in figures are annotated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Data are represented as mean with error bars representing SD.
Acknowledgments
We thank members of the Wilson laboratory and the Murphy lab at the Center for Regenerative Medicine for helpful discussions. Thanks to Shreya Nakhawa for quantification assistance. We also thank Greg Miller (CReM laboratory manager) and Marianne James (iPSC core manager) for invaluable support. We thank Brian Tilton and the BUMC Flow Cytometry Core for technical assistance with cell sorting (supported by NIH grant 1S10OD021587-01A1) and Yuriy Aleksyeyev, Ashley LeClerc, and the Single Cell Sequencing Core Facility for performing scRNA-seq. This work was supported by a CJ Martin Early Career Fellowship from the Australian National Health and Medical Research Council awarded to R.B.W. and NIH grants U01TR001810 and R01DK101501 and grant support from Beam Therapeutics awarded to A.A.W. The graphical abstract was created using BioRender.com.
Author contributions
R.B.W. and A.A.W. designed experiments; M.S.P., Y.A.-S., and F.G. designed the base editors; R.B.W., J.E.K., and M.S.P performed experiments; R.B.W., J.L.-V., C.V.-M., and L.E.Y. performed bioinformatics analyses; R.B.W. and A.A.W. wrote the manuscript.
Declaration of interests
Support for this work was provided in the form of a research grant from Beam Therapeutics to A.A.W. M.S.P., L.E.Y., Y.A.-S., and F.G. are employed by Beam Therapeutics and contributed to data collection, data analysis, and manuscript preparation.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.021.
Supplemental information
References
- 1.Schultz H.E., Heremans J.F. Elsevier; New York: 1966. Synthesis of the Plasma Proteins; pp. 321–349. [Google Scholar]
- 2.Brantly M., Nukiwa T., Crystal R.G. Molecular basis of alpha-1-antitrypsin deficiency. Am. J. Med. 1988;84(6A):13–31. doi: 10.1016/0002-9343(88)90154-4. [DOI] [PubMed] [Google Scholar]
- 3.Wewers M.D., Casolaro M.A., Sellers S.E., Swayze S.C., McPhaul K.M., Wittes J.T., Crystal R.G. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N. Engl. J. Med. 1987;316:1055–1062. doi: 10.1056/NEJM198704233161704. [DOI] [PubMed] [Google Scholar]
- 4.Brantly M.L., Paul L.D., Miller B.H., Falk R.T., Wu M., Crystal R.G. Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am. Rev. Respir. Dis. 1988;138:327–336. doi: 10.1164/ajrccm/138.2.327. [DOI] [PubMed] [Google Scholar]
- 5.Lomas D.A., Evans D.L., Finch J.T., Carrell R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- 6.Eriksson S., Carlson J., Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N. Engl. J. Med. 1986;314:736–739. doi: 10.1056/NEJM198603203141202. [DOI] [PubMed] [Google Scholar]
- 7.Chapman K.R., Burdon J.G., Piitulainen E., Sandhaus R.A., Seersholm N., Stocks J.M., Stoel B.C., Huang L., Yao Z., Edelman J.M., McElvaney N.G., RAPID Trial Study Group Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386:360–368. doi: 10.1016/S0140-6736(15)60860-1. [DOI] [PubMed] [Google Scholar]
- 8.Brantly M.L., Chulay J.D., Wang L., Mueller C., Humphries M., Spencer L.T., Rouhani F., Conlon T.J., Calcedo R., Betts M.R. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. U S A. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller C., Gernoux G., Gruntman A.M., Borel F., Reeves E.P., Calcedo R., Rouhani F.N., Yachnis A., Humphries M., Campbell-Thompson M. 5 Year expression and neutrophil defect repair after gene therapy in alpha-1 antitrypsin deficiency. Mol. Ther. 2017;25:1387–1394. doi: 10.1016/j.ymthe.2017.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen S., Sanchez M.E., Blomenkamp K., Corcoran E.M., Marco E., Yudkoff C.J., Jiang H., Teckman J.H., Bumcrot D., Albright C.F. Amelioration of alpha-1 antitrypsin deficiency diseases with genome editing in transgenic mice. Hum. Gene Ther. 2018;29:861–873. doi: 10.1089/hum.2017.227. [DOI] [PubMed] [Google Scholar]
- 11.Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordóñez A., Hannan N.R., Rouhani F.J. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaserman J.E., Hurley K., Dodge M., Villacorta-Martin C., Vedaie M., Jean J.C., Liberti D.C., James M.F., Higgins M.I., Lee N.J. A highly phenotyped open access repository of alpha-1 antitrypsin deficiency pluripotent stem cells. Stem Cell Reports. 2020;15:242–255. doi: 10.1016/j.stemcr.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi S.M., Kim Y., Shim J.S., Park J.T., Wang R.H., Leach S.D., Liu J.O., Deng C., Ye Z., Jang Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology. 2013;57:2458–2468. doi: 10.1002/hep.26237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson A.A., Ying L., Liesa M., Segeritz C.P., Mills J.A., Shen S.S., Jean J., Lonza G.C., Liberti D.C., Lang A.H. Emergence of a stage-dependent human liver disease signature with directed differentiation of alpha-1 antitrypsin-deficient iPS cells. Stem Cell Reports. 2015;4:873–885. doi: 10.1016/j.stemcr.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joung K.J., Kleinstiver B. 2019. Engineered CRISPR-Cas9 nucleases with altered pam specificity. US patent US20190106687, filed August 22, 2018, and granted April 11, 2019. [Google Scholar]
- 20.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 21.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laurent L.C., Ulitsky I., Slavin I., Tran H., Schork A., Morey R., Lynch C., Harness J.V., Lee S., Barrero M.J. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng X., Ying L., Lu L., Galvão A.M., Mills J.A., Lin H.C., Kotton D.N., Shen S.S., Nostro M.C., Choi J.K. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell. 2012;10:371–384. doi: 10.1016/j.stem.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy J.M., Yeh W.H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeh W.-H., Chiang H., Rees H.A., Edge A.S.B., Liu D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018;9:2184. doi: 10.1038/s41467-018-04580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borel F., Tang Q., Gernoux G., Greer C., Wang Z., Barzel A., Kay M.A., Shultz L.D., Greiner D.L., Flotte T.R. Survival advantage of both human hepatocyte xenografts and genome-edited hepatocytes for treatment of α-1 antitrypsin deficiency. Mol. Ther. 2017;25:2477–2489. doi: 10.1016/j.ymthe.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miranda E., Pérez J., Ekeowa U.I., Hadzic N., Kalsheker N., Gooptu B., Portmann B., Belorgey D., Hill M., Chambers S. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology. 2010;52:1078–1088. doi: 10.1002/hep.23760. [DOI] [PubMed] [Google Scholar]
- 28.Ordóñez A., Snapp E.L., Tan L., Miranda E., Marciniak S.J., Lomas D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology. 2013;57:2049–2060. doi: 10.1002/hep.26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carroll T.P., Greene C.M., O’Connor C.A., Nolan A.M., O’Neill S.J., McElvaney N.G. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J. Immunol. 2010;184:4538–4546. doi: 10.4049/jimmunol.0802864. [DOI] [PubMed] [Google Scholar]
- 30.Segeritz C.P., Rashid S.T., de Brito M.C., Serra M.P., Ordonez A., Morell C.M., Kaserman J.E., Madrigal P., Hannan N.R.F., Gatto L. hiPSC hepatocyte model demonstrates the role of unfolded protein response and inflammatory networks in α1-antitrypsin deficiency. J. Hepatol. 2018;69:851–860. doi: 10.1016/j.jhep.2018.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hidvegi T., Schmidt B.Z., Hale P., Perlmutter D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005;280:39002–39015. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 32.Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L., Wang J., Liu Y., Xie L., Su B., Mou D., Wang L., Liu T., Wang X., Zhang B. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N. Engl. J. Med. 2019;381:1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 34.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 35.Orthwein A., Noordermeer S.M., Wilson M.D., Landry S., Enchev R.I., Sherker A., Munro M., Pinder J., Salsman J., Dellaire G. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528:422–426. doi: 10.1038/nature16142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grisham J.W. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res. 1962;22:842–849. [PubMed] [Google Scholar]
- 37.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H.K., Smith H.E., Liu C., Willi M., Hennighausen L. Cytosine base editor 4 but not adenine base editor generates off-target mutations in mouse embryos. Commun. Biol. 2020;3:19. doi: 10.1038/s42003-019-0745-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amps K., Andrews P.W., Anyfantis G., Armstrong L., Avery S., Baharvand H., Baker J., Baker D., Munoz M.B., Beil S., International Stem Cell Initiative Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji J., Ng S.H., Sharma V., Neculai D., Hussein S., Sam M., Trinh Q., Church G.M., McPherson J.D., Nagy A., Batada N.N. Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells. 2012;30:435–440. doi: 10.1002/stem.1011. [DOI] [PubMed] [Google Scholar]
- 41.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 43.Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L. Calling somatic SNVs and indels with Mutect2. bioRxiv. 2019 doi: 10.1101/861054. [DOI] [Google Scholar]
- 44.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilm A., Aw P.P., Bertrand D., Yeo G.H., Ong S.H., Wong C.H., Khor C.C., Petric R., Hibberd M.L., Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koboldt D.C., Larson D.E., Wilson R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinform. 2013;44:15.14.11–15.14.17. doi: 10.1002/0471250953.bi1504s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.